

Abstract
The mechanism of direct displacement of alkoxy groups in vinylogous and aromatic esters by Grignard reagents, a reaction that is not observed with expectedly better tosyloxy leaving groups, is elucidated computationally. The mechanism of this reaction has been determined to proceed through the inner-sphere attack of nucleophilic alkyl groups from magnesium to the reacting carbons via a metalaoxetane transition state. The formation of a strong magnesium chelate with the reacting alkoxy and carbonyl groups dictates the observed reactivity and selectivity. The influence of ester, ketone and aldehyde substituents was investigated. In some cases, the calculations predicted the formation of products different than those previously reported; these predictions were then verified experimentally. The importance of studying the actual system, and not simplified models as computational systems, is demonstrated.
Introduction
The substitution of alkoxy groups on aromatics (SNAr) activated by ortho-carbonyl substitution1 has been known for a number of early examples in the literature,2–21 but this reaction has been shown to be quite general in recent studies by our group. A general example with 1-methoxynaphthalenes is shown in Scheme 1a. Related reactions are found in non-aromatic systems (Scheme 1b). While the reactions shown are tolerant to other alkoxy groups, the normally better leaving group tosyloxy is not displaced. In addition, while many Grignard reagents are effective, organolithium and organozinc reagents give poor yields at best. We report a computational study of this reaction that provides a thorough mechanistic rationale of these facts. Furthermore, in some cases, theory predicts the formation of different products from those originally assigned, and these predictions have been verified experimentally.
Scheme 1.

Displacement of alkoxy groups by Grignard reagents in aromatic (a) and non-aromatic (b) substrates.
Results and Discussion
Reaction mechanism
While metallic chelates of 1,3 and 1,4-alkoxycarbonyl compounds have been detected by NMR spectroscopy22–24 we were unable to identify these complexes in solution using magnesium halides as chelating agents, due to their insolubility in the deuterated reaction solvents. Instead, we have performed a computational study of all conceivable bidentate complexation modes of CH3MgCl to methyl 1-methoxy-2-naphthoate 1a. Although most experiments were performed with ethyl or larger Grignards, our initial calculations involved methyl Grignard reagents for simplicity. The well-known μ-dichloro dimer form of Grignard reagent25–28 including two solvent molecules (dimethyl ether, DME, in our model) was selected as the starting material, and several dimer and monomer isomers of the reactant complex, denoted as 1a•CH3MgCl (a-m), were calculated. Figure 1 shows the geometries and formation energies of the most significant structures for these complexes (see Figure S1 in Supporting Information for further details). The Schlenk equilibrium 29 (i.e. formation of dialkyl magnesium compounds and magnesium halide salts) was not considered to take place significantly under the experimental conditions. The monomeric magnesium chelate 1a•CH3MgCl (a) formed after scission of two Mg–Cl bonds upon substrate coordination was by far the most stable complex in solution (ΔGcomplex = −7.2 kcal mol−1), whereas the formation of complexes with Grignard dimer species was always endergonic (ΔGcomplex = +2.4 to +10.9 kcal mol−1). This complex was considered the reference minimum for the estimation of the free energy barriers calculated here.
Figure 1.

a) Proposed coordination of Grignard reagents to vinylogous 1,2-alkoxy carbonyl compounds leading to 1:1 and 2:1 complexes (eq. 1 and eq. 2, respectively). Additional coordination modes are described in the Supporting Information. b) Structures and complexation free energies (ΔGcomplex) calculated at the PCM(CH2Cl2)/B3LYP/6-31G(d) level. Energies are in kcal mol−1 and distances are in Angstrom.
The classical mechanism of Grignard reagents addition to carbonyl compounds, especially ketones, was first proposed by Ashby.25–28,30 This mechanism involves the participation of a μ-chloro organomagnesium dimer in which one Mg is coordinated to the carbonyl oxygen, and the other Mg delivers the alkyl or aryl nucleophile to the carbonyl carbon through a concerted six-membered cyclic transition state (TS) (Figure 2). When we calculated this mechanism starting from complex 1a•CH3MgCl (d), it was first necessary to cleave one Mg–Cl bond and coordinate one ether molecule to provide a reasonable coordination environment for the Mg transferring the nucleophile. This rearrangement barely stabilized the new reactant complex 1a•CH3MgCl(DME) (d') by 0.4 kcal mol−1 over the unsolvated μ-dichloro species. In the proposed six-membered TS geometry, both for the addition to the carbonyl or the alkoxy displacement, the alkyl group suffers pyramidal inversion at the reacting carbon during the C–C bond formation, at a significant energy cost. Accordingly, quite high energy barriers for the addition to ipso position in the naphthalene ring and 1,2-addition to the ester carbonyl were calculated (ΔG‡ OMe = +32.7 for 1a-TS'OMe and ΔG‡ C=O = +28.2 kcal mol−1 for 1a-TS'C=O, respectively). Moreover, the favored addition to the carbonyl group by 4.5 kcal mol−1 completely disagrees with the experimental observations.
Figure 2.

a) Proposed mechanisms for the reaction of vinylogous 1,2-alkoxy carbonyl compounds with Grignard reagents through six-membered (TS') and four-membered (TS) cyclic transition states. b) Most relevant structures and activation free energies (ΔG‡) calculated at the PCM(CH2Cl2)/B3LYP/6-31G(d) level. Energies are in kcal mol−1 and distances are in Angstrom.
These results prompted us to investigate alternative pathways for the studied reactions. Looking at the structure of the most stable magnesium chelate 1a•CH3MgCl (a), in which the nucleophilic alkyl group is located proximal to both reacting carbons at the methoxy and ester groups (3.85 and 4.04 Å respectively), we envisaged a direct transfer of the methyl group to either the carbonyl or ipso aromatic carbons through a four-membered metalaoxetane TS (Figure 2). Despite the apparently more strained arrangement of these TS with respect to the aforementioned six-membered TS, the alkyl group is transferred from the Grignard reagent without pyramidal inversion and with minimal rehybridization of the reacting carbon. This smaller distortion of the reactants in the TS provides a significantly lower activation barrier for the both alkyl addition processes (ΔG‡ OMe = +25.2 for 1a-TSOMe and ΔG‡ C=O = +24.8 kcal mol−1 for 1a-TSC=O, respectively). More importantly, both competitive pathways get much closer to the observed preferential displacement of the methoxy group leading to compound 2a.
In order to test the validity of our model, the reaction of ester 1a with CH3MgCl was assayed experimentally (Scheme 2). In agreement with our calculations, this reaction yields both 1,2- and 1,4-conjugate addition products together with a small amount of hydroxylated compound 18. The intermediate methyl ketone was not observed and tertiary alcohol 15a was obtained as the minor reaction product. Despite the major proportion of 2a in the reaction mixture, 2 equiv. of Grignard reagent are consumed in the two successive 1,2-additions per equiv. consumed in the 1,4-conjugate addition; this limits the proportion of tertiary alcohol in the final mixture when stoichiometric amounts of CH3MgCl are used and suggests that the 1,2-addition to the carbonyl group must be competitive with 1,4-addition’. Hence, the formation of tertiary alcohol 15a with methyl Grignard reagents, not reported previously for methoxynaphthyl esters, was successfully predicted by our calculations and demonstrated experimentally.
Scheme 2.

Reaction of aromatic β-alkoxy esters with methyl Grignard reagents.
The influence of the nature of the Grignard reagent on the rate-limiting step of the reaction was investigated computationally.
In good agreement with experimental results1 (Scheme 1), the simple substitution of MeMgCl (yielding 2a) by EtMgBr as a closer model to n-BuMgCl (yielding 2b) had a quite beneficial effect on both the calculated reactivity and chemoselectivity, providing lower activation energies and shifting to a 98:2 ratio towards methoxy group displacement (lowest energy barriers: ΔG‡ OMe = +22.1 for 1a-TSOMe and ΔG‡ C=O = +23.7 kcal mol−1 for 1a-TSC=O, respectively). We decided to use EtMgCl as a more appropriate Grignard reagent model henceforth, since the majority of experiments were conducted using primary nucleophiles such as n-BuMgCl and i-BuMgCl.
In order to determine the rate-limiting step of each competing reaction, the whole mechanisms were calculated for both processes. As depicted in Figure 3, both reactions follow stepwise mechanisms in which the initial addition to the carbonyl or alkoxy groups is clearly the rate-limiting step. This step is moderately exergonic in both cases and leads to quite stable but very reactive tetrahedral intermediates.
Figure 3.

Reaction pathways for the addition of Grignard reagents to aromatic 1,2-alkoxy esters calculated at the PCM(CH2Cl2)/B3LYP/6-31G(d) level. Free energies (ΔG) relative to the reactant complex are in kcal mol−1.
Such intermediates derived from 1,2- and 1,4-conjugate addition can be visualized as magnesium alkoxides and enolates, respectively. These transient species evolve through an almost barrierless second step to cleave the methoxy leaving group and recover either naphthalene aromaticity or the carbonyl sp2 hybridization. The restoration of π-delocalization along the aromatic and carbonyl systems, and the formation of covalent Mg-alkoxy species after leaving group departure are the driving forces for such hyper fast and highly exergonic processes. Interestingly, the formation of a tetrahedral intermediate at the ipso carbonyl of naphthalene does not disrupt the aromaticity and π-conjugation with the ortho carbonyl group significantly, as demonstrated by the slight exergonicity of the first reaction step. As will be discussed below, this is not the case with other position isomers of this substrate.
In our previous study,1 we reported that non-aromatic vinylogous esters such us Z-ethyl 3-methoxycinnamate 3 and methyl 1-methoxy-3,4-dihydro-2-naphthoate 5 (Scheme 3) readily react with Grignard reagents in a similar manner to the naphthalene counterparts, namely with complete selectivity towards alkoxy group displacement. However, treatment of the corresponding 1H-indene analogue 7 with 1 equiv of i-PrMgCl only yielded starting unreacted starting material after extractive workup, hence we decided to shed some light onto this behavior computationally. Our calculations predicted a complete selectivity towards methoxy group substitution and activation barriers similar to those obtained for the analogous 1-methoxy-2-naphthoate (lowest energy barriers: ΔG‡ OMe = +19.9 for 7-TSOMe and ΔG‡ C=O = +27.1 kcal mol−1 for 7-TSC=O, respectively).
Scheme 3.

Reactivity of non-aromatic vinylogous esters with Grignard reagents.
Based on previous reports,31 we envisaged the possible in situ formation of indenylmagnesium bromide32 or bis(indenyl)magnesium33,34 as the reason for this lack of reactivity with stoichiometric amount of Grignard reagent (the pKa of 1H-indene is 20.1 in dimethylsulfoxide35). According to this hypothesis and our predicted reactivity, we were glad to demonstrate that the treatment of 7 with 3 equiv of i-PrMgCl cleanly gave displacement of the methoxy group with no detectable attack to the carbonyl group in excellent yield (90%, Scheme 3). Interestingly, the activation barriers for the same reactions starting from the deprotonated and fully aromatic indenyl anion 7', are exceedingly high to be overcome at the reaction temperature (lowest energy barriers: ΔG‡ OMe = +44.5 for 7'-TSOMe and ΔG‡ C=O = +33.9 kcal mol−1 for 7'-TSC=O, respectively), pointing to the remaining protonated 1Hindene in solution as the reactive species in this reaction.
The source of such a great difference between the two addition pathways in the 1H-indene derivative 7, lies on the interruption of π-delocalization along the well-defined α,β-unsaturated system when the addition takes place to the carbonyl group, an issue that is alleviated in much more delocalized systems such as naphthalenes. The greater conjugation with respect to reactants achieved in the ipso-disubstituted adduct makes the 1,4-addition step more exergonic (ΔG = −14.7 kcal mol−1) and the 1,2-addition less exergonic (ΔG = −10.0 kcal mol−1) compared to the aromatic ester 1a (ΔG = −7.3 and −12.7 kcal mol−1, respectively).
As a general trend observed here, the lower energy TS for methoxy group displacement are earlier (i.e. close in geometry and energy to the reactants), in good agreement with Hammond’s postulate36 for exergonic processes. Thus, the COMe–CEt forming bond distance in the more stable 7-TSOMe derived from 1H-indene is longer (2.40 Å) than in the naphthalene-derived 1a-TSOMe (2.30 Å) whereas the CC=O–CEt forming bond distance is slightly shorter in less stable in 7-TSC=O than in 1a-TSC=O (2.26 and 2.27 Å, respectively).
Functional groups effects
The influence of both the nature and position of the reacting groups was demonstrated in our previous report.1 Thus, a simple change in the location of the alkoxy group to provide other ortho isomer, namely methyl 3-methoxy-2-naphthoate 9, disrupted its reactivity as demonstrated by competition experiments. In the course of our computational investigations to rationalize this reactivity, we found that although the methoxy displacement pathway was clearly disfavored using EtMgCl as Grignard reagent model (lowest energy barrier: ΔG‡ OMe = +30.8 for 9-TSOMe), the reactivity at the carbonyl group remained essentially unchanged (lowest energy barrier: ΔG‡ OMe = +22.1 for 9-TSC=O), providing a complete chemoselectivity towards 1,2-addition.
The change of primary EtMgGl by secondary i-PrMgCl, which was the Grignard reagent used experimentally, did not change this trend (lowest energy barriers: ΔG‡ OMe = +30.4 for 9'-TSOMe and ΔG‡ C=O = +22.4 kcal mol−1 for 9'-TSC=O, respectively). These theoretical results were not in accordance with the reported reactivity for this substrate, since a slower but still complete chemoselectivity towards methoxy group displacement was previously published.1 Intrigued by these observations, we repeated the same reactions and re-examined the NMR data of the isolated compounds carefully. Thereafter, we concluded that our previous assessment of reactivity for this compound was wrong, and the ester carbonyl group indeed reacts exclusively to provide the corresponding isopropyl ketone 10 in good yield (85%, Scheme 4), which did not undergo further 1,2-addition.
Scheme 4.

Structural revision of the reaction product between of naphthyl ester 9 and Grignard reagents. As predicted computationally, 1,2-addition to the carbonyl group takes place exclusively.
These new results predicted by the computations gave rise to a correction of the original manuscript and stress the great benefits of integrating experiments and calculations.
The reduced reactivity of the methoxy group at the 3 position in compound 9 is again reflected by the late character (structurally close to the product) of the corresponding 1,4-addition TS illustrated by the short COMe– Calkyl forming bond distance (2.17 Å with EtMgCl and 2.23 Å with i-PrMgCl, Figure 4). Moreover, this methoxy group displacement proceeded in an asynchronous but concerted manner without the formation of a stable tetrahedral intermediate, as revealed by IRC calculations. The origin of this lower reactivity is the poorer ability of 3-methoxy-2-naphthoates to stabilize the formal negative charge generated after 1,4-addition of the alkyl group with respect to 1-methoxy-2-naphthoates (Figure S2 in Supporting Information). The lack of resonance structures preserving the aromaticity of one fused benzene ring precludes the formation of stable adducts after 1,4-addition and drives the concerted elimination of the methoxy group in order to recover naphthalene aromaticity. Additionally, and as can be seen in Figure 4, the contribution of the ipso carbon bearing the methoxy group to the LUMO of 3-methoxy-2-naphthoate 9 is almost negligible in opposition to 1-methoxy isomer 1a, justifying the complete selectivity towards 1,2-carbonyl addition with Grignard reagents of the former.
Figure 4.

a) Lowest-energy transition structures and activation free energies (ΔG‡) for the reaction of naphthyl ester 9 and EtMgCl calculated at the PCM(CH2Cl2)/B3LYP/6-31G(d) level. Energies are in kcal mol−1 and distances are in Angstrom. b) Isosurface representation of LUMO of reactant complexes of isomers 1a and 9. Potentially reactive positions are marked with red (methoxy group) and blue (carbonyl group) arrows.
As described in our previous study,1 the presence of a theoretically better leaving group than alkoxide (such as tosylate) completely inhibits the displacement reaction at low temperature, even with primary nucleophiles like n-BuMgCl and i-BuMgCl. This apparently counterintuitive result was rationalized computationally by a higher calculated activation barrier for mesylate (simpler model of tosylate) displacement using EtMgCl as Grignard model (lowest energy barrier: ΔG‡ OMe = +25.6 for 11-TSOMe) with respect to methoxy substitution in analogous compounds. Reversing the trend calculated for similar alkoxy derivatives, the nucleophilic addition of the same reactant to the carbonyl group is now ca. 4 kcal mol−1 lower in energy (lowest energy barrier: ΔG‡ C=O = +21.8 for 11-TSC=O), which provides complete chemoselectivity towards this pathway. To our delight, when the reaction mixture was allowed to reach room temperature overnight, only addition to the ester carbonyl group of 11 towards the corresponding tertiary alcohol 12 was observed, as predicted by our calculations, albeit with low conversion and in low yield (20%, Scheme 5).
Scheme 5.

Reactivity of (tosyl)naphthyl esters with Grignard reagents.
The decreased propensity of tosylate group to be displaced lies in its inability to form a stable six-membered chelate involving the oxygen attached to the ipso carbon (11•EtMgCl_a) as in the methoxy-substituted analogues. Instead, the carbonyl oxygen and a sulfonyl oxygen form an eight-membered chelate (11•EtMgCl_b), which promotes the activation of only the ester group towards nucleophilic addition (Figure 5). Due to the weaker coordinating character of sulfonate esters compared to ethers, these chelates are significantly less stable (ΔGcomplex = +7.5 and −0.2 kcal mol−1, respectively) than the 1-methoxysubstitued analogue.
Figure 5.

a) Six-membered (left) and eight-membered (right) chelate reactant complexes of (mesyl)naphthyl ester 11 with EtMgCl together with their respective complexation free energies (ΔGcomplex). b) Lowest-energy transition structures and activation free energies (ΔG‡) for the reaction of 11 and EtMgCl calculated at the PCM(CH2Cl2)/B3LYP/6-31G(d) level.
Although ketones are generally viewed as incompatible with the high nucleophilicity of Grignard reagents, we have shown that the displacement reaction is facile at temperatures below which the ester or ketone products will undergo addition (Table 1, entries 2–5)1. As an exception, MeMgI in CH2Cl2 undergoes complete addition to the carbonyl group as previously reported31 and confirmed in our lab (Table 1, entry 1). In this sense, computations on the reaction of methylketone 13a with EtMgCl reproduced this trend quantitatively by estimating a 77:23 ratio favoring the displacement of the methoxy group with EtMgCl (lowest energy barriers: ΔG‡ OMe = +19.0 for 13a-TSOMe and ΔG‡ C=O = +19.5 kcal mol−1 for 13a-TSC=O, respectively). Interestingly, the simple change of methyl group in the ketone by a n-butyl group in 13b, produces an increase in the calculated selectivity, providing a calculated ratio of 95:5 towards 1,4-conjugate addition, in very good agreement with experimental observations (lowest energy barriers: ΔG‡ OMe = +19.6 for 13b-TSOMe and ΔG‡ C=O = +20.5 kcal mol−1 for 13b-TSC=O, respectively). Moreover, and somewhat unexpectedly, the computations on the reaction of methyl ketone 13a with a slightly harder nucleophile such as MeMgCl also reproduced the experimentally observed inversion of chemoselectivity, providing a calculated 9:91 ratio towards the formation of the corresponding tertiary alcohol (lowest energy barriers: ΔG‡ OMe = +22.0 for 13a-TS'OMe and ΔG‡ C=O = +21.1 kcal mol−1 for 13a-TS'C=O, respectively).
Table 1.
Comparison of carbonyl reactivity toward alkoxide displacement.
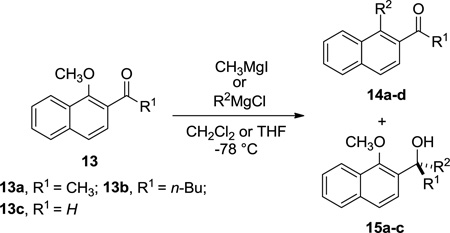 | |||||
|---|---|---|---|---|---|
| Entry | Substrate (R1) |
R2 | Ratio of 14 : 15 | Isolated yield, % (product) |
|
| Predict.a | Exp.b | ||||
| 1 | 13a | Me | 8:91 | <5:>95 | 35 (15a) |
| 2 | 13a | n-Bu | 77:23 | 73:27 | 66 (14a) |
| 3 | 13a | i-Pr | – | >95:<5 | 61 (14c) c |
| 4 | 13b | n-Bu | 95:5 | >95:<5 | 85 (14b) |
| 5 | 13b | i-Pr | – | >95:<5 | 90 (14d) |
| 6 | 13c | n-Bu | 3:97 | <5:>95 | 85 (15c) |
Calculated through a Maxwell–Boltzmann distribution at −78 °C of all computed TS.
Determined by 1H NMR in the reaction mixture.
The corresponding secondary alcohol, resulting from carbonyl reduction, was isolated in 20% yield.
As mentioned before, the more stable TS´s for 1,4-addition on 1-methoxy-2-naphthyl methyl ketone 13a are earlier as reflected by the COMe–CEt forming bond distances (2.38 Å and 2.28 Å with EtMgCl and MeMgCl, respectively).
These results stress the great importance of properly choosing the substrate models for the calculations, because very similar EtMgCl and MeMgCl provide very different reactivity patterns.
The reaction with more reactive carbonyls such as aldehydes completely changes the general selectivity trend observed with less electrophilic vinylogous esters and ketones. According to the experimental results (Table 1, entry 6),1 the chemoselectivity calculated for the addition of EtMgCl (computational model n-BuMgCl) to 1-methoxy-2-naphthaldehyde 13c is reversed with respect to analogous ester 1a with a calculated ratio of 2:98 favoring the attack towards the carbonyl group. As can be seen in Figure 7, the contribution of the carbonyl carbon to the LUMO is larger in naphthyl aldehyde 13c than in the analogous methyl ester 1a, justifying the higher selectivity towards 1,2 carbonyl addition with Grignard reagents of the former. It is noteworthy that the activation energies for both competitive pathways decrease significantly in the case of aromatic aldehydes (lowest energy barriers: ΔG‡ OMe = +16.8 kcal mol−1 for 13c-TSOMe and ΔG‡ C=O +15.4 kcal mol−1 for 13c-TSC=O, respectively). Interestingly, this decrease in the activation barriers matches the lower stability of the reactant complex with weaker coordinating aldehydes (13c•EtMgCl, ΔGcomplex = +0.1 kcal mol−1), which are essentially thermoneutral and 6–7 kcal mol−1 less stable that those of the corresponding vinylogous esters and ketones. The TS’s for both nucleophilic additions in 13c, are the earliest of all calculated species, exhibiting the largest COMe–CEt and CC=O–CEt forming bonds distances (2.42 Å and 2.55 Å, respectively).
Figure 7.

a) Lowest-energy transition structures and activation free energies (ΔG‡) for the reaction of nahphyl aldehyde 13c and EtMgCl calculated at the PCM(CH2Cl2)/B3LYP/6-31G(d) level. Energies are in kcal mol−1 and distances are in Angstrom. b) Isosurface representation of LUMO of reactant complex derived from ester 1a and aldehyde 13c. Potentially reactive positions are marked with red (methoxy group) and blue (carbonyl group) arrows.
The role of magnesium chelate
In previously reported reactions similar to those described herein, the formation of stable transient chelate complexes with organoalkyl reagents prior to nucleophilic akyl transfer has been proposed.22–24
The key role of magnesium in this type of reactivity is reflected by the numerous examples reported, for instance, for the conversion of esters into ketones in the presence of alkoxy groups susceptible of displacement.37–41
In our calculations, satisfactory activation barriers and chemoselectivities were obtained from quite stable complexes between alkyl Grignards and bidentate ortho-methoxycarbonyl species. To further investigate the role of these complexes in the reaction outcome, we calculated the same reaction profiles starting from alternative monodentate structures with CH3MgCl, adding a solvent molecule (DME) to achieve a comparable coordination environment around magnesium and prevent chelation (Figure 8a). Any attempt to locate methoxy-coordinated non-chelated structures was unsuccessful even if an alternate ester rotamer was considered. Notably, the reactant complex in which the magnesium adopts a pentacoordinated environment by coordinating to both the methoxy and carboxyl oxygen atoms, is significantly less stable than the tetrahedral monodentante complex with the carbonyl group (ΔGcomplex = +2.9 kcal mol−1 for 1’a•CH3MgCl(DME)), which in turn is much less stable than the Mg-carbonyl-ether chelated species discussed above. The energy barrier for methoxy displacement increases by ca. 13 kcal mol−1 with respect to the unsolvated chelate, but the activation energy for 1,2 addition to the carbonyl group remains essentially unperturbed.
Figure 8.

a) Lowest-energy reactant complexes and transition structures for the reaction of nahphyl ester 1a and CH3MgCl(DME). b) Lowest-energy reactant complexes and transition structures for the reaction of 1-methoxynaphthalene 16 and methyl 2-naphthoate 17 with CH3MgCl(DME). Complexation (ΔG) and activation free energies (ΔG‡) were calculated at the PCM(CH2Cl2)/B3LYP/6-31G(d) level and are related to the isolated reactants. Energies are in kcal mol−1 and distances are in Angstrom.
Finally, in an attempt to completely dissect the contribution of the methoxy and ester groups to the observed reactivity, we calculated the energy profiles starting from monosubstituted 1-methoxynaphthalene 16 and methyl 2-naphthoate 17, including again a solvent molecule to provide tetracoordination for magnesium (Figure 8b). The monodentate complexes of these species with CH3MgCl resulted to be quite unstable compared to the chelated ones, specially the methoxy-derived species (ΔGcomplex = +14.4 kcal mol−1 for 16•CH3MgCl(DME) and ΔGcomplex = +2.6 kcal mol−1 for 17•CH3MgCl(DME)). Moreover, the activation barrier for methoxy group displacement is totally unfeasible (ΔG‡ OMe = +55.9 for 16-TSOMe (DME)), and virtually identical to those previously calculated for 1,2 ester carbonyl attack (ΔG‡ C=O = +25.1 for 17-TSC=O (DME)).
Altogether, these results clearly indicate that the presence of a carbonyl group is required to achieve alkoxy group displacement with Grignard reagents, as ethers themselves are not able to efficiently coordinate to the Lewis acid. The presence of a chelate complex does not appear to be mandatory for carbonyl activation, whereas the methoxy group is completely unreactive in the absence of the chelate even if magnesium is (poorly) coordinated to the alkoxy group oxygen. The true 1,4-conjugate addition character of the initial steps of methoxy group displacement is revealed through these results.
Thus, the presence of an acyl group in ortho position of the aromatic system allows both the Lewis acid-enhanced activation of the β position of the α,β-unsaturated carbonyl system, and the stabilization of the negative charge generated upon alkyl addition in the form of magnesium enolates, which ultimately triggers the displacement reaction.
Conclusions
An inner-sphere mechanism through metalaoxetane-like TS has been determined computationally for the displacement of alkoxy groups of vinylogous esters and ketones by Grignard reagents. The formation of a stable organomagnesium chelate between both oxygenated groups allows this reaction to take place, which is otherwise inaccessible, even in the presence of expectedly better leaving groups such as tosylate. The calculated chemoselectivities towards 1,4-conjugate (with esters and ketones) or 1,2-addition (with aldehydes) are in excellent agreement with the experimental results and rationalize the influence of the nature and position of both the leaving alkoxy and adjacent carbonyl groups. The reasons behind the different reactivities are the modification of the substrate LUMO by the ortho carbonyl group, and the ability of the substrates to stabilize the negative charge by π-delocalization after nucleophilic addition. Our computational predictions were verified experimentally, leading in some cases to the reinterpretation of previous experimental results as in the reaction of two position isomers of the same methyl alkoxynaphthanoate.
Experimental Section
Computational Details
All calculations were carried out with the B3LYP hybrid functional42,43 and 6-31G(d) basis set. Full geometry optimizations and transition structure (TS) searches were carried out with the Gaussian 09 package.44 The possibility of different conformations was taken into account for all structures. The theoretical ratio of reaction products was obtained through the energy of the different transition states using a Maxwell–Boltzmann distribution at −78 °C, temperature at which thermal and entropic corrections to energy were calculated. Frequency analyses were carried out at the same level used in the geometry optimizations, and the nature of the stationary points was determined in each case according to the appropriate number of negative eigenvalues of the Hessian matrix. The harmonic oscillator approximation in the calculation of vibration frequencies was replaced by the quasiharmonic approximation developed by Cramer and Truhlar.45 Scaled frequencies were not considered since significant errors in the calculated thermodynamic properties are not found at this theoretical level.46,47 Where necessary, mass-weighted intrinsic reaction coordinate (IRC) calculations were carried out by using the Gonzalez and Schlegel scheme48,49 in order to ensure that the TSs indeed connected the appropriate reactants and products. Bulk solvent effects were considered implicitly by performing single-point energy calculations on the gas-phase optimized geometries, through the SMD polarizable continuum model of Cramer and Thrular50 as implemented in Gaussian 09. The internally stored parameters for tetrahydrofuran were used to calculate solvation free energies (ΔGsolv). Gibbs free energies (ΔG) were used for the discussion on the relative stabilities of the considered structures. Cartesian coordinates, electronic energies, entropies, enthalpies, Gibbs free energies, lowest frequencies of the different conformations of all structures considered are available as Supporting Information.
Materials
Unless otherwise specified, all commercially available reagents were used as received. All reactions using dried solvents were carried out under an atmosphere of argon in flame-dried glassware with magnetic stirring. Dry solvent was dispensed from a solvent purification system that passes solvent through two columns of dry neutral alumina. Silica gel chromatographic purifications were performed by flash chromatography with silica gel (sigma, grade 62, 60–200 mesh) packed in glass columns; the eluting solvent for each purification was determined by thin layer chromatography (TLC). Analytical TLC was performed on glass plates coated with 0.25 mm silica gel using UV for visualization.
Instrumentation
1H NMR spectra were obtained on a 300 MHz, 400 MHz or a 600 MHz NMR spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) relative to residual solvent (CHCl3, s, δ 7.26). Multiplicities are given as: s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), sep (septuplet), m (multiplet). Proton-decoupled 13C NMR spectra were obtained on a 75 MHz, 101 MHz, or a 150 MHz NMR spectrometer. 13C chemical shifts are reported relative to CDCl3 (δ 77.2 ppm). IR frequencies are given in cm−1 and spectra were obtained on a FT-IR spectrometer equipped with a DTGS detector and diamond ATR accessory. High-resolution mass spectra were obtained on an Orbitrap FTMS.
Synthesis of methyl 1-methyl-2-naphthoate (2a)
To a cooled solution (−78 °C) of 1a (1.55 g, 7.19 mmol) in 36 mL of CH2Cl2 was added a solution of CH3MgI in Et2O (3.0 M, 2.4 mL). The reaction was monitored until complete by TLC and quenched with saturated ammonium chloride (5 mL). The resulting layers were separated, and the aqueous layer was extracted with 2×10 mL of EtOAc. The combined organic layers were washed with 20 mL of brine, dried (MgSO4), and concentrated in vacuo to afford the crude product mixture. Integration of the crude NMR indicated a 83:17 ratio of 2a to 15a. The product was purified by flash chromatography (5:95 EtOAc/hexanes) to afford the product 2a as a white solid (0.72 g, 50%): m.p. = 55-56 °C; 1H NMR (600 MHz, CDCl3) δ 8.19 (d, J = 9.0 Hz, 1H), 7.84 (m, 2H), 7.72 (d, J = 8.5 Hz, 1H), 7.57 (m, 2H), 3.97 (s, 3H), 2.95 (s, 3H); 13C NMR (151 MHz, CDCl3) 169.2, 137.1, 134.6, 132.8, 128.5, 127.8, 127.3, 126.5, 126.1, 125.8, 125.3, 52.1, 15.8; HRMS (ammonia CI) m/z calcd for C13H16NO2 (M + NH4)+ 218.1181, found 218.1183; IR (thin film) 3090, 2954, 1685 and demethylated byproduct methyl 1-hydroxy-2-naphthoate 18 (0.07 g, 5%) 1H NMR values matched those previously reported. The tertiary alcohol 15a was unable to be chromatographically separated from a small amount of residual starting material. An analytically pure sample used a reference for integration of the crude NMR was prepared from the corresponding ketone 13a.
Synthesis of methyl 3-isopropyl-1H-indene-2-carboxylate (8)
To a cooled solution (−78 °C) of 7 (1.14 g, 5.58 mmol) in 28 mL of THF was added a solution of i-PrMgCl in Et2O (2.0 M, 8.37 mL) The reaction was monitored until complete by TLC and quenched with saturated ammonium chloride (5 mL). The resulting layers were separated, and the aqueous layer was extracted with 2×10 mL of EtOAc. The combined organic layers were washed with 20 mL of brine, dried (MgSO4), and concentrated in vacuo to afford the crude displacement product. The product was purified by flash chromatography (5:95 EtOAc/hexanes) to afford the product 8 as a yellow solid (1.08 g , 90%): m.p. = 79-80 °C; 1H NMR (600 MHz, CDCl3) δ 7.78 (m, 1H), 7.50 (m, 1H), 7.33 (m, 2H), 4.31(m, 1H), 3.84 (s, 3H), 3.66 (s, 2H), 1.45 (d, J = 7.2 Hz, 6H); 13C NMR (151 MHz, CDCl3) δ 166.3, 161.2, 144.4, 143.0, 128.3, 127.2, 126.1, 124.4, 123.7, 51.2, 39.2, 26.7, 20.9; HRMS (ammonia CI) m/z calcd for C14H17O2 (M + H)+ 217.1223, found 217.1233; IR (thin film) 3075, 2975, 1710.
Synthesis of 2-(4-hydroxy-2,6-dimethylheptan-4-yl)naphthalen-1-yl 4-methylbenzenesulfonate (12)
To a cooled solution (−78 °C) of tosylate 11 (0.23 g, 0.63 mmol) in 3.15 mL of CH2Cl2 was added a solution of i-BuMgCl in Et2O (2.0 M, 0.32 mL) The reaction was then stirred for 18 h during which time the solution was allowed to warm to room temperature. The reaction was quenched with saturated ammonium chloride (10 mL). The resulting layers were separated, and the aqueous layer was extracted with 2×10 mL of CH2Cl2. The combined organic layers were washed with 20 mL of brine, dried (MgSO4), and concentrated in vacuo to afford the crude product. The product was purified by flash chromatography (5:95 EtOAc/hexanes) to afford the product 12 as a colorless oil (0.056 g, 20%). 1H NMR (600 MHz, CDCl3) δ 7.98 (d, J = 8.3 Hz, 2H), 7.80 – 7.76 (m, 2H), 7.72 (d, J = 8.8 Hz, 1H), 7.53 (d, J = 8.7 Hz, 1H), 7.44 (ddd, J = 8.1, 6.8, 1.1 Hz, 1H), 7.41 (d, J = 8.1 Hz, 2H), 7.34 (ddd, J = 8.3, 6.8, 1.2 Hz, 1H), 2.51 (s, 3H), 2.10 (dd, J = 14.5, 5.2 Hz, 2H), 1.78 (dd, J = 14.5, 6.6 Hz, 2H), 1.69 (m, 2H), 0.94 (d, J = 6.6 Hz, 6H), 0.75 (d, J = 6.6 Hz, 6H). 13C NMR (151 MHz, CDCl3) δ 145.4, 142.5, 137.1, 134.0, 133.6, 129.9, 128.9, 128.3, 127.3, 126.8, 126.6, 126.3, 123.2, 79.8, 51.9, 24.5, 24.3, 21.8, 18.6; HRMS (ESI) m/z calcd for C26H32NaO4S (M + Na)+ 463.1919, found 463.1921; IR (thin film) 3415, 3065, 2950, 1580.
Synthesis of 2-(1-methoxynaphthalen-2-yl)propan-2-ol (15a)
To a cooled solution (−78 °C) of 13a (1.86 g, 9.30 mmol) in 50 mL of CH2Cl2 was added a solution of MeMgI in Et2O (3.0 M, 3.1 mL). The reaction was monitored until complete by TLC and quenched with saturated ammonium chloride (5 mL). The resulting layers were separated, and the aqueous layer was extracted with 2×10 mL of EtOAc. The combined organic layers were washed with 20 mL of brine, dried (MgSO4), and concentrated in vacuo to afford the crude product mixture. The product was purified by flash chromatography (5:95 EtOAc/hexanes) to afford the product 15a as a clear oil (0.71 g, 35%): 1H NMR (600 MHz, CDCl3) δ 8.07 (d, J = 8.3 Hz, 1H), 7.83 (d, J = 8.1 Hz, 1H), 7.60 (d, J = 8.6 Hz, 1H), 7.51(m, 3H), 4.07 (s, 3H), 1.73 (s, 6H); (600 MHz, CDCl3) δ 152.7, 136.1, 134.3, 128.1, 127.9, 126.0, 125.9, 124.6, 124.2, 122.1, 73.6, 63.3, 31.8; HRMS (ammonia CI) m/z calcd for C14H15O (M + H –H2O)+ 199.1123, found 199.1132; IR (thin film) 3420, 3066, 2957, 1586.
Supplementary Material
Figure 6.

a) Lowest-energy transition structures and activation free energies (ΔG‡) for the reaction of nahphyl ketone 13a with EtMgCl (top) and MeMgCl (bottom) calculated at the PCM(CH2Cl2)/B3LYP/6-31G(d) level. Energies are in kcal mol−1 and distances are in Angstrom.
ACKNOWLEDGMENT
This research was supported by National Science Fundation (CHE-1059084 to K.N.H.) the Petroleum Research Fund (administered by the American Chemical Society) and by the National Institutes of Health (R56AI80931-01 and R01AI080931-01 to J.T.S.). G.J.O acknowledges Ministerio de Economía y Competitividad (EX2010-1063 postdoctoral grant) and A.J.B for support in the form of a Bradford Borge Graduate Research Fellowship from UC Davis. Calculations were performed on the Hoffman2 cluster at UCLA and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation (OCI-1053575). We thank Dr. Jesús García-López for useful discussions.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Additional figures, cartesian coordinates, electronic energies, entropies, enthalpies, Gibbs free energies, lowest frequencies of the different conformations of all structures considered. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Brockway A, González-López M, Fettinger JC, Shaw JT. J. Org. Chem. 2011;76:3515–3518. doi: 10.1021/jo102537n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fuson RC, Speck SB. J. Am. Chem. Soc. 1942;64:2446–2448. [Google Scholar]
- 3.Fuson RC, Wassmundt FW. J. Am. Chem. Soc. 1956;78:5409–5413. [Google Scholar]
- 4.Rice JE, He ZM. J. Org. Chem. 1990;55:5490–5494. [Google Scholar]
- 5.Rice JE, Shih HC, Hussain N, LaVoie EJ. J. Org. Chem. 1987;52:849–855. [Google Scholar]
- 6.Hotta H, Suzuki T, Miyano S. Chem. Lett. 1990;19:143–144. [Google Scholar]
- 7.Parker JS, Smith NA, Welham MJ, Moss WO. Org. Process Res. Dev. 2003;8:45–50. [Google Scholar]
- 8.Hattori T, Suzuki T, Miyano S. J. Chem. Soc. Chem. Commun. 1991:1375–1376. [Google Scholar]
- 9.Hattori T, Suzuki T, Hayashizaka N, Koike N, Miyano S. Bull. Chem. Soc. Jpn. 1993;66:3034–3040. [Google Scholar]
- 10.Hattori T, Koike N, Miyano S. J. Chem. Soc. Perkin Trans. 1994;1:2273–2282. [Google Scholar]
- 11.Hattori T, Shimazumi Y, Goto H, Yamabe O, Morohashi N, Kawai W, Miyano S. J. Org. Chem. 2003;68:2099–2108. doi: 10.1021/jo026747k. [DOI] [PubMed] [Google Scholar]
- 12.Meyers AI, Mihelich ED. J. Am. Chem. Soc. 1975;97:7383–7385. [Google Scholar]
- 13.Meyers AI, Gabel R, Mihelich ED. J. Org. Chem. 1978;43:1372–1379. [Google Scholar]
- 14.Meyers AI, Avila WB. J. Org. Chem. 1981;46:3881–3886. [Google Scholar]
- 15.Meyers AI, Lutomski KA. J. Am. Chem. Soc. 1982;104:879–881. [Google Scholar]
- 16.Meyers AI, Lutomski KA. Synthesis. 1983;1983:105–107. [Google Scholar]
- 17.Patten AD, Nguyen Nhan H, Danishefsky SJ. J. Org. Chem. 1988;53:1003–1007. [Google Scholar]
- 18.Wilson JM, Cram DJ. J. Am. Chem. Soc. 1982;104:881–884. [Google Scholar]
- 19.Meyers AI, Flisak JR, Aitken RA. J. Am. Chem. Soc. 1987;109:5446–5452. [Google Scholar]
- 20.Rizzacasa MA, Sargent MV. J. Chem. Soc. Chem. Commun. 1990:894–896. [Google Scholar]
- 21.Meyers AI, Willemsen JJ. Tetrahedron. 1998;54:10493–10511. [Google Scholar]
- 22.Reetz MT, Keßeler K, Schmidtberger S, Wenderoth B, Steinbach R. Angew. Chem. Int. Ed. Engl. 1983;22:1511–1526. [Google Scholar]
- 23.Tamai Y, Hattori T, Date M, Koike S, Kamikubo Y, Akiyama M, Seino K, Takayama H, Oyama T, Miyano S. ChemInform. 1999;30:1–1. [Google Scholar]
- 24.Keck GE, Castellino S. J. Am. Chem. Soc. 1986;108:3847–3849. [Google Scholar]
- 25.Ashby EC, Smith MB. J. Am. Chem. Soc. 1964;86:4363–4370. [Google Scholar]
- 26.Spek AL, Voorbergen P, Schat G, Blomberg C, Bickelhaupt F. J. Organomet. Chem. 1974;77:147–151. [Google Scholar]
- 27.Avent AG, Caro CF, Hitchcock PB, Lappert MF, Li Z, Wei XH. Dalton Trans. 2004:1567–1577. doi: 10.1039/b316695n. [DOI] [PubMed] [Google Scholar]
- 28.Ellison JJ, Power PP. J. Organomet. Chem. 1996;526:263–267. [Google Scholar]
- 29.Schlenk W, Schlenk W. Berichte d. D. Chem. Gesellschaft. 1929;62:920–924. [Google Scholar]
- 30.Ashby EC. Q. Rev. Chem. Soc. . 1967;21:259–285. [Google Scholar]
- 31.Watanabe N, Nagamatsu K, Mizuno T, Matsumoto M. Luminescence. 2005;20:63–72. doi: 10.1002/bio.799. [DOI] [PubMed] [Google Scholar]
- 32.Ford WT, Radue R, Walker JA. J. Chem. Soc. D. 1970:966–967. [Google Scholar]
- 33.Gritzo H, Schaper F, Brintzinger HH. Acta Crystallogr. Sect. E: Struct. Rep. Online. 2004;60:m1108–m1110. [Google Scholar]
- 34.Atwood JL, Smith KD. J. Am. Chem. Soc. 1974;96:994–998. [Google Scholar]
- 35.Bordwell FG. Acc. Chem. Res. 1988;21:456–463. [Google Scholar]
- 36.Hammond GS. J. Am. Chem. Soc. 1955;77:334–338. [Google Scholar]
- 37.Yamaguchi M, Nakamura S, Okuma T, Minami T. Tetrahedron Lett. 1990;31:3913–3916. [Google Scholar]
- 38.Jeong JH, Weinreb SM. Org. Lett. 2006;8:2309–2312. doi: 10.1021/ol060556c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kraus GA, Wie J, Thite A. Synthesis. 2008;2008:2427–2431. [Google Scholar]
- 40.Laatsch H, Pudleiner H. Liebigs Ann. Chem. 1989;1989:863–881. [Google Scholar]
- 41.Carpenter TA, Evans GE, Leeper FJ, Staunton J, Wilkinson MR. J. Chem. Soc. Perkin Trans. 1984;1:1043–1051. [Google Scholar]
- 42.Lee C, Yang W, Parr RG. Phys. Rev. B: Condens. Matter Mater. Phys. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 43.Becke AD. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- 44.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, JA Montgomery J, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision C.1. Wallingford CT: Gaussian, Inc.; 2009. [Google Scholar]
- 45.Ribeiro RF, Marenich AV, Cramer CJ, Truhlar DG. J. Phys. Chem. B. 2011;115:14556–14562. doi: 10.1021/jp205508z. [DOI] [PubMed] [Google Scholar]
- 46.Bauschlicher CW., Jr Chem. Phys. Lett. 1995;246:40–44. [Google Scholar]
- 47.Merrick JP, Moran D, Radom L. J. Phys. Chem. A. 2007;111:11683–11700. doi: 10.1021/jp073974n. [DOI] [PubMed] [Google Scholar]
- 48.Gonzalez C, Schlegel HB. J. Chem. Phys. 1989;90:2154–2161. [Google Scholar]
- 49.Gonzalez C, Schlegel HB. J. Phys. Chem. 1990;94:5523–5527. [Google Scholar]
- 50.Marenich AV, Cramer CJ, Truhlar DG. J. Phys. Chem. B. 2009;113:6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.