

Abstract
Background
The mechanisms of ventilator-induced lung injury, an iatrogenic inflammatory condition induced by mechanical ventilation, are not completely understood. Toll-like receptor 4 (TLR4) signaling via the adaptor protein, myeloid differentiation factor 88 (MyD88) is proinflammatory and plays a critical role in host immune response to invading pathogen and noninfectious tissue injury. The role of TLR4-MyD88 signaling in ventilator-induced lung injury remains incompletely understood.
Methods
Mice were ventilated with low or high tidal volume (HTV), 7 or 20 ml/kg, after tracheotomy for 4 h. Control mice were tracheotomized without ventilation. Lung injury was assessed by: alveolar capillary permeability to Evans blue albumin, wet/dry ratio, bronchoalveolar lavage analysis for cell counts, total proteins and cytokines, lung histopathology, and plasma cytokine levels.
Results
Wildtype mice subjected to HTV had increased: pulmonary permeability; inflammatory cell infiltration/lung edema; and interleukin-6/macrophage-inflammatory protein-2 in the lavage compared to control. In HTV, inhibitor of κB alpha decreased whereas phosphorylated extracellular signal-regulated kinases increased. TLR4 mutant and MyD88−/− mice showed markedly attenuated response to HTV, including less lung inflammation; pulmonary edema; and cell number, protein content, and the cytokines in the lavage. Furthermore, compared to wildtype, both TLR4 mutant and MyD88−/− mice had significantly higher inhibitor of κB alpha and reduced extracellular signal-regulated kinases phosphorylation following HTV.
Conclusions
TLR4-MyD88 signaling plays an important role in the development of ventilator-induced lung injury in mice, possibly through mechanisms involving nuclear factor-κB and mitogen-activated protein kinase pathways.
INTRODUCTION
Mechanical ventilation is a widely used life-saving supportive measure in the management of a variety of critically ill patients. However, it is well known that such therapy may produce an iatrogenic condition referred to as ventilator-induced lung injury (VILI).1–3 Although the exact underlying mechanisms of VILI remain unclear, recent emerging evidence suggests that mechanical ventilation may activate an inflammatory response in the lung that may contribute to VILI.4–7 Recently, a critical role for Toll-like receptor in mediating the effects of mechanical ventilation on lung inflammation and injury has been reported.8,9
Toll-like receptors (TLRs) are pattern recognition receptors and are considered key mediators in inflammation and play an essential role in innate and adaptive immune responses.10 TLR4 is the first identified and one of the most studied TLR family members. TLR4 recognizes both pathogen-associated molecular pattern (e.g., lipopolysaccharide) and damage-associated molecular pattern (e.g., high-mobility group box 1 and heat shock proteins). Upon stimulation, TLR4 signals through two downstream pathways: myeloid differentiation factor 88 (MyD88-) and Toll/Interleukin-1 receptor-domain containing adaptor-inducing interferon-β (TRIF-) dependent pathways, ultimately leading to the activation of nuclear factor-κB (NF-κB) and the production of proinflammatory cytokines.11–14 It has been suggested that activation of NF-κB and activator protein 1 controls inflammatory responses through the induction of proinflammatory cytokines, while NF-κB activation is associated with phosphorylation of inhibitor of κB alpha (IκBα) and activator protein 1 activation depends upon activation of mitogen-activated protein kinases (MAPKs).10 The TLR4-TRIF pathway has been identified as a key genetic pathway in acid aspiration-induced lung injury.15 More recently, the role of the TLR4-TRIF signaling pathway has been suggested in a mouse model of VILI.16 Given that MyD88-dependent signaling mediates early phase activation of NF-κB and is a major proinflammatory pathway, we sought to test the hypothesis that in addition to TLR4-TRIF pathway that TLR4-MyD88-dependent pathway plays a key role in a mouse model of VILI and further explore the role of NF-κB and MAPKs in TLR4-MyD88 signaling pathway.
Materials and Methods
Animals
TLR4-functional C3H/HeOuJ (TLR4-wildtype (WT)), TLR4-inactive mutated C3H/HeJ (TLR4-mutant), and MyD88-sufficient (C57BL/6J, the background strain, MyD88-WT) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). MyD88-knockout or null (MyD88-KO, MyD88−/−) mice were generated by Kawai et al.17 and were backcrossed > 10 generations into the C57BL/6J strain. All mice were female, 8–12 weeks of age, and weighed 20–30 g. Mice were fed with normal diet and water ad libitum and were housed in accordance with guidelines from the American Association for Laboratory Animal Care. All experimental animal protocols were performed in accordance with guidelines approved by the Animal Care and Use Committee at the University of Pittsburgh, Pittsburgh, Pennsylvania
VILI Animal Model
Mice were anesthetized with ketamine (100 mg/kg body weight) and xylazine (10 mg/kg) intraperitoneally. Anesthesia was maintained by supplementing with one-third of the initial dose of anesthetic agents regularly at approximately every 45 min during the experimental period. The mice were placed in the supine position on an adjustable warming pad (FHC Inc., Bowdoinham, ME) to maintain at 37 ± 1°C by continuously monitoring with a rectal temperature probe. The trachea was exposed with a neck midline incision under sterile conditions and a 20-gauge, 1-inch-long sterilized metal catheter with smooth beveled tip was inserted and sutured, then connected to a small animal ventilator (Inspira ASV, Harvard Apparatus, Holliston, MA). Mice with mechanical ventilation were ventilated either at 7 ml/kg (low tidal volume, LTV) with 140 breaths/min or 20 ml/kg (high tidal volume, HTV) with 100 breaths/min, 0 positive end-expiratory pressure for 4 h after tracheotomy, while control mice underwent tracheotomy but breathed spontaneously. Both spontaneous and ventilated mice were supplemented with oxygen (approximately 35–45% the fraction of inspired oxygen as determined by an oxygen monitor and always maintained less than 50%). During ventilation, intraperitoneal administration of saline (0.01 ml/g body weight) at 45-min intervals was supplied to maintain intravascular volume status. The oxygen saturation and heart rate in the anesthetized mice were continuously monitored with MouseOx (Starr Life Science Corp., Oakmont, PA) and maintained within physiological range with supplement of oxygen and intraperitoneal administration of glycoppyrolate (0.01 μg/g body weight). Animals were given a lethal dose of the anesthetic agent at the end of 4 h mechanical ventilation prior to harvesting samples.
Evans Blue Albumin Permeability Measurement
Lung injury was assessed by the alveolar capillary permeability to Evans blue albumin (EBA) as previous described.18 Briefly, Evans blue (EB, Sigma-Aldrich, St. Louis, MO) was dissolved in Ca2+-Mg2+ free phosphate-buffered saline (Invitrogen, Carlsbad, CA) in a concentration of 0.5% (5.2 mM). Evans blue dye conjugated to albumin (EBA) was prepared by adding bovine serum albumin (Fraction V, Sigma) to 0.5% Evans blue to a final concentration of 4% (0.6 mM). After dissolving thoroughly by gently stirring with a magnet bar, this EBA solution was then filtered sterilely through a 0.22-μm syringe filter and stored in aliquots at −80°C until use. Each aliquot was used only once for each animal to prevent cross contamination. To evaluate the alveolar capillary barrier function, EBA (20 mg/kg) was administered via the internal jugular vein one hour before euthanasia and tissue harvesting. At the termination of each experiment, all animals were euthanized and blood samples were obtained via the right heart for plasma EBA measurement. The pulmonary vasculature was then flushed with phosphate buffered saline to remove blood-borne elements. The right lung was ligated at the level of the right mainstem bronchus, excised, and weighed and stored in liquid nitrogen for subsequent Evans blue assay. After freeze/thaw, the lung tissue was homogenized in 2 ml phosphate buffered saline and incubated with additional 2 ml formamide (Sigma) at 60°C for 18 h. Formamide extracts were centrifuged (Beckman TLX, Fullerton, CA) at 15,000 × g for 30 min at 4°C and the centrifuged supernatants were collected to quantify lung EBA content by a dual wavelength spectrophotometric method (Model Du-640, Beckman) at 620 nm and 740 nm. EBA permeability index was calculated by dividing the corrected pulmonary tissue EBA absorbance at 620 nm per gram of lung tissue by the corrected plasma EBA absorbance at 620 nm.
Lung Wet/Dry Weight Ratio
Lung wet-to-dry weight ratio was used as an index of pulmonary edema formation. At the end of experimental period, mice were sacrificed and the right heart was flushed with phosphate buffered saline. Left lungs were weighed immediately after removal (wet weight) and again after drying in an oven at 65°C for 48 h (dry weight).
Lung Histopathology
Lungs were fixed by inflation with 10% formalin through intra-tracheal instillation. After fixation, the lungs were embedded in paraffin, 4 μm sections were stained with hematoxylin and eosin, and sections were examined at light microscopic level.
Bronchoalveolar Lavage Analysis
Lung inflammation was evaluated by cell counts and protein content in bronchoalveolar lavage (BAL) fluid. The BAL procedure was performed with instillation of sterile phosphate buffered saline in 1.0 ml volume with four replicates. Approximately 80% of the instilled volume was retrieved. All samples were kept on ice until processed. Recovered BAL fluids were centrifuged (5 min, 2,000 rpm, 4°C). The pellet was resuspended in phosphate buffered saline and total cell numbers in BAL fluid were counted using a hemocytometer. The supernatants were frozen immediately on dry ice and stored at −80°C for total protein concentration measurement with BCA Protein Assay kit (Pierce Chemical Co., Rockford, IL) and analysis for cytokines.
Cytokine Analysis in BAL fluid and Plasma
Tumor necrosis factor-alpha, interleukin-6 (IL-6), interleukin-1 beta (IL-1β), interleukin-10 (IL-10), macrophage-inflammatory protein-2 (MIP-2) in BAL fluid and in plasma were analyzed by enzyme-linked immunosorbent assay (ELISA) according to the instructions of commercial ELISA kits (R&D systems, Minneapolis, MN). The detection threshold of the assay was: a) Tumor necrosis factor-α: 5.1 pg/ml; b) IL-6: 1.6 pg/ml; c) IL-1β: 3.0 pg/ml; d) IL-10: 4.0 pg/ml; and e) MIP-2: 1.5 pg/ml.
Protein Extraction and Immunoblotting
Frozen lung tissues were thawed and suspended in 10 μl/mg ice-cold RIPA lysis buffer with protease inhibitors (Santa Cruz Biotechnology, Santa Cruz, CA), homogenized, and centrifuged at 13,000 rpm for 10 min at 4°C to remove insoluble materials. Supernatant protein concentrations were determined with BCA Protein Assay kit (Pierce Chemical Co.). After addition of 2× sodium dodecyl sulfate loading buffer (Invitrogen), equivalent amounts of protein were separated by electrophoresis using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels (Invitrogen). The proteins were then transferred onto polyvinylidene difluoride membrane (Invitrogen). Blots were blocked overnight at 4°C with Tris buffered saline containing 0.1% Tween-20 and 5% nonfat milk, then incubated with primary antibody for 1 h followed by horseradish peroxidase-conjugated secondary antibody for chemiluminescent visualization using enhanced chemiluminescence reagent (Perkin-Elmer Life Science, Boston, MA). Blots were stripped with stripping buffer (Pierce Chemical Co.) and reprobed with β-actin (Sigma). Films were scanned at 600 dpi in 16-bit gray-scale on an Epson Precision 4180 flatbed scanner (Epson America, Long Beach, CA). Densitometric analysis was performed using Image J software from the National Institutes of Health (Bethesda, MD). Antibodies against extracellular signal-regulated kinase (ERK), phosphorylated-ERK, IκBα were purchased from Cell Signaling Technology (Beverly, MA). Horseradish peroxidase-conjugated anti-rabbit, and anti-mouse antibodies were purchased from Santa Cruz Biotechnology.
Statistical Analysis
Data were presented as mean ± SD from 4–6 animals for each experimental condition as indicated in the legends. Two-way or one-way analysis of variance was used to compare differences within groups followed by pairwise multiple comparison procedure (Holm-Sidak method). SigmaPlot statistical software ver. 10.0 (Systat Software Inc., Chicago, IL) was used for statistical analysis. Statistical significance was defined as P < 0.05. All P values were two-tailed.
Results
TLR4-mutant mice prevent increased pulmonary permeability and edema following HTV
As illustrated in figure 1A, in TLR4-WT mice subjected to HTV (20 ml/kg), but not LTV (7 ml/kg), there was marked increase in permeability compared to tracheotomized and spontaneously breathing mice. In contrast, in TLR4-mutant mice, there were no differences in EBA permeability among the three groups. Similarly, there was less pulmonary edema, as assessed by excess lung water measured by wet/dry weight ratio in TLR4-mutant mice compared to TLR4-WT mice following HTV (fig. 1B).
Figure 1. TLR4 is a susceptible gene of increased pulmonary permeability and edema in VILI.
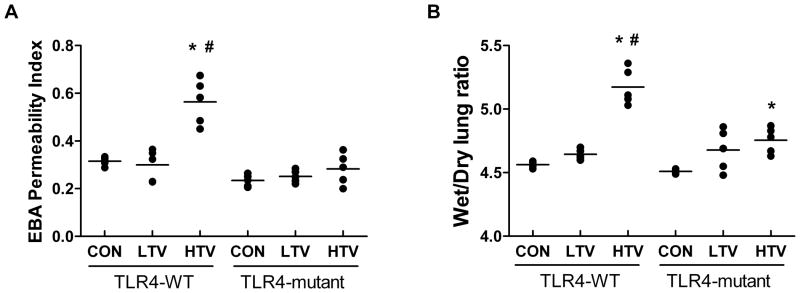
(A): Pulmonary permeability, as quantified by the amount of albumin conjugated to Evans Blue dye (EBA) fluxing across pulmonary vascular barriers, by using mouse lung turbidity correction factor and expressed as permeability index calculated by dividing the EBA absorbance in lung tissue over plasma (see Materials and Methods). In TLR4-WT mice, high tidal volume ventilation (HTV) elicited significant increase in permeability compared to both control (CON) and low tidal volume ventilation (LTV) mice. In contrast, in TLR4-mutant mice, HTV did not increase pulmonary permeability compared to both LTV and CON mice groups. (B): Pulmonary edema, as measured by lung Wet/Dry ratio, was significantly elevated in TLR4-WT mice following 4 h of HTV compared to LTV and unventilated CON mice. In contrast, HTV increased Wet/Dry ratio in TLR4-mutant mice was significantly attenuated compared to HTV in TLR4-WT mice. All data are presented as mean ± SD. *P < 0.05 compared to CON; #P < 0.05 compared to LTV. n = 5 in all groups.
TLR4-mutant mice elicit attenuated inflammatory cell infiltration and cytokine levels following HTV
Figure 2 shows representative lung tissue histology following VILI that illustrates a marked increase in inflammatory cell infiltration, alveolar septal thickening, and pulmonary edema in the lung subjected to HTV in TLR4-WT mice compared to TLR4-mutant mice. Although both BAL total protein concentration and total cell counts were significantly higher in both TLR4-WT and TLR4-mutant mice in response to HTV mechanical ventilation compared to LTV and CON, TLR4-mutant mice showed significantly attenuated elevation of protein content and cell counts in BAL compared to TLR4-WT mice during HTV (table 1). As shown in table 1, in TLR4-WT mice, BAL fluid levels of IL-6 and MIP-2 increased significantly after 4 h of HTV compared with unventilated controls. In contrast to TLR4-WT mice, TLR4-mutant mice elicited significant but much less pronounced increase in IL-6 and MIP-2 after 4 h of HTV. LTV did not affect the BAL levels of IL-6 and MIP-2. Interestingly, 4 hours of LTV and HTV also induced a significant increase in the plasma IL-6 but not MIP-2 in both TLR4-WT and TLR4-mutant mice. Plasma MIP-2 levels were below the level of detection (the detection threshold for MIP-2 was 1.5 pg/ml) in both TLR4-WT and TLR4-mutant control mice although there were somewhat increase in plasma MIP-2 levels following HTV. Other cytokines (e.g., Tumor necrosis factor-α, IL-1β, IL-10) were undetectable in both BAL fluid and plasma in both TLR4-WT and TLR4-mutant mice following 4 h of HTV.
Figure 2. Lung tissue histopathology in TLR4-WT and TLR4-mutant mice following VILI.
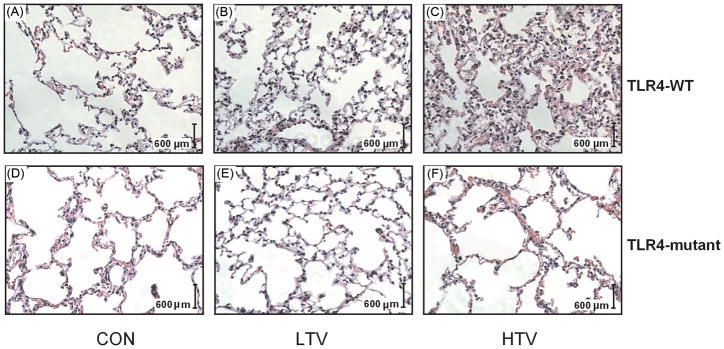
Representative photomicrographs of hematoxylin and eosin staining (× 20 objective) of lungs from TLR4-WT (A, B, C) and TLR4-mutant (D, E, F) mice subjected to unventilated and tracheotomized control (CON), low tidal volume ventilation (LTV), and high tidal volume ventilation (HTV). HTV illustrates a marked increase in inflammatory cell infiltration, alveolar septal thickening, and pulmonary edema in the lung in TLR4-WT mice (C) compared to TLR4-mutant mice (F).
Table 1.
BAL Cell Counts, Protein Content, IL-6 and MIP-2 Levels in VILI
| TLR4-WT | TLR4-mutant | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| CON | LTV | HTV | CON | LTV | HTV | |
| Protein content (μg/ml) | 378.9 ± 71.4 | 386.0 ± 18.1 | 657.2 ± 25.4* | 333.6 ± 22.3 | 332.4 ± 59.3 | 475.7 ± 82.4*, # |
| Cell counts (× 104) | 26.4 ± 2.2 | 27.5 ± 2.6 | 51.2 ± 1.1* | 17.3 ± 3.5 | 19.2 ± 3.4 | 28.3 ± 3.9*, # (n=4) |
| IL-6 (BAL) (pg/ml) | 133.4 ± 2.4 | 321.1 ± 103.9 | 452.5 ± 51.4* (n=6) | 92.6 ± 17.2 | 125.2 ± 3.9 | 265.7 ± 28.1*, # |
| MIP-2 (BAL) (pg/ml) | 16.5 ± 6.0 (n=6) | 20.9 ± 1.8 | 68.3 ± 0.9* | 9.6 ± 3.3 | 17.7 ± 14.6 | 37.9 ± 3.7*, # (n=4) |
| IL-6 (Plasma) (pg/ml) | 30.6 ± 9.8 | 83.7 ± 13.8† | 110.5 ± 23.4† | 34.8 ± 13.6 | 71.1 ± 2.8† | 111.3 ± 39.4* |
| MIP-2 (Plasma) (pg/ml) | 1.5§ | 1.6 ± 0.3 | 2.4 ± 0.9 | 1.5 § | 2.2 ± 0.7 (n=4) | 3.1 ± 1.5 (n=4) |
Data are presented as mean ± SD.
P < 0.05 compared to strain-matched CON and LTV;
P < 0.05 compared to strain-matched CON;
P < 0.05 compared to HTV in TLR4-WT mice. n = 5 in all groups except groups indicated in the table. Levels of MIP-2 in plasma were under detectable limits in unventilated control mice
The detection threshold of the ELISA assay
BAL = bronchoalveolar lavage; CON = control; HTV = high tidal volume ventilation; IL-6 = interleukin 6; LTV = low tidal volume ventilation; MIP-2 = macrophage inflammatory protein 2; TLR4 = Toll-like receptor 4; VILI = ventilator-induced lung injury; WT = wildtype.
TLR4-mutant mice attenuate NF-kB and MAPK activation induced by HTV
Since most TLR signaling pathways culminate in activation of the transcription factor NF-κB, we examined if HTV would lead to NF-κB activation and if TLR4 status affected such a change. Under normal conditions, NF-κB and IκBα form NF-κB/IκBα complexes in the cytoplasm that prevent NF-κB from translocating to the nucleus, thereby rendering it inactive. Phosphorylation and degradation of IκBα result in release of NF-κB from the complexes, enabling NF-κB to move into the nucleus where it activates the induction of proinflammatory cytokines. Thus, IκBα functions as an inhibitor of NF-κB, and NF-κB activation is associated with IκBα degradation.19–21 We therefore used IκBα degradation as an index of NF-κB activation. As shown in figure 3A, the lung IκBα protein level was decreased in response to HTV in TLR4-WT mice but maintained at the same level in TLR-mutant mice. Moreover, the mitogen-activated protein kinase (MAPK-ERK) in lung tissue became significantly more phosphorylated in TLR4-WT mice in response to HTV. Importantly, phosphorylated ERK (P-ERK) was significantly lower in TLR4-mutant mice compared to TLR4-WT mice following 4 h of HTV (fig. 3B).
Figure 3. Activation of NF-κB and MAPK in TLR4-WT mice following HTV.
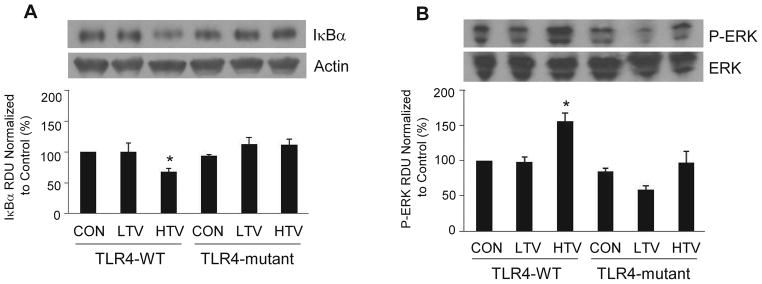
A representative of western blots of expression of inhibitor of κB alpha, IκBα (A) and MAPK-ERK phsphorylation, P-ERK (B) in lungs isolated from TLR4-wildtype (WT) and TLR4-mutant mice from three independent experiments 4 h after spontaneous breathing with tracheotomy (CON), mechanical ventilation with low tidal volume (LTV, 7 ml/kg), and high tidal volume (HTV, 20 ml/kg). All data are presented as mean ± SD. *P < 0.05 compared to CON in TLR4-WT mice. RUD = relative densitometry units.
MyD88 −/− mice have attenuated permeability and pulmonary inflammation
As shown in table 2, WT mice showed a modest but significant increase in pulmonary permeability compared with the unventilated control mice and there was no significant change in alveolar permeability in MyD88−/− mice. Similarly, lung histology (fig. 4) showed that HTV elicited marked pulmonary edema, alveolar septal thickening, and increased inflammatory cell infiltration in MyD88-WT mice compared to MyD88−/− and control mice. Furthermore, HTV increased BAL protein content and cell number significantly in MyD88-WT mice compared to control. In contrast, MyD88−/− mice did not increase either protein content or total cell number in BAL following 4 hours of HTV compared to control spontaneously unventilated mice. Finally, MyD88−/− mice had much lower levels of cytokines in BAL compared to WT mice subjected to HTV (table 2).
Table 2.
EBA Permeability and BAL Cell Counts, Protein Content, IL-6 and MIP-2 Levels in VILI
| CON | HTV (MyD88-WT) | HTV (MyD88-KO) | |
|---|---|---|---|
| EBA Permeability | 0.18 ± 0.01 (n=4) | 0.22 ± 0.02* (n=6) | 0.19 ± 0.02# |
| Protein Content (μg/ml) | 412.1 ± 74.8 (n=6) | 636.0 ± 99.0* | 469.4 ± 3.4# |
| Cell Counts (× 104) | 17.9 ± 0.9 (n=6) | 47.0 ± 3.3* | 21.9 ± 6.9# (n=4) |
| IL-6 (pg/ml) | 542.5 ± 58.1 (n=6) | 1010.2 ± 97.5* | 556.1 ± 215.3# (n=4) |
| MIP-2 (pg/ml) | 8.5 ± 2.5 | 86.3 ± 8.8* | 39.8 ± 10.9*, # (n=4) |
Data are presented as mean ± SD.
P < 0.05 compared to CON;
P < 0.05 compared to HTV (MyD88-WT). n = 5 in all groups except groups indicated in the table.
BAL = bronchoalveolar lavage; CON = control; EBA = Evans blue albumin; HTV = high tidal volume ventilation; IL-6 = interleukin 6; KO = knockout; MIP-2 = macrophage inflammatory protein 2; MyD88 = myeloid differentiation factor 88; VILI = ventilator-induced lung injury; WT = wildtype.
Figure 4. Lung histopathology in MyD88-WT and MyD88-KO mice following HTV.

Representative photomicrographs of hematoxylin and eosin staining from ventilator-induced lungs. High (× 40 objective, A, B, C) and low power (× 20 objective, D, E, F) views of mouse lungs from MyD88-wildtype (WT) and MyD88-knockout (KO) mice subjected to tracheotomized but unventilated control (CON) and ventilated with high tidal volume (HTV). MyD88-KO mice (C, F) show much less inflammatory cell infiltration, alveolar septal thickening, and pulmonary edema in the lung compared to the MyD88-WT mice (B, E) following HTV.
MyD88 −/− mice attenuate NF-kB and MAPK activation following HTV
Figure 5A shows that there was minimal degradation of IκBα and figure 5B shows MAPK-ERK phosphorylation, P-ERK in MyD88 −/− mice whereas there was significant degradation of IκBα and P-ERK in MyD88-WT mice following 4 h of HTV compared to control mice, suggesting both NF-κB and MAPK are involved in MyD88 signaling pathway in VILI.
Figure 5. Activation of NF-κB and MAPK in MyD88-WT mice following HTV.
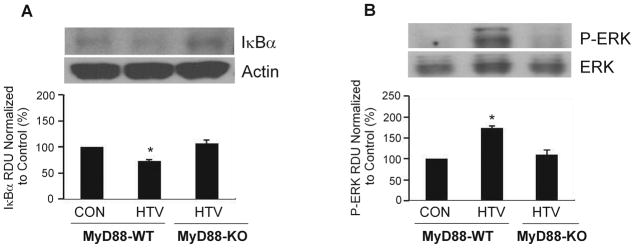
A representative of western blots of expression of inhibitor of κB alpha, IκBα (A) and MAPK-ERK phsphorylation, P-ERK (B) in lungs isolated from MyD88-wildtype (WT) and MyD88-knockout (KO) mice 4 h after spontaneous breathing with tracheotomy (CON) and mechanical ventilation with high tidal volume (HTV) from three independent experiments. All data are presented as mean ± SD. *P < 0.05 compared to CON mice. RUD = relative densitometry units.
Discussion
In the present study, we tested the role of TLR4-MyD88 signaling in the development of VILI. In a mouse VILI model, HTV at 20 ml/kg led to a series of pathological changes consistent with VILI. These included increased alveolar permeability (fig. 1) with elevated cells and protein content in alveolar space (table 1), significant inflammatory cell infiltration and cytokine production in lung tissues (table 1), and activation of the proinflammatory NF-κB and MAPK-ERK pathways (fig. 3). We demonstrate that TLR4 is essential in the development of VILI and that the critical role of TLR4 is most likely mediated via MyD88 signaling since MyD88−/− mice have attenuated pulmonary inflammation and were protected from increased pulmonary vascular permeability (table 2). These results suggest that TLR4-MyD88 signaling pathway may be critical for the development of VILI.
A hallmark of acute lung injury (ALI) is structural impairment in the alveolar-capillary membrane barrier with subsequent increased pulmonary vascular permeability and inflammation. Although extensive research with numerous animal models has attempted to identify effective therapeutic strategies for ALI for almost 35 yr, at present, research on the effects of HTV mechanical ventilation has had perhaps the most significant impact on clinical practice, and mechanical ventilation with reduced tidal volume ventilation strategies is the only definitive treatment modality to improve survival in patients with ALI.3,22,23 However, emerging evidence suggests that mechanical ventilation, even at protective low tidal volume, may activate an inflammatory response in the lung and may cause or predispose to VILI.6–8,16 For example, data from computed tomography demonstrate that even current protective LTV may produce tidal hyperinflation in patients with acute respiratory distress syndrome due to its anatomical heterogeneity of damaged lung,24,25 from which VILI may be considered a regional phenomenon, and thus additional information on the effect of high (and low) tidal volume mechanical ventilation in experimental animals remains an important translational effort. Further improvements of this model may involve additional injurious stimuli to mechanical ventilation, as suggested by the importance of “two-hit” animal model of ALI that has been emphasized by the National Institutes of Health,26 to simulate comorbidities and complexities of ALI. Nonetheless, the standard VILI model that we adapted has been an important contributor to insights into VILI. It is noteworthy that we took great care to continuously monitor oxygen saturation and heart rate in the anesthetized mice. In addition, mice were supplemented with oxygen under anesthesia with the fraction of inspired oxygen less than 50% to prevent possible hypoxia while purposefully minimizing chances of hyperoxia.
Lung inflammation, induced by alveolar over-distention during injurious mechanical ventilation, is considered to contribute to VILI.27,28 Toll-like receptors have long been recognized to play a crucial role in innate immune response and adaptive immune response to pathogens and to non-infectious tissue injury.10–14 TLR4 is unique among the TLRs because it is the only known TLR able to activate both MyD88-dependent and TRIF-dependent signaling pathways.11,29 More importantly, it has been demonstrated that TLR4 is activated after mechanical ventilation with low tidal volume ventilation7 and plays a critical role in ALI induced by high tidal volume ventilation,9 lipopolysaccharide,30 acid aspiration,15 hemorrhage,31 and ischemia and reperfusion injury.32 It is generally considered that TLR4 induces two downstream signaling pathways: MyD88-dependent and TRIF-dependent pathways, simultaneously from the plasma membrane.13,14 Upon binding to endogenous activators, TLR4 forms a dimmer and recruits the downstream adaptor molecule MyD88, ultimately leading to the activation of NF-κB and activator protein 1, inducing transcription of proinflammatory genes, and resulting in cytokine production (fig. 6). However, a recent study also showed that TLR4 may activate these two signaling pathways in a sequential manner; the MyD88 pathway is induced from the plasma membrane, whereas the TRIF pathway is induced from endosomes.33 Lately, Vaneker and colleagues16 demonstrated that lung inflammation was induced by mechanical ventilation with a low tidal volume (8 ml/kg) and that the effect is TRIF-dependent. In general, we did not detect significant pulmonary inflammation with our LTV protocol (7 ml/kg) but it is noteworthy that we instrumented (e.g., tracheostomy) our spontaneously breathing controls whereas Veneker et al16 used uncatheterized spontaneous breathing mice as control. As such, it is possible that the tracheostomy itself produced a sufficient inflammatory response to obscure the effect of LTV and/or to predispose the mice to injury from additional insult such as HTV mechanical ventilation. Nevertheless, LTV mechanical ventilation did not change pulmonary function such as EBA permeability, BAL cell counts and protein content as well as TLR4 downstream signaling pathways such as degradation of IκBα and MAPK/ERK phosphorylation. However, there was two-fold increase in plasma level of IL-6 with LTV compared to spontaneously tracheotomized control mice, a similar finding with recent study7 using mechanical ventilation at a tidal volume of 10 ml/kg for 6 h, indicating a relative small extent increase of systemic levels of IL-6 may not correlate to pulmonary function. In the current study, we also demonstrated that MyD88−/− mice elicited marked attenuation in the lung injury, including pulmonary capillary leak, pulmonary protein and cell emigration, and IL-6 production following HTV mechanical ventilation. Moreover, we found that both TLR4-mutant and MyD88−/− mice had attenuated NF-κB and MAPK/ERK activation in the lung, suggesting that the TLR4-MyD88 signaling contributes to the pro-inflammatory response during VILI. Taken together, these data suggest that pulmonary TLR4-MyD88 signaling may play a pivotal role in the pathogenesis of VILI and is responsible for the inflammatory changes observed in the injured lung.
Figure 6. TLR4-MyD88 signaling pathway following VILI.

Two pathways are involved referring as TLR4-MyD88 pathway and TLR4-TRIF pathway. Upon binding to endogenous activators (e.g., oxidized phospholipids, high mobility group box1, fragmented hyaluronan, heat shock proteins), the adaptor molecule MyD88, which is associated with TIRAP, is recruited to the TLR4 complex. Binding of MyD88 promotes association with IRAKs and TRAF6. The complex IRAKs/TRAF6 dissociates from the receptor and then interacts with another complex,TAK1. TAK1 results in phosphorylation and degradation of IκBα, consequent release of NF-κB, translocation of NF-κB into the nucleus, and transcription of pro-inflammatory genes inducing cytokine production. In the meantime, TAK1 also leads to activation of MAPK such as ERK that activates AP-1 transcription factor, which induces the transcription of inflammatory cytokines. In addition to MyD88, other adaptor proteins such as TRIF, which is associated with TRAM, mediate induction of IRF3, which in turn phosphorylates IκBα, leading to release of NF-κB to the nucleus. Unregulated proinflammatory cytokines may predispose VILI. TLR4 = Toll/interleukin-1 like receptor 4; MyD88 = myeloid differentiation factor 88; TIRAP = Toll/Interleukin-1 receptor (TIR)-domain-containing adaptor protein; TRIF = Toll/Interleukin-1 receptor-domain containing adaptor-inducing interferon-beta; TRAM = TRIF-related adaptor molecule; RIP1 = receptor interacting protein-1; IRAKs = interleukin (IL)-1 receptor-associated kinases; IRF3 = interferon regulatory factor 3; TRAF6 = tumor necrosis factor receptor associated factor 6; TAK1 = transforming growth factor activated kinase 1; MAPK = mitogen-activated protein kinases; ERK = extracellular signal-regulated kinases; AP1 = activator protein 1; IκBα = inhibitor of κB alpha; VILI = ventilator-induced lung injury.
It is unclear how TLR4-MyD88 (and TLR4-TRIF) signaling is activated in response to VILI. Lipopolysaccharide, itself, the pathogenic ligand of TLR4 is responsible for TLR4 activation in Gram-negative bacterial infection, and might have been a contributing factor. Although its potential role was minimized in a recent study16 in which Vaneker et al carefully monitored the presence of lipopolysaccharide, we are uncertain of any contaminating levels during our protocols. Nonetheless, all our surgical procedures were done in a sterile environment. In addition, the sham control mice underwent the same surgical intervention and were used to offset the possible effect of lipopolysaccharide in the animal group that underwent mechanical ventilation. In addition to lipopolysaccharide, recent evidence suggests an important role of endogenous ligands released in the setting of noninfectious tissue in activating TLR4. Examples of these potential endogenous ligands include high-mobility group box 1 protein released from necrotic cells, oxidized phospholipids from local generation of reactive oxygen species, low molecular weight hyaluran and fibrinogen from degraded extracellular matrix, heat shock proteins from necrotic cells, and surfactant protein-A.34–42 In light of some controversy between the detrimental effects of oxidized phospholipids via intratracheal administration15 and the protective effects of oxidized phospholipids via intravenous route 43,44 in VILI, it may be particularly fruitful to clarify the role of oxidized phospholipids as an endogenous ligand and a potential future therapeutic agent, especially through intratracheal administration. It is possible that the extent and duration of cyclic stretch by mechanical ventilation may trigger different quantitative and qualitative oxidized phospholipids that may produce either antiinflammatory or proinflammatory effects to regulate pulmonary permeability.
In the Acute Respiratory Distress Syndrome Network study,3 plasma IL-6 levels had a positive correlation to patients’ mortality when comparing patients with ALI supported by mechanical ventilation with LTV to that with HTV. In addition, lower tidal volume ventilation was associated with a decrease in plasma IL-6 levels.45 In our study, we confirmed the clinical studies as HTV elicited significant increase in IL-6 levels in both BAL and plasma following 4 h of HTV (table 1). The extent of IL-6 elevation correlates to increased BAL cell counts, indicating greater neutrophil migration and accumulation in the airspace. In the current study, we also found that HTV increased MIP-2 production in the bronchoalveolar lavage fluid (table 1 and 2). MIP-2 has been reported to be elevated in VILI and shown to augment migration of neutrophils into the alveoli.46 Although our data showed that LTV did not increase pulmonary permeability and pulmonary edema, LTV did not prevent increase of IL-6 in BAL and plasma, indicating that IL-6 may not be a causative factor for VILI, and increased level of IL-6 may be an adaptive host defense to potential injurious MV. Indeed in a recent study,47 IL-6 actually played a protective role in VILI and VILI is considered as a neutrophil-dependent process. The data from our group48 supports such a notion, which showed that the absence of neutrophil elastase potentially resulted in greater instances of VILI.
In the current study, we did not try to distinguish between the magnitude of contributions of downstream pathways of TLR4 (e.g., MyD88- or TRIF-dependent) signaling in VILI, as previous studies16 have also shown the role of TLR4-TRIF pathway in the development of inflammatory response in VILI. The strain of mouse used is a major confounding variable to contrast studies performed in different laboratories and we have systematically described this with respect to VILI.* In this regard, C57BL/6J are relatively resistant to HTV-induced lung injury (table 2). Nonetheless, within this strain (with MyD88 null mice being backbred on C57BL/6J background), it was apparent that MyD88−/− mice were resistant to VILI-induced increases in permeability, histopathologic damage, and cytokine production, consistent with an important role for this limb of TLR4 signaling.
The role of gender to VILI remains unclear and may have contributed to any differences from other existing reports. We chose female mice because they have been reported to have increased sensitivity to hyperoxia49, bleomycin50 and ozone51 induced acute lung injury.
No single animal model reproduces all of the characteristics of acute lung injury, and discrepancies exist between effective therapeutic approaches in animal studies and unsuccessful therapies in human clinical trials. Small animals, such as mice, are a very powerful research tool because they can be genetically modified to facilitate the detailed mechanistic study of complex pathways. It is technically challenging, however, to reproduce long-term mechanical ventilation in mice52 (compared to human patients) and thus in this regard, results from most animal studies need to be interpreted with caution. Nonetheless, short-term mechanical ventilation in rodents remains the most common model used and information derived from these studies has been useful in influencing clinical practice in humans.53,54 The mechanical ventilation model we used here combined high tidal volume ventilation and tracheotomy to reproduce clinical characteristics of acute lung injury in humans, namely increased pulmonary permeability and inflammation, although other factors may influence the magnitude of lung injury such as anesthetic use (e.g., sevoflurane, ketamine),55,56 variability of pressure support57 and positive end-expiratory pressure.58 The insights gained from the study may still have some important clinical implications. Although lung-protective ventilation strategies using low tidal volume with limited airway pressure and appropriate positive end-expiratory pressure has been proposed as a current recommendation for the management of critically ill patients, however, a world-wide survey has been found that it is not uncommon for critically ill patients to still be ventilated with high tidal volume.59 In addition, recent computed tomography images for patients with lung injury have demonstrated nonhomogeneous distribution of pulmonary aeration and thus normally relatively small aerated lung regions may receive the largest part of tidal volume and thereby be exposed to excessive alveolar wall tension and stress due to overdistention.25,60 From a clinical point of view, the high tidal volume mechanical ventilation mouse model is still of value in clinical relevance. The emerging evidence of clinical trials and animal models of VILI, including our current study, has shown that routine use of low tidal volume seems beneficial in all patients requiring mechanical ventilation especially for those patients with preexisting lung injury and inflammation.
In summary, our results demonstrate a critical role of TLR4 and its adaptor MyD88 in the development of VILI. The TLR4-MyD88 signaling pathway may possibly act via the mechanisms involving activation of NF-κB and MAPK. Strategies to modulate NF-κB and MAPK activation and routine use of low tidal volume mechanical ventilation may have potential therapeutic benefits in patients suffering from VILI.
Summary Statement.
The current study shows Toll-like receptor 4 via myeloid differentiation factor 88 signaling plays an important role in the development of ventilator-induced lung injury possibly through mechanisms involving nuclear factor-κB and mitogen-activated protein kinase pathways.
Acknowledgments
Supported in part by funding from the National Institutes of Health and National Heart, Lung, and Blood Institute (Bethesda, Maryland), Grants HL79456 (to L.M.Z) and HL 65697 (to B.R.P) and the Seed grant from the Department of Anesthesiology, University of Pittsburgh Medical Center.
The authors would like to thank Yulin Liu, M.D, Ph.D., Associate Professor and Director of Cytology in the Department of Pathology, Allegheny General Hospital, Pittsburgh, Pennsylvania for his technical assistance in pathohistology.
Footnotes
Presented at the Annual Meeting of the American Society of Anesthesiologists, New Orleans, Louisiana, October 20, 2009, A1278.
Presented in part at the Annual Meeting of the American Society of Anesthesiologists, New Orleans, Louisiana, October 20, 2009.
Received from the Department of Anesthesiology, University of Pittsburgh School of Medicine and University of Pittsburgh Medical Center.
References
- 1.Dos Santos CC, Slutsky AS. Invited review: Mechanisms of ventilator-induced lung injury: A perspective. J Appl Physiol. 2000;89:1645–55. doi: 10.1152/jappl.2000.89.4.1645. [DOI] [PubMed] [Google Scholar]
- 2.Dreyfuss D, Saumon G. Ventilator-induced lung injury: Lessons from experimental studies. Am J Respir Crit Care Med. 1998;157:294–323. doi: 10.1164/ajrccm.157.1.9604014. [DOI] [PubMed] [Google Scholar]
- 3.The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342:1301–8. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 4.Dos Santos CC, Slutsky AS. The contribution of biophysical lung injury to the development of biotrauma. Annu Rev Physiol. 2006;68:585–618. doi: 10.1146/annurev.physiol.68.072304.113443. [DOI] [PubMed] [Google Scholar]
- 5.Ma SF, Grigoryev DN, Taylor AD, Nonas S, Sammani S, Ye SQ, Garcia JG. Bioinformatic identification of novel early stress response genes in rodent models of lung injury. Am J Physiol Lung Cell Mol Physiol. 2005;289:L468–77. doi: 10.1152/ajplung.00109.2005. [DOI] [PubMed] [Google Scholar]
- 6.Ranieri VM, Suter PM, Tortorella C, De Tullio R, Dayer JM, Brienza A, Bruno F, Slutsky AS. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA. 1999;282:54–61. doi: 10.1001/jama.282.1.54. [DOI] [PubMed] [Google Scholar]
- 7.Gharib SA, Liles WC, Klaff LS, Altemeier WA. Noninjurious mechanical ventilation activates a proinflammatory transcriptional program in the lung. Physiol Genomics. 2009;37:239–48. doi: 10.1152/physiolgenomics.00027.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaneker M, Joosten LA, HeunVaneker M, Joosten LA, Heunks LM, Snijdelaar DG, Halbertsma FJ, van Egmond J, Netea MG, van der Hoeven JG, Scheffer GJ. Low-tidal-volume mechanical ventilation induces a toll-like receptor 4-dependent inflammatory response in healthy mice. Anesthesiology. 2008;109:465–72. doi: 10.1097/ALN.0b013e318182aef1. [DOI] [PubMed] [Google Scholar]
- 9.Hu G, Malik AB, Minshall RD. Toll-like receptor 4 mediates neutrophil sequestration and lung injury induced by endotoxin and hyperinflation. Crit Care Med. 2010;38:194–201. doi: 10.1097/CCM.0b013e3181bc7c17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388:621–5. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- 11.Chao W. Toll-like receptor signaling: A critical modulator of cell survival and ischemic injury in the heart. Am J Physiol Heart Circ Physiol. 2009;296:H1–12. doi: 10.1152/ajpheart.00995.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beutler BA. TLRs and innate immunity. Blood. 2009;113:1399–407. doi: 10.1182/blood-2008-07-019307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 14.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signaling. Nat Rev Immunol. 2007;7:353–64. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 15.Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, Liu H, Sun Y, Pasparakis M, Kopf M, Mech C, Bavari S, Peiris JS, Slutsky AS, Akira S, Hultqvist M, Holmdahl R, Nicholls J, Jiang C, Binder CJ, Penninger JM. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–49. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaneker M, Heunks LM, Joosten LA, van Hees HW, Snijdelaar DG, Halbertsma FJ, van Egmond J, Netea MG, van der Hoeven JG, Scheffer GJ. Mechanical ventilation induces a Toll/interleukin-1 receptor domain-containing adapter-inducing interferon beta-dependent inflammatory response in healthy mice. Anesthesiology. 2009;111:836–43. doi: 10.1097/ALN.0b013e3181b76499. [DOI] [PubMed] [Google Scholar]
- 17.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–22. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 18.Moitra J, Sammani S, Garcia JG. Re-evaluation of Evans Blue dye as a marker of albumin clearance in murine models of acute lung injury. Transl Res. 2007;150:253–65. doi: 10.1016/j.trsl.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 19.Prigent M, Barlat I, Langen H, Dargemont C. IkappaBalpha and IkappaBalpha/NF-kappa B complexes are retained in the cytoplasm through interaction with a novel partner, RasGAP SH3-binding protein 2. J Biol Chem. 2000;275:36441–9. doi: 10.1074/jbc.M004751200. [DOI] [PubMed] [Google Scholar]
- 20.Chao W, Shen Y, Li L, Zhao H, Meiler SE, Cook SA, Rosenzweig A. Fas-associated death-domain protein inhibits TNF-alpha mediated NF-kappaB activation in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2005;289:H2073–80. doi: 10.1152/ajpheart.01216.2004. [DOI] [PubMed] [Google Scholar]
- 21.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13:460–9. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 22.Matthay MA, Zimmerman GA. Acute lung injury and the acute respiratory distress syndrome: Four decades of inquiry into pathogenesis and rational management. Am J Respir Cell Mol Biol. 2005;33:319–27. doi: 10.1165/rcmb.F305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matthay MA. Treatment of acute lung injury: Clinical and experimental studies. Proc Am Thorac Soc. 2008;5:297–9. doi: 10.1513/pats.200708-141DR. [DOI] [PubMed] [Google Scholar]
- 24.Terragni PP, Rosboch G, Tealdi A, Corno E, Menaldo E, Davini O, Gandini G, Herrmann P, Mascia L, Quintel M, Slutsky AS, Gattinoni L, Ranieri VM. Tidal hyperinflation during low tidal volume ventilation in acute respiratory distress syndrome. Am J Respir Crit Care Med. 2007;175:160–6. doi: 10.1164/rccm.200607-915OC. [DOI] [PubMed] [Google Scholar]
- 25.Bellani G, Messa C, Guerra L, Spagnolli E, Foti G, Patroniti N, Fumagalli R, Musch G, Fazio F, Pesenti A. Lungs of patients with acute respiratory distress syndrome show diffuse inflammation in normally aerated regions: A [18F]-fluoro-2-deoxy-D-glucose PET/CT study. Crit Care Med. 2009;37:2216–22. doi: 10.1097/CCM.0b013e3181aab31f. [DOI] [PubMed] [Google Scholar]
- 26.Matthay MA, Zimmerman GA, Esmon C, Bhattacharya J, Coller B, Doerschuk CM, Floros J, Gimbrone MA, Jr, Hoffman E, Hubmayr RD, Leppert M, Matalon S, Munford R, Parsons P, Slutsky AS, Tracey KJ, Ward P, Gail DB, Harabin AL. Future research directions in acute lung injury: Summary of a National Heart, Lung, and Blood Institute working group. Am J Respir Crit Care Med. 2003;167:1027–35. doi: 10.1164/rccm.200208-966WS. [DOI] [PubMed] [Google Scholar]
- 27.Matthay MA, Bhattacharya S, Gaver D, Ware LB, Lim LH, Syrkina O, Eyal F, Hubmayr R. Ventilator-induced lung injury: In vivo and in vitro mechanisms. Am J Physiol Lung Cell Mol Physiol. 2002;283:L678–82. doi: 10.1152/ajplung.00154.2002. [DOI] [PubMed] [Google Scholar]
- 28.Belperio JA, Keane MP, Lynch JP, III, Strieter RM. The role of cytokines during the pathogenesis of ventilator-associated and ventilator-induced lung injury. Semin Respir Crit Care Med. 2006;27:350–64. doi: 10.1055/s-2006-948289. [DOI] [PubMed] [Google Scholar]
- 29.Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300:1524–5. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- 30.Tanimura N, Saitoh S, Matsumoto F, Akashi-Takamura S, Miyake K. Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem Biophys Res Commun. 2008;368:94–9. doi: 10.1016/j.bbrc.2008.01.061. [DOI] [PubMed] [Google Scholar]
- 31.Lv T, Shen X, Shi Y, Song Y. TLR4 is essential in acute lung injury induced by unresuscitated hemorrhagic shock. J Trauma. 2009;66:124–31. doi: 10.1097/TA.0b013e318181e555. [DOI] [PubMed] [Google Scholar]
- 32.Zanotti G, Casiraghi M, Abano JB, Tatreau JR, Sevala M, Berlin H, Smyth S, Funkhouser WK, Burridge K, Randell SH, Egan TM. Novel critical role of Toll-like receptor 4 in lung ischemia-reperfusion injury and edema. Am J Physiol Lung Cell Mol Physiol. 2009;297:L52–63. doi: 10.1152/ajplung.90406.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008;9:361–8. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, Fenton MJ, Tracey KJ, Yang H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26:174–9. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 35.Hazen SL. Oxidized phospholipids as endogenous pattern recognition ligands in innate immunity. J Biol Chem. 2008;283:15527–31. doi: 10.1074/jbc.R700054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–9. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 37.Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, Stolz DB, Geller DA, Rosengart MR, Billiar TR. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med. 2007;204:2913–23. doi: 10.1084/jem.20070247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mollen KP, Anand RJ, Tsung A, Prince JM, Levy RM, Billiar TR. Emerging paradigm: Toll-like receptor 4-sentinel for the detection of tissue damage. Shock. 2006;26:430–7. doi: 10.1097/01.shk.0000228797.41044.08. [DOI] [PubMed] [Google Scholar]
- 39.Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol Rev. 2005;204:27–42. doi: 10.1111/j.0105-2896.2005.00239.x. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Z, Schluesener HJ. Mammalian toll-like receptors: From endogenous ligands to tissue regeneration. Cell Mol Life Sci. 2006;63:2901–7. doi: 10.1007/s00018-006-6189-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsan MF, Gao Baochong. Pathogen-associated molecular pattern contamination as putative endogenous ligands of Toll-like receptors. J Endotoxin Res. 2007;13:6–14. doi: 10.1177/0968051907078604. [DOI] [PubMed] [Google Scholar]
- 42.Zhai Y, Qiao B, Shen XD, Gao F, Busuttil RW, Cheng G, Platt JL, Volk HD, Kupiec-Weglinski JW. Evidence for the pivotal role of endogenous toll-like receptor 4 ligands in liver ischemia and reperfusion injury. Transplantation. 2008;85:1016–22. doi: 10.1097/TP.0b013e3181684248. [DOI] [PubMed] [Google Scholar]
- 43.Nonas S, Birukova AA, Fu P, Xing J, Chatchavalvanich S, Bochkov VN, Leitinger N, Garcia JG, Birukov KG. Oxidized phospholipids reduce ventilator-induced vascular leak and inflammation in vivo. Crit Care. 2008;12:R27. doi: 10.1186/cc6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singleton PA, Chatchavalvanich S, Fu P, Xing J, Birukova AA, Fortune JA, Klibanov AM, Garcia JG, Birukov KG. Akt-mediated transactivation of the S1P1 receptor in caveolin-enriched microdomains regulates endothelial barrier enhancement by oxidized phospholipids. Circ Res. 2009;104:978–86. doi: 10.1161/CIRCRESAHA.108.193367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parsons PE, Eisner MD, Thompson BT, Matthay MA, Ancukiewicz M, Bernard GR, Wheeler AP. Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Crit Care Med. 2005;33:1–6. doi: 10.1097/01.ccm.0000149854.61192.dc. [DOI] [PubMed] [Google Scholar]
- 46.Quinn DA, Moufarrej RK, Volokhov A, Hales CA. Interactions of lung stretch, hyperoxia, and MIP-2 production in ventilator-induced lung injury. J Appl Physiol. 2002;93:517–25. doi: 10.1152/japplphysiol.00570.2001. [DOI] [PubMed] [Google Scholar]
- 47.Wolters PJ, Wray C, Sutherland RE, Kim SS, Koff J, Mao Y, Frank JA. Neutrophil-derived IL-6 limits alveolar barrier disruption in experimental ventilator-induced lung injury. J Immunol. 2009;182:8056–62. doi: 10.4049/jimmunol.0801323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaynar AM, Houghton AM, Lum EH, Pitt BR, Shapiro SD. Neutrophil elastase is needed for neutrophil emigration into lungs in ventilator-induced lung injury. Am J Respir Cell Mol Biol. 2008;39:53–60. doi: 10.1165/rcmb.2007-0315OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prows DR, Winterberg AV, Gibbons WJ, Jr, Burzynski BB, Liu C, Nick TG. Reciprocal backcross mice confirm major loci linked to hyperoxic acute lung injury survival time. Physiol Genomics. 2009;38:158–68. doi: 10.1152/physiolgenomics.90392.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gharaee-Kermani M, Hatano K, Nozaki Y, Phan SH. Gender-based differences in bleomycin-induced pulmonary fibrosis. Am J Pathol. 2005;166:1593–606. doi: 10.1016/S0002-9440(10)62470-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vancza EM, Galdanes K, Gunnison A, Hatch G, Gordon T. Age, strain, and gender as factors for increased sensitivity of the mouse lung to inhaled ozone. Toxicol Sci. 2009;107:535–43. doi: 10.1093/toxsci/kfn253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mokres LM, Parai K, Hilgendorff A, Ertsey R, Alvira CM, Rabinovitch M, Bland RD. Prolonged mechanical ventilation with air induces apoptosis and causes failure of alveolar septation and angiogenesis in lungs of newborn mice. Am J Physiol Lung Cell Mol Physiol. 2010;298:L23–35. doi: 10.1152/ajplung.00251.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295:L379–99. doi: 10.1152/ajplung.00010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bastarache JA, Blackwell TS. Development of animal models for the acute respiratory distress syndrome. Dis Model Mech. 2009;2:218–23. doi: 10.1242/dmm.001677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Conno E, Steurer MP, Wittlinger M, Zalunardo MP, Weder W, Schneiter D, Schimmer RC, Klaghofer R, Neff TA, Schmid ER, Spahn DR, Z’graggen BR, Urner M, Beck-Schimmer B. Anesthetic-induced improvement of the inflammatory response to one-lung ventilation. Anesthesiology. 2009;110:1316–26. doi: 10.1097/ALN.0b013e3181a10731. [DOI] [PubMed] [Google Scholar]
- 56.Wu GJ, Chen TL, Ueng YF, Chen RM. Ketamine inhibits tumor necrosis factor-alpha and interleukin-6 gene expressions in lipopolysaccharide-stimulated macrophages through suppression of toll-like receptor 4-mediated c-Jun N-terminal kinase phosphorylation and activator protein-1 activation. Toxicol Appl Pharmacol. 2008;228:105–13. doi: 10.1016/j.taap.2007.11.027. [DOI] [PubMed] [Google Scholar]
- 57.Spieth PM, Carvalho AR, Güldner A, Pelosi P, Kirichuk O, Koch T, de Abreu MG. Effects of different levels of pressure support variability in experimental lung injury. Anesthesiology. 2009;110:342–50. doi: 10.1097/ALN.0b013e318194d06e. [DOI] [PubMed] [Google Scholar]
- 58.Villar J, Herrera-Abreu MT, Valladares F, Muros M, Pérez-Méndez L, Flores C, Kacmarek RM. Experimental ventilator-induced lung injury: Exacerbation by positive end-expiratory pressure. Anesthesiology. 2009;110:1341–7. doi: 10.1097/ALN.0b013e31819fcba9. [DOI] [PubMed] [Google Scholar]
- 59.Putensen C, Theuerkauf N, Zinserling J, Wrigge H, Pelosi P. Meta-analysis: Ventilation strategies and outcomes of the acute respiratory distress syndrome and acute lung injury. Ann Intern Med. 2009;151:566–76. doi: 10.7326/0003-4819-151-8-200910200-00011. [DOI] [PubMed] [Google Scholar]
- 60.Terragni PP, Rosboch G, Tealdi A, Corno E, Menaldo E, Davini O, Gandini G, Herrmann P, Mascia L, Quintel M, Slutsky AS, Gattinoni L, Ranieri VM. Tidal hyperinflation during low tidal volume ventilation in acute respiratory distress syndrome. Am J Respir Crit Care Med. 2007;175:160–6. doi: 10.1164/rccm.200607-915OC. [DOI] [PubMed] [Google Scholar]