

Abstract
Background/aims
Dendritic cells (DCs) act as a portal for virus invasion as well as potent antigen-presenting cells (APCs) involved in the antiviral host response. Interferons (IFNs) are produced in response to bacterial and viral infection and activate innate immune responses to efficiently counteract and remove pathogenic invaders. Respiratory syncytial virus (RSV) could inhibit IFN-mediated signaling pathway in epithelial cells; however, the effects of RSV on IFN signaling in the dendritic cells (DCs) are still unknown.
Methods
Mouse bone marrow derived DCs (BMDCs) were mock or infected with RSV at different multiplicity of infection (MOI) for 24 h, and then treated with different cytokines such as interferon-β (IFN-β), IFN-γ or interleukin-10 (IL-10). The mRNA expression of RSV nonstructural protein-1 (NS-1) and NS-2 was detected by RT-PCR. The expression of Janus family kinase-signal transducer and activator of transcription (JAK/STAT) signaling proteins was assessed by immunoblotting assays. The nuclear localization of specific signaling proteins was determined by immunofluorescence assay.
Results
Increasing amounts of NS-1 or NS-2 mRNA expression in BMDCs were observed with infected RSV at increasing MOI, suggesting BMDCs were permissive for viral gene expression. Further examination of the IFN-β signaling cascade showed RSV infection increased the total cellular levels of STAT1 and STAT2 in BMDCs, but impaired the IFN-β-dependent phosphorylation and nuclear localization of STAT1 and STAT2. The inhibitory effects of RSV on STAT1 and STAT2 phosphorylation and translocation were abolished by UV inactivation. In contrast, RSV did not inhibit the IFN-γ-stimulated STAT1 phosphorylation and nuclear localization. IL-10-stimulated STAT3 phosphorylation was also unaffected by RSV.
Conclusions
As well as RSV inhibiting STAT protein levels through degradation mechanisms in epithelial cells, these findings demonstrate that RSV also can specifically inhibit the type I interferon response in BMDCs through regulation of STAT1 and STAT2 phosphorylation and nuclear translocation.
Keywords: Bone marrow derived dendritic cells, Respiratory syncytial virus, Type I interferon, STAT, Signal pathway
1. Introduction
Human respiratory syncytial virus (RSV) is the most important viral agent of serious respiratory tract illness in infants and children worldwide (Collins and Graham, 2008). It is estimated by the CDC that up to 125,000 pediatric hospitalizations in the United States each year are due to RSV, at an annual cost of over USD 300,000,000 (Openshaw et al., 2003). RSV is also widely becoming recognized as an important pathogen in the elderly, transplant recipients, patients with chronic obstructive pulmonary disease (COPD), as well as chronic asthmatics (Falsey, 2007). Despite the generation of RSV-specific adaptive immune responses, RSV does not confer protective immunity and recurrent infections throughout life are common (Welliver, 2003). It is generally taken as evidence that the immune response to RSV infection is deficient, but this important and complex issue is poorly understood (Collins and Graham, 2008). Interferon-α (IFN-α), IFN-β (type I) and IFN-γ (type II) are produced in the lung in response to microbial infection and are potent activators of innate antimicrobial immunity. IFN-α/IFN-β ligation of the receptor results in the phosphorylation and activation of the accessory protein kinases Jak1 and Tyk2 that subsequently phosphorylate STAT2 (p-STAT2) and STAT1 (p-STAT1), leading to STAT2-STAT1 heterotrimerization with interferon regulatory factor (IRF) 9 and nuclear localization (Takaoka and Yanai, 2006). In the nucleus this complex transactivates the IFN-stimulated response element (ISRE) found in the promoter of IFN-stimulated genes (ISGs). Similarly, interaction of IFN-γ with the IFN-γ receptor initiates the Jak1/Jak2 dependent phosphorylation of STAT1. P-STAT1 dimerizes, translocates to the nucleus, binds to the gamma-activated sequence (GAS) DNA response element, and initiates transcription of IFN-γ responsive genes (Schroder et al, 2004). Viruses have evolved numerous unique mechanisms to inhibit the host type I and type II antiviral IFN signaling pathways (Goodbourn et al., 2000). Numerous Paramyxovirus family members inhibit IFN signaling in epithelial cells through inhibition of STAT phosphorylation (Goodbourn et al., 2000; Dinwiddie and Harrod, 2008), proteasomal degradation of STAT proteins (Ramaswamy et al., 2004; Didcock et al, 1999; Kubota et al., 2001; Elliott et al., 2007), sequestration of STAT proteins in high molecular weight complexes (Rodriguez et al., 2002, 2003), and inhibition of nuclear localization of STAT proteins (Takeuchi et al., 2003). Specifically, RSV inhibits type I IFN signaling in the respiratory epithelium through the proteasomal degradation of STAT2, a mechanism that is dependent on the expression of the RSV nonstructural protein-2 (NS-2) protein (Ramaswamy et al, 2004, 2006).
Dendritic cells (DCs) act as a portal for virus invasion as well as potent antigen presenting cells (APCs) involved in the antiviral host response (Rinaldo and Piazza, 2004). Respiratory tract dendritic cells are present at high frequency within airway epithelium, submucosa, and associated lung parenchymal tissue under resting conditions (Stumbles et al., 2003). After detection, uptake, and degradation of viruses, immature DCs undergo profound changes, called maturation, that greatly enhance their ability to present antigen to naive antigen specific T cells. Plasmacytoid DCs (pDCs) are known to be the major producers of type I IFNs in many tissues (Cella et al., 1999). Interestingly, a recent study also suggested that myeloid DCs (mDCs), along with macrophages, rather than pDCs, are the major source of type I IFN during pulmonary viral infections (Kumagai et al., 2007). We investigated whether RSV also could inhibit IFN signaling pathway in DCs, similar to that of epithelial cells. In the present study, we used murine bone marrow derived DCs (BMDCs), which are higher yield than primary lung DCs. Here, we report that RSV infection increased the total cellular levels of STAT1 and STAT2 in BMDCs, but impaired the IFN β-dependent phosphorylation and nuclear localization of STAT1 and STAT2. In contrast, RSV did not inhibit the IFN γ-stimulated STAT1 phosphorylation and IL-10-stimulated STAT3 phosphorylation.
2. Methods
2.1. Animals
Adult BALB/c mice (6–8 weeks old) were bred and maintained in the animal facility of Lovelace Respiratory Research Institute (LRRI). Animal studies were performed in accordance with AAALAC guidelines and approved by the Institutional Animal Care and Use Committee (IACUC). All study amendments and deviations were approved by the IACUC or by the study veterinarian. All experiments were repeated at least three times and each experimental group consisted of two to three mice. Samples were evaluated individually.
2.2. The isolation and culture of bone marrow-derived DCs
Bone marrow (BM) cells were collected and cultured using a protocol modified from that described previously (Inaba et al., 1992). Briefly, BALB/c mice were anaesthetized and euthanized; the femurs, tibiae, and humerus of mice were removed and cleaned of tissue, and sterilized in 70% ethanol. Both ends of each bone were cut, and marrow was flushed with RPMI-2 (RPMI1640 [Invitrogen, Carlsbad, CA] with 2% heat inactivated fetal Bovine serum [FBS; Hyclone, Logan, UT]). The BM cells were depleted of RBC by ACK lysis buffer (0.15 M NH4Cl, 1.0 mM KHCO3, and 0.1 mM EDTA), and filtered (70 μm) to remove large cell aggregates. The remaining cells were washed again, counted by trypan blue exclusion, and resuspended in complete medium ([cRPMI]; 10% FBS, RPMI 1640 medium, 400 mM l-glutamine, 100 U of penicillin/streptomycin, 5 × 10−7 M 2-mercaptoethanol [2-ME; Invitrogen, Carlsbad, CA]) at a final concentration of 5 × 105 cells/ml. The BM cells were cultured in 6-well plates (2.0 ml/well) with recombinant murine granulocyte macrophage colony stimulating factor (GM-CSF; PeproTech, Rocky Hill, NJ) at a final concentration of 10 ng/ml for 3 days. On day 3 of culture, the majority of the non-adherent cells were removed by aspiration of the culture media from each well. Then fresh media containing 10 ng/ml of rmGM-CSF and 10 ng/ml of recombinant murine interlukin-4 (rmIL-4; PeproTech, Rocky Hill, NJ) was added and cultured for another 3 days. On day 6 of culture, the loosely adherent cells, most of which (>80%) are BMDCs as identified by DC markers (CD11c+, Ia+ MHC class II) using FACS analysis, were harvested using cold Hank's solution. The cells were then washed, resuspended, and counted.
2.3. Preparation of RSV stocks
The A2 strain of RSV was plaque-purified three times under agarose. Following selection, one plaque was used to inoculate a subconfluent HEp-2 cell monolayer. After adsorption for 1 h at room temperature, 10% MEM was added and the infection was allowed to proceed for 3 days at 37°C until the entire monolayer showed cytopathic effects. The contents of the flask were resuspended and distributed in 1 ml aliquots, snap-frozen with alcohol/dry ice, and stored at −80 °C. Virus was derived from this master stock by infecting subconfluent HEp-2 monolayers at multiplicity of infection (MOI) of 0.1, and harvesting the monolayer when it appeared to be completely infected. The cells and media were sonicated on ice and then the suspension was clarified by centrifugation at 1000 × g for 10 min. The supernatant was frozen and stored at −80 °C and thawed rapidly at 37 °C for use. Viral titers were determined by plaque assay. To inactivate RSV, aliquots of the virus were exposed to ultraviolet (UV) light using UV1800 Stratalinker (Stratagene). UV irradiation was titrated to show a lack of infectivity while retaining the ability to visualize RSV proteins by immunoblotting at the appropriate protein size.
2.4. Infection of BMDCs with RSV
BMDCs (1 × 106) were incubated with RSV at different multiplicity of infection (MOI) in 200 μl plain RPMI for 1–2 h at 37°C (viral adsorption phase). Then, cRPMI was added to a total volume of 2 ml and BMDCs were cultured for 6–24 h at 37 °C in 5% CO2. At different time points of infection, cells were collected for subsequent analysis. In some experiments, RSV was inactivated by exposure to UV light prior to inoculation with BMDCs as control.
2.5. RSV RT-PCR
RNA was isolated from BMDCs cultures following RSV- or mock-infection using the RNeasy Kit (Qiagen, Valencia, CA) with optional on-column DNase digestion per the manufacturer's directions. The concentration and quality of RNA was determined by spectrophotometry and equal amounts of RNA were transcribed into cDNA using an Omniscript kit (Qiagen). The mRNA of RSV NS-1 and NS-2 was detected by PCR. The primer sequences for PCR were synthesized by Invitrogen and were as follows:
RSV NS-1, forward 5′-TTTGGCTAAGGCAGTGA-3′
reverse 5′-CCATTAGGTTGAGAGCA-3′
RSV NS-2, forward 5′-ATAATAACATCACTAACCAGAGAC-3′
reverse, 5′- ATAGTTATGCATAGAGTTGTTGTT-3′
The PCR was cycled 30 times after initial denaturation (94°C, 5 min), with the following parameters: denaturation, 94°C, 45 s; annealing, 52°C, 45 s; and extension, 72°C, 45 s.
2.6. Immunoblotting assays
BMDCs (1 × 106) were cultured in 6-wells plate overnight. Then the cells were mock- or RSV-infected for 24 h prior to subsequent treatment with different cytokines (IFN-γ 100 U/ml for 45 min, IFN-β100 U/ml for 30 min, or IL-10 20 ng/ml for 2 h). BMDC lysates were prepared in lysis buffer (10 mM Tris-HCl, pH 7.5, 15 mM NaCl, 0.5% Nonidet P-40, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, pH 8.0, 0.2 mM sodium orthovanadate, and 0.4 mM PMSF). Protein concentrations were measured using the BCA protein assay kit (Pierce, Rockford, IL) per the manufacturer's directions. Equal amounts of protein samples were resolved on 8–16% SDS–Tris–glycine polyacrylamide gels (Invitrogen) and transferred to polyvinylidene difluoride (PVDF) membrane (Invitrogen). The membrane was blocked with 5% powdered milk in Tris-buffered saline with 0.1% Tween 20 (TBST) and incubated overnight at 4°C with rabbit anti-mouse STAT1 (Cell Signaling, Danvers, MA) diluted 1:1000, rabbit anti-mouse STAT2 (Cell Signaling) diluted 1:1000, rabbit anti-mouse STAT3 (Cell Signaling) diluted 1:1000, rabbit anti-mouse phospho-specific STAT1 (Cell Signaling) diluted 1:1000, rabbit anti-mouse phospho-specific STAT2 (Cell Signaling) diluted 1:1000, rabbit anti-mouse phospho-specific STAT3 (Cell Signaling) diluted 1:1000, or mouse anti-human β-actin (Sigma, St. Louis, MO) diluted 1:20,000 in TBST containing 5% powdered milk. Blots were washed with TBST and incubated with peroxidase-conjugated goat anti-mouse IgG or goat anti-rabbit IgG Ab (Pierce) diluted 1:10,000 in TBST containing 1% powdered milk. After washing, blots were developed with a chemiluminescence detection system (Millipore, Billerica, MA).
2.7. Immunofluorescence microscopy
BMDCs were grown and infected with RSV on glass chamber slides (Nalgene Nunc) for 24 h. Following treatment, cells were fixed in 200 μl of 4% paraformaldehyde in PBS for 30 min, washed 3 times with PBS and permeablized using 0.2% Triton X-100 in PBS for 30 min. Following 3 washes with PBS, cells were blocked with PBS containing 10% goat serum (Vector) for 1 h at room temperature, and then incubated with a mouse anti-RSV-F (Serotec) at a dilution of 1:200 and rabbit anti-STAT1, STAT2 at a dilution of 1:100 (Cell Signaling) overnight at 4°C in PBS containing 2% normal goat serum. Primary antibodies were removed and cells were washed 3 times in PBS before addition of secondary antibodies. The nuclei of all cells were stained using 1:400 dilution of Hoescht (Molecular Probes, Invitrogen), while RSV was visualized using a goat anti-mouse Cy5 conjugated antibody (Vector) and STAT proteins were visualized using a goat anti-rabbit Cy3 conjugated antibody (Vector) both at a dilution of 1:200 in 2% goat serum PBS for 1 h at room temperature. Fluorescence microscopy was carried out on a Zeiss Axioplan 2 with an Intelligent Image Innovations CCD Camera using Slidebook software version 4.1.0.7.
3. Results
3.1. The replication of RSV viruses on BMDCs
Dendritic cell cultures were generated from harvested bone marrow. DC populations were verified by FACS analysis for CD11c+ and Ia+ MHC class II cell surface markers. To determine whether cultured DCs can support RSV gene expression, NS-1 or NS-2 mRNA in BMDCs after incubation with different MOIs of RSV for 24 h were analyzed by non-quantitative RT-PCR (Fig. 1). NS-1 and NS-2 gene expression was observed even at the lowest MOI of 0.05. Increasing amounts of NS-1 or NS-2 mRNA expression in BMDCs were found with increasing MOI of RSV infection, suggesting BMDCs were permissive for viral gene expression.
Fig. 1.
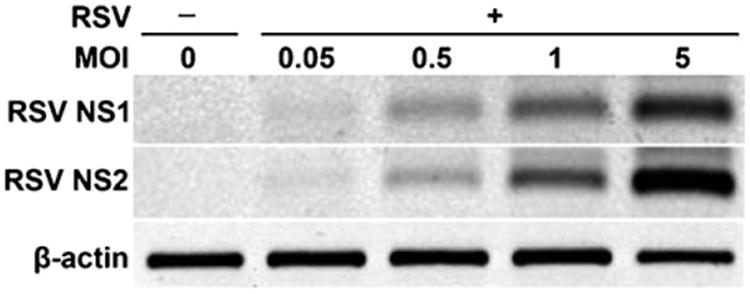
The replication of RSV viruses on BMDCs. The replication of RSV viruses was identified by detecting the mRNA expression of RSV non-structure protein 1 (RSV NS-1) and RSV non-structure protein 2 (RSV NS-2) by RT-PCR on BMDC that were uninfected or were infected with RSV at the indicated MOI for 24 h.
3.2. RSV inhibits IFN-β- but not IFN-γ-dependent STAT1 phosphorylation
Multiple members of the Paramyxovirus family inhibit type I IFN signaling by interfering with the normal activities of STAT1 in the Jak/STAT signal transduction pathway. Despite evidence that some paramyxoviruses diminish STAT protein levels through degradation mechanisms, STAT1 levels were markedly increased in BMDCs infected with RSV at MOI of 1 or 5 (Fig. 2A). STAT1 phosphorylation was assessed using phosphorylated STAT1-specific antibodies to determine the activation of STAT1 in response to IFN-β. In mock-infected BMDCs, IFN-β induced easily discernable STAT1 phosphorylation. Moreover, infection with RSV before IFN-β treatment completely eliminated the induction of STAT1 phosphorylation. However, inactivation of RSV by UV treatment restored the ability of IFN-β to phosphorylate STAT1. Importantly, UV-inactivated RSV inoculum contained all components of viral propagation, suggesting that the inhibition of STAT1 phosphorylation is not due to a cellular product produced during viral propagation (Fig. 2A). Infection by RSV alone did not induce STAT1 phosphorylation in the absence of IFN-β (data not shown) as has been shown previously (Senft et al, 2010).
Fig. 2.
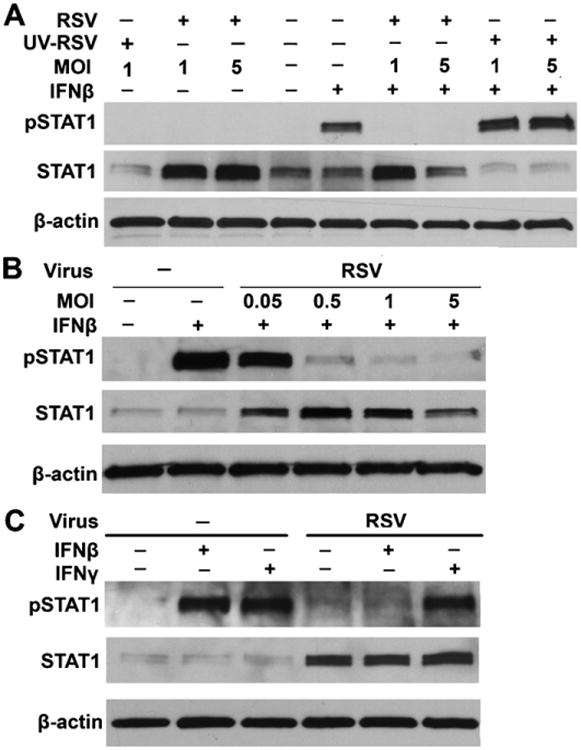
RSV infection impairs phosphorylation of STAT1 induced by IFN-β, but not IFN-γ. (A) BMDCs were infected with mock, live or UV-killed RSV for 24 h at an MOI of 1 and 5, followed by incubation with or without IFN-β (100 U/ml) for 30 min before cell harvesting. Phosphorylated STAT1 and total levels of STAT1 protein were detected by immunoblotting. β-Actin was used a control for equal loading of protein. (B) The inhibition of phosphorylation of STAT1 in response to 30 min of IFN-β (100 U/ml) stimulation and total levels of STAT1 protein were assessed in mock- or RSV-infected (MOIs of 0.05, 0.5, 1.0, and 5.0) BMDCs. (C) BMDCs were infected with RSV for 24 h at an MOI of 1 and treated with IFN-β (100 U/ml) for 30 min or IFN-γ (10 ng/ml) for 45 min before cell harvesting. Phosphorylated STAT1, total levels of STAT1 protein and β-actin protein levels were detected by immunoblotting.
Infection with increasing MOIs of RSV was used to determine the level of infection required for inhibition of IFN-β-mediated STAT1 phosphorylation. BMDCs were infected with increasing MOIs of RSV followed by stimulation with IFN-β for 30 min. The lowest MOI of 0.05 of RSV was sufficient to cause increased total levels of STAT1 in BMDCs (Fig. 2B). However, the phosphorylation of STAT1 in response to IFN-β was decreased in a dose-dependent manner, consistent with previous findings (Dinwiddie and Harrod, 2008). Infection with MOIs of 0.05 did not affect STAT1 phosphorylation when compared with mock-infected and IFN-β-stimulated BMDCs, whereas infection with MOIs of 0.5 or higher resulted in inhibition of STAT1 phosphorylation (Fig. 2B). To determine if inhibition of STAT1 phosphorylation by RSV was specific to the type I IFN signaling pathway, IFN-γ-mediated STAT1 phosphorylation was analyzed. Mock- and RSV-infected BMDCs were treated with IFN-γ (100 U/ml) for 45 min. Total cell lysates were collected and STAT1 phosphorylation was assessed by immunoblotting. IFN-β-mediated phosphorylation of STAT1 was inhibited in BMDCs infected with RSV; however, IFN-γ-mediated STAT1 phosphorylation was unaffected by RSV infection (Fig. 2C). These results suggest that the inhibition of STAT1 phosphorylation by RSV is limited to type I IFN signaling.
3.3. RSV infection inhibits type I IFN-mediated STAT2 phosphorylation
STAT2 facilitates the IFN-β-stimulated phosphorylation of STAT1 by serving as a docking site for STAT1; STAT2 must be phosphorylated in response to IFN-β prior to recruitment and subsequent phosphorylation of STAT1 (Brierley and Fish, 2005). RSV prevents STAT1 phosphorylation in lung epithelial cells by targeting STAT2 for proteasomal degradation (Ramaswamy et al., 2004). Because RSV inhibited the phosphorylation of STAT1 in response to IFN-β, we investigated whether RSV also impairs IFN-β-mediated STAT2 phosphorylation and transcriptional activation in BMDCs. RSV alone did not substantially induce STAT2 phosphorylation in the absence of IFN treatment. Interestingly, the total levels of STAT2 were increased as assessed by Western blot analysis, suggesting mouse BMDC STAT2 is not targeted for proteasomal degradation in response to RSV infection (Fig. 3A). STAT2 phosphorylation was assessed using phospho-specific STAT2 antibodies following IFN-β stimulation of mock- and RSV-infected BMDCs. As with phosphorylation of STAT1, RSV infection significantly inhibited IFN-β-mediated phosphorylation of STAT2 in BMDCs (Fig. 3A). Moreover, infection with RSV at MOI of 0.05 did not affect STAT2 phosphorylation when compared with mock-infected and IFN-β-stimulated BMDCs, whereas infection with MOIs of 0.5 or higher resulted in inhibition of STAT2 phosphorylation (Fig. 3B).
Fig. 3.
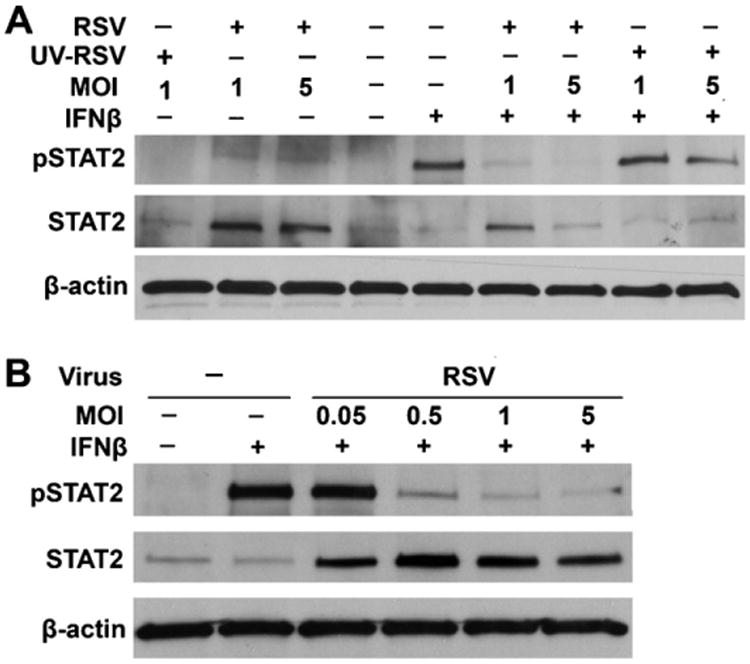
RSV infection also inhibits phosphorylation of STAT2 induced by IFN-β. (A) BMDCs were infected with mock, live or UV-killed RSV for 24 h at an MOI of 1 and 5, followed by incubation with or without IFN-β (100 U/ml) for 30 min before cell harvesting. Phosphorylated STAT2, total levels of STAT2 protein and β-actin were detected by immunoblotting. (B) The inhibition of phosphorylation of STAT2 in response to 30 min of IFN-β (100 U/ml) stimulation and total levels of STAT2 protein were assessed in mock- or RSV-infected (MOIs of 0.05, 0.5, 1.0, and 5.0) BMDCs.
3.4. RSV does not inhibit IL-10-dependent STAT3 phosphorylation
IL-10 mediates its diverse activities via high affinity cell surface receptors composed of two chains, IL-10R1 and CRF2-4/IL-10R2 (Lutfalla et al., 1992; Kotenko et al., 1997). Both chains are members of the type I IFN receptor subgroup of cytokine receptor. IL-10 activates Janus Kinases, Jak-1 and Tyk-2 (Kotenko et al., 1997), and as a consequence, induces the activation of STAT transcription factors, in particular STAT3 (Finbloom and Winestock, 1995; Williams et al., 2004; Lang et al., 2002). To determine if inhibition ofJAK/STAT by RSV was specific to the type I IFN signaling pathway, IL-10 mediated STAT3 phosphorylation was assessed. Mock- and RSV-infected BMDCs were treated with IL-10 (20 ng/ml) for 2 h. Total cell lysates were collected and STAT3 phosphorylation was assessed by immunoblotting. Compared with mock-infected BMDCs, STAT3 levels were markedly increased in BMDCs infected with RSV at MOI of 1. In mock-infected BMDCs, IL-10 induced STAT3 phosphorylation. Interestingly, infection with RSV before IL-10 treatment did not affect the STAT3 phosphorylation (Fig. 4). These results suggest that RSV does not impair the IL-10 signaling pathway.
Fig. 4.
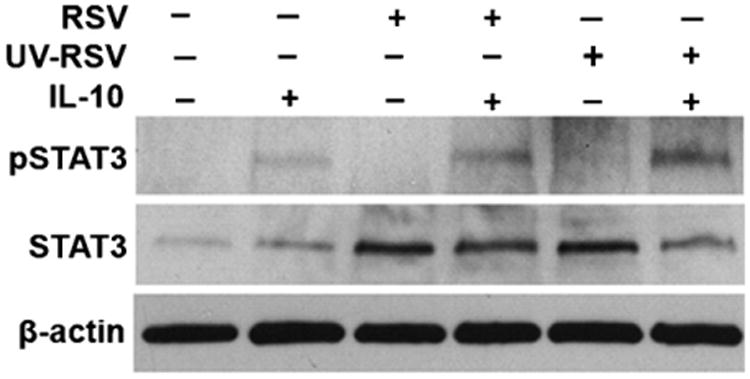
RSV does not inhibit IL-10-mediated STAT3 phosphorylation. BMDCs were infected with mock, live or UV-killed RSV for 24 h at a MOI of 1, followed by incubation without or with IL-10 (20 ng/ml) for 2 h before cell harvesting. Phosphorylated STAT3, total levels of STAT3 protein and β-actin were detected by immunoblotting.
3.5. Nuclear translocation of STAT1 and STAT2 in response to IFN-β is blocked in RSV-infected BMDCs
Nuclear localizations of phosphorylated STAT1 and STAT2 are key cellular events in mediating IFN-β-transduced signaling. Therefore, immunofluorescence (IF) microscopy was used to further characterize the cellular localizations of STAT1 and STAT2 during RSV infection and in response to IFN-β signaling. Mock-infected, untreated BMDCs exhibited the characteristic cytoplasmic staining of STAT1 and STAT2 (Fig. 5A and B). However, treatment with IFN-β for 30 min resulted in marked redistribution of STAT1 and STAT2 to the nucleus as observed by co-localization of STAT1 or STAT2 and DAPI-stained nuclei in nearly 100 percent of cells visualized (Fig. 5A and B). Consistent with the phosphorylation data obtained by immunoblotting analysis, treatment of RSV-infected BMDCs with IFN-β did not result in redistribution of STAT1 and STAT2 into the nucleus as was seen in the mock-infected and IFN-β-treated cells (Fig. 5A and B). However, when RSV was inactivated by UV treatment before infection, the abilities of STAT1 and STAT2 to translocate to the nucleus in response to IFN-β treatment were unaltered (Fig. 5). Merging of IF for RSV-F and STAT proteins often masked the red staining for RSV-F, likely due to the greater abundance of STAT proteins than RSV-F in BMDCs. At high magnification and full fluorescent illumination, RSV-F staining could be observed in greater than 90% of cells. These results further confirm that IFN-β-mediated signaling is interrupted by RSV through inhibition of STAT1 and STAT2 phosphorylation and nuclear translocation.
Fig. 5.
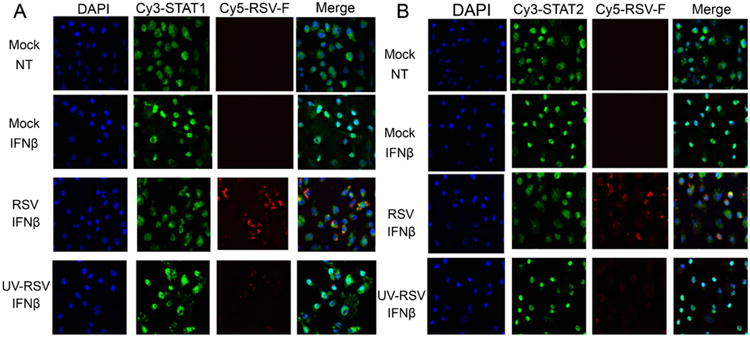
Nuclear translocations of STAT1 and STAT2 in response to IFN-β are blocked in RSV-infected BMDCs. Cellular localization of STAT1 in mock-, RSV-, or UV-killed RSV-infected BMDCs was analyzed by immunofluorescence microscopy in IFN-β (100 U/ml for 30 min)-treated cells. Nuclei (blue) were stained with Hoechst (DAPI). STAT1 (5A) and STAT2 (5B) were detected with a rabbit anti-STAT1 and anti-STAT2 antibody followed by incubation with a goat anti-rabbit-conjugated Cy3 antibody (green), while RSV (red) infection was detected using a mouse anti-RSV-F protein antibody and a goat anti-mouse Cy5 antibody. Magnification: 400×. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
3.6. Nuclear translocation of STAT1 in response to IFN-y is not affected in RSV-infected BMDCs
To determine if inhibition of STAT1 and STAT2 nuclear translocation by RSV was specific to the type I IFN signaling pathway, the cellular localization of STAT1 in response to IFN-γ was also assessed by IF microscopy. Treatment with IFN-γ for 45 min in infected BMDCs resulted in marked redistribution of STAT1 to the nucleus as observed by co-localization of STAT1 and DAPI-stained nuclei (Fig. 6). Consistent with the phosphorylation data obtained by immunoblotting analysis, treatment of RSV infected BMDCs with IFN-γ did not affect the redistribution of STAT1 into the nucleus (Fig. 6). These results show that RSV does not affect IFN-γ-mediated STAT1 nuclear translocation.
Fig. 6.
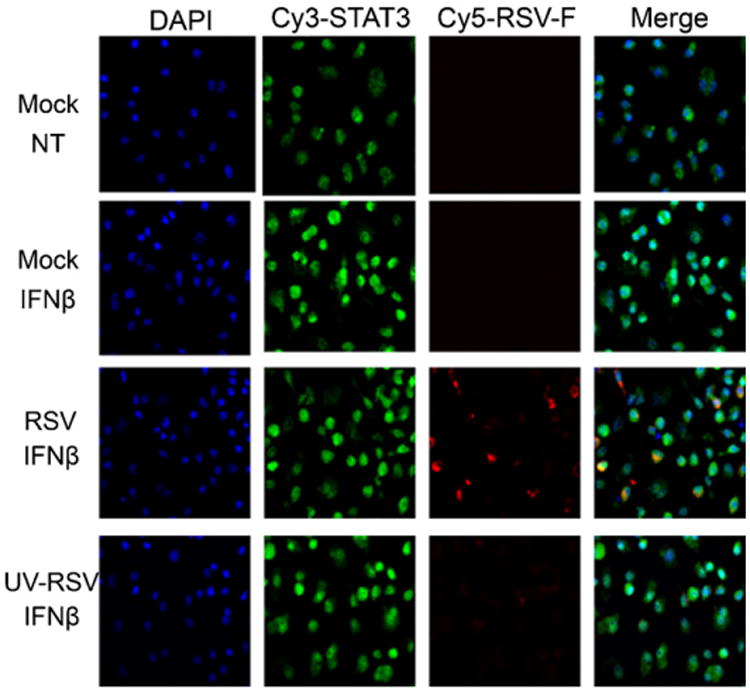
Nuclear translocation of STAT1 in response to IFN-γ is not affected in RSV Infected BMDCs. Cellular localization of STAT1 in mock-, RSV-, or UV-killed RSV-infected BMDCs was analyzed by immunofluorescence microscopy in IFN-γ (100 U/ml for 45 min)-treated cells. Nuclei (blue) were stained with Hoechst (DAPI). STAT1 (green) were detected with a rabbit anti-STAT1 antibody followed by incubation with a goat anti-rabbit-conjugated Cy3 antibody, while RSV (red) infection was detected using a mouse anti-RSV-F protein antibody and a goat anti-mouse Cy5 antibody. Magnification: 400×. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
4. Discussion
It is clear that most, if not all, viruses have evolved mechanisms to avert the interferon response mounted by cells during infection. These mechanisms are needed to ensure that the virus has sufficient time to successfully replicate, package, and release from host cells. Our study demonstrates that RSV can infect and replicate in BMDCs by detecting RSV NS-1 and NS-2 mRNA. Herein, infection with RSV in murine BMDCs was able to prevent IFN-β-mediated phosphorylation and subsequent nuclear translocation of STAT1 and STAT2. In contrast, RSV did not inhibit the phosphorylation of STAT1 or the subsequent nuclear translocation of pSTAT1 in response to IFN-γ stimulation. RSV infection also did not alter IL-10 induced STAT3 phosphorylation. These results suggest that RSV can specifically inhibit type IIFN signaling in BMDCs through regulating STAT1 and STAT2 phosphorylation and nuclear translocation.
Inhibition of IFN pathways by paramyxoviruses is well documented. Interestingly, members of the Paramyxovirus family have evolved separate and distinct mechanisms to target the same components of the IFN pathways. Specifically, SV5, mumps, and type II human parainfluenza viruses (PIV-2) use their V and C proteins to target STAT1 and STAT2 for proteasome degradation by polyubiquitination (Didcock et al, 1999; Parisien et al., 2001). Similarly, RSV inhibits type I IFN signaling and transcriptional activation in human tracheobronchiolar epithelial cells through a mechanism that involves the proteasome-dependent destruction of STAT2 and NS-2 protein (Ramaswamy et al., 2004, 2006; Lo et al., 2005). As a result of the proteasomal degradation the levels of the targeted STAT protein are markedly decreased or undetectable in RSV-infected cells. In contrast, our results clearly indicate that RSV infection increases the cellular level of STAT1 and STAT2 in the mouse BMDCs and, therefore, do not suggest that degradation of STAT2 serves as the inhibitory mechanism in BMDCs. RSV only inhibited the phosphorylation and nuclear translocation of STAT1 and STAT2. In macrophages, we also found RSV infection increased the total cellular levels of STAT1 and STAT2, but impaired the IFN-α-and IFN-β-dependent phosphorylation of STAT1 and STAT2 (Senft et al, 2010). Multiple paramyxoviruses also prevent IFN signaling not by causing degradation of the STAT proteins, but instead by binding, sequestering, and preventing either their phosphorylation or nuclear translocation. Nipah and Hendra viruses are able to sequester STAT1 and STAT2 in high-molecular complexes, preventing their translocation into the nucleus via binding by their V proteins (Rodriguez et al., 2002, 2003). Likewise, measles virus causes a portion of STAT1 and STAT2 to be redistributed to cytoplasmic aggregates (Palosaari et al, 2003) and can inhibit both IFN-α and IFN-γ-dependent STAT1 and STAT2 nuclear localization (Takeuchi et al, 2003). The C protein of Sendai virus has been shown to bind STAT1 (Takeuchi et al., 2001) and inhibit both STAT1 and STAT2 phosphorylation (Garcin et al, 2003). Closely related to RSV, human metapneumovirus (hMPV) is a member of the Metapneumovirus genus within the Pneumovirinae subfamily of the Paramyxoviridae family. hMPV has also been found to prevent IFN-α-induced phosphorylation and nuclear translocation of STAT1 (Dinwiddie and Harrod, 2008). An intriguing finding herein was the increased levels of both STAT1 and STAT2 during RSV infection. However, it is currently unclear if the mechanistic differences regarding RSV inhibition of type I IFN signaling in this study using mouse BMDCs and previous studies in human tracheobronchiolar epithelial cells are due to organismal or cell lineage differences. Interestingly, Ramaswamy et al. (2004) also reported that RSV infection of human tracheobronchiolar epithelial cells increased cellular STAT1 protein levels and did not inhibit the IFN-γ-dependent phosphorylation of STAT1. These findings are consistent with our observations in mouse BMDCs and may suggest that the differences in type I IFN signaling are attributable to inherent differences between cells of epithelial and myeloid origin. The consequences of the inhibition of type I IFN signaling by RSV are not completely understood, but likely play a role in pathogenesis and severity of infection and could impact the adaptive immune response. Previous reports suggest that RSV is able to persist in the lungs of infected mice (Beyer et al, 2004). The ability of RSV to inhibit IFN signaling likely plays an important role in facilitating this persistence. Furthermore, inhibition of IFN signaling might play an important role in modulating the function of DCs. Munir et al. (2008) recently found that deletion of the NS-1 and NS-2 protein of RSV resulted in increased expression of cell surface markers of DC maturation and an increase in the expression of multiple cytokines and chemokines. These up regulations were largely inhibited by pretreatment with a blocking antibody against the type I IFN receptor (IFNAR2-blocking antibody), suggesting that suppression of DC maturation by NS-1 and NS-2 is due, at least in part, to the suppression of type I IFN signaling.
5. Conclusions
In summary, in this study we have reported the findings that RSV is able to inhibit type I IFN signaling on DCs by suppressing the STAT1 and STAT2 phosphorylation and translocation. Additional understanding of how RSV inhibits the type I IFN pathway, the effects on DCs' maturation, and the consequences with regard to adaptive immune responses will be crucial in improving approaches for treatment and the development of vaccines.
Acknowledgments
This study was supported by the National Institute of Health HL-66994 (KSH) and National Natural Science Foundation of China (30800499).
Abbreviations
- RSV
respiratory syncytial virus
- BMDCs
bone marrow derived dendritic cells
- IFN
interferon
- STAT
signal transducer and activator of transcription
- MOI
multiplicity of infection
Contributor Information
Zhijun Jie, Email: jiezjlxh@gmail.com.
Darrell L. Dinwiddie, Email: ddinwiddie@lrri.org.
Albert P. Senft, Email: asenft@lrri.org.
Kevin S. Harrod, Email: kharrod@lrri.org.
References
- Beyer M, Bartz H, Hörner K, Doths S, Koerner-Rettberg C, Schwarze J. Sustained increases in numbers of pulmonary dendritic cells after respiratory syncytial virus infection. J Allergy Clin Immunol. 2004;113:127–133. doi: 10.1016/j.jaci.2003.10.057. [DOI] [PubMed] [Google Scholar]
- Brierley MM, Fish EN. STATs: multifaceted regulators of transcription. J Interferon Cytokine Res. 2005;25(12):733–744. doi: 10.1089/jir.2005.25.733. [DOI] [PubMed] [Google Scholar]
- Cella MD, Jarrossay F, Facchetti O, Alebardi H, Nakajima A, Colonna M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. 1999;5:919–923. doi: 10.1038/11360. [DOI] [PubMed] [Google Scholar]
- Collins PL, Graham BS. Viral and host factors in human respiratory syncytial virus pathogenesis. J Virol. 2008;82:2040–2055. doi: 10.1128/JVI.01625-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didcock L, Young DF, Goodbourn S, Randall RE. The V protein of simian virus 5 inhibits interferon signaling by targeting STAT1 for proteasome-mediated degradation. J Virol. 1999;73(12):9928–9933. doi: 10.1128/jvi.73.12.9928-9933.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinwiddie DL, Harrod KS. Human metapneumovirus inhibits IFN-alpha signaling through inhibition of STAT1 phosphorylation. Am J Respir Cell Mol Biol. 2008;38:661–670. doi: 10.1165/rcmb.2007-0285OC. [DOI] [PubMed] [Google Scholar]
- Elliott J, Lynch OT, Suessmuth Y, Qian P, Boyd CR, Burrows JF, Buick R, Stevenson NJ, Touzelet O, Gadina M, Power UF, Johnston JA. Respiratory syncytial virus NS1 protein degrades STAT2 by using the Elongin-Cullin E3 ligase. J Virol. 2007;81(7):3428–3436. doi: 10.1128/JVI.02303-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsey AR. Respiratory syncytial virus infection in adults. Semin Respir Crit Care Med. 2007;28(2):171–181. doi: 10.1055/s-2007-976489. [DOI] [PubMed] [Google Scholar]
- Finbloom DS, Winestock KD. IL-10 induces the tyrosine phosphorylation of tyk2 and Jak1 and the differential assembly of STAT1a and STAT3 complexes in human T cells and monocytes. J Immunol. 1995;155(3):1079–1090. [PubMed] [Google Scholar]
- Garcin D, Marq JB, Goodbourn S, Kolakofsky D. The amino-terminal extensions of the longer Sendai virus C proteins modulate pY701-Stat1 and bulk Stat1 levels independently of interferon signaling. J Virol. 2003;77:2321–2329. doi: 10.1128/JVI.77.4.2321-2329.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodbourn S, Didcock L, Randall RE. Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures. J Gen Virol. 2000;81:2341–2364. doi: 10.1099/0022-1317-81-10-2341. [DOI] [PubMed] [Google Scholar]
- Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotenko SV, Krause CD, Izotova LS, Pollack BP, Wu W, Pestka S. Identification and functional characterization of a second chain of the interleukin-10 receptor complex. EMBO J. 1997;16(19):5894–5903. doi: 10.1093/emboj/16.19.5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota T, Yokosawa N, Yokota S, Fujii N. C terminal CYS-RICH region of mumps virus structural V protein correlates with block of interferon alpha and gamma signal transduction pathway through decrease of STAT 1-alpha. Biochem Biophys Res Commun. 2001;283(1):255–259. doi: 10.1006/bbrc.2001.4764. [DOI] [PubMed] [Google Scholar]
- Kumagai YO, Takeuchi H, Kato H, Kumar K, Matsui E, Morii K, Aozasa T, Akira KS. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity. 2007;27:240–252. doi: 10.1016/j.immuni.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression inactivated and resting primary macrophages by IL-10. J Immunol. 2002;169(5):2253–2263. doi: 10.4049/jimmunol.169.5.2253. [DOI] [PubMed] [Google Scholar]
- Lo MS, Brazas RM, Holtzman MJ. Respiratory syncytial virus nonstructural proteins NS1 and NS2 mediate inhibition of Stat2 expression and alpha/beta interferon responsiveness. J Virol. 2005;79:9315–9319. doi: 10.1128/JVI.79.14.9315-9319.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutfalla G, Gardiner K, Proudhon D, Vielh E, Uzé G. The structure of the human interferon a/β receptor gene. J Biol Chem. 1992;267(4):2802–2809. [PubMed] [Google Scholar]
- Munir S, Le Nouen C, Luongo C, Buchholz UJ, Collins PL, Bukreyev A. Nonstructural proteins 1 and 2 of respiratory syncytial virus suppress maturation of human dendritic cells. J Virol. 2008;82(17):8780–8796. doi: 10.1128/JVI.00630-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Openshaw PJ, Dean GS, Culley FJ. Links between respiratory syncytial virus bronchiolitis and childhood asthma: clinical and research approaches. Pediatr Infect Dis J. 2003;22(2 Suppl):S58–64. doi: 10.1097/01.inf.0000053887.26571.eb. [DOI] [PubMed] [Google Scholar]
- Palosaari H, Parisien JP, Rodriguez JJ, Ulane CM, Horvath CM. STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J Virol. 2003;77:7635–7644. doi: 10.1128/JVI.77.13.7635-7644.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien JP, LauJ F, Rodriguez JJ, Sullivan BM, Moscona A, Parks GD, Lamb RA, Horvath CM. The V protein of human parainfluenza virus 2 antagonizes type I interferon responses by destabilizing signal transducer and activator of transcription 2. Virology. 2001;283:230–239. doi: 10.1006/viro.2001.0856. [DOI] [PubMed] [Google Scholar]
- Ramaswamy M, Shi L, Monick MM, Hunninghake GW, Look DC. Specific inhibition of type I interferon signal transduction by respiratory syncytial virus. Am J Respir Cell Mol Biol. 2004;30(6):893–900. doi: 10.1165/rcmb.2003-0410OC. [DOI] [PubMed] [Google Scholar]
- Ramaswamy M, Shi L, Varga SM, Barik S, Behlke MA, Look DC. Respiratory syncytial virus nonstructural protein 2 specifically inhibits type I interferon signal transduction. Virology. 2006;344(2):328–339. doi: 10.1016/j.virol.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Rinaldo CR, Jr, Piazza P. Virus infection of dendritic cells: portal for host invasion and host defense. Trends Microbiol. 2004;12(7):337–345. doi: 10.1016/j.tim.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Rodriguez JJ, Parisien JP, Horvath CM. Nipah virus V protein evades alpha 1 and gamma interferonsbypreventing STAT1 and STAT2activation and nuclear accumulation. J Virol. 2002;76(22):11476–11483. doi: 10.1128/JVI.76.22.11476-11483.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JJ, Wang LF, Horvath CM. Hendra virus V protein inhibits interferon signaling by preventing STAT1 and STAT2 nuclear accumulation. J Virol. 2003;77(21):11842–11845. doi: 10.1128/JVI.77.21.11842-11845.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75(2):163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- Senft AP, Taylor RH, Lei W, Campbell SA, Tipper JL, Martinez MJ, Witt TL, Clay CC, Harrod KS. Respiratory syncytial virus impairs macrophage IFN-alpha/beta- and IFN-gamma-stimulated transcription by distinct mechanisms. Am J Respir Cell Mol Biol. 2010;42(4):404–414. doi: 10.1165/rcmb.2008-0229OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumbles PA, Upham JW, Holt PG. Airway dendritic cells: Coordinators of immunological homeostasis and immunity in the respiratory tract. APMIS. 2003;111:741–755. doi: 10.1034/j.1600-0463.2003.11107806.x. [DOI] [PubMed] [Google Scholar]
- Takaoka A, Yanai H. Interferon signaling network in innate defence. Cell Microbiol. 2006;8(6):907–922. doi: 10.1111/j.1462-5822.2006.00716.x. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Kadota SI, Takeda M, Miyajima N, Nagata K. Measles virus V protein blocks interferon (IFN)-alpha/beta but not IFN-gamma signaling by inhibiting STAT1 and STAT2 phosphorylation. FEBS Lett. 2003;545(2–3):177–182. doi: 10.1016/s0014-5793(03)00528-3. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Komatsu T, Yokoo J, Kato A, Shioda T, Nagai Y, Gotoh B. Sendai virus C protein physically associates with Stat1. Genes Cells. 2001;6:545–557. doi: 10.1046/j.1365-2443.2001.00442.x. [DOI] [PubMed] [Google Scholar]
- Welliver RC. Respiratory syncytial virus and other respiratory viruses. Pediatr Infect Dis J. 2003;22(2 Suppl):S6–10. doi: 10.1097/01.inf.0000053880.92496.db. [DOI] [PubMed] [Google Scholar]
- Williams L, Bradley L, Smith A, Foxwell B. Signal transducer and activator of transcription 3 is the dominant mediator of the anti-inflammatory effects of IL-10 in human macrophages. J Immunol. 2004;172(1):567–576. doi: 10.4049/jimmunol.172.1.567. [DOI] [PubMed] [Google Scholar]