

Abstract
Background
VATER association was first described in 1972 by Quan and Smith as an acronym which identifies a non-random co-occurrence of Vertebral anomalies, Anal atresia, Tracheoesophageal fistula and/or Esophageal atresia, Radial dysplasia. It is even possible to find out Cardiovascular, Renal and Limb anomalies and the acronym VACTERL was adopted, also, embodying Vascular, as single umbilical artery, and external genitalia anomalies.
Methods
Data on patients with esophageal atresia (EA) with or without tracheoesophageal fistula (TEF) admitted in the Neonatal Intensive Care Unit (NICU) between January 2003 and January 2013 were evaluated for the contingent occurrence of typical VACTERL anomalies (VACTERL-type) and non tipical VACTERL anomalies (non-VACTERL-type). The inclusion criterion was the presence of EA with or without TEF plus two or more of the following additional malformations: vertebral defects, anal atresia, cardiovascular defects, renal anomalies and lower limb deformities, like radial dysplasia.
Results
Among 52 patients with EA/TEF, 20 (38,4%) had isolated EA and 7 (21,8%) had a recognized etiology such a syndrome and therefore were excluded. Among 32 infants with EA and associated malformations, 15 (46,8%) had VACTERL association. The most common anomalies were congenital heart defects (73,3%), followed by vertebral anomalies (66,6%). Many patients also had additional non-VACTERL-type defects. Single umbilical artery was the most common one followed by nervous system abnormalities and anomalies of toes. Between the groups of infants with VACTERL type and non-VACTERL-type anomalies, there are several overlapping data regarding both the tipically described spectrum and the most frequently reported non-VACTERL-type malformations. Thus, it is possible to differentiate infants with a full phenotype (VACTERL full phenotype) and patients that do not meet all the criteria mentioned above, but with some homologies with the first group (VACTERL partial phenotype).
Conclusion
The high frequency of non-VACTERL-type anomalies encountered in full and partial phenotype patients would suggest the need for an extension of the clinical criteria for the diagnosis of VACTERL association and also for pre- and post-operative management and follow-up in the short and long term.
Keywords: Esophageal atresia, Tracheo-esophageal fistula, VATER, VACTERL, Association, Congenital malformations, Anomalies
Background
VATER association is a specific pattern of multiple congenital malformations [1] and was first described in 1972 by Quan and Smith as a non-random co-occurrence of Vertebral anomalies, Anal atresia, Tracheoesophageal fistula and/or Esophageal atresia, Radial dysplasia. All single cases, did not show history of teratogenic etiologies or chromosomal aberrations [2-4]. The original acronym VATER has been widened into VACTERL, including Cardiovascular, Renal and Limb anomalies associated with the former ones [2,5-7]. Later, other authors suggested that diagnostic criteria should include Vascular anomalies (such as single umbelical artery) as part of the V in the acronym and other malformations (such as external genitalia and laryngotracheal anomalies) [2,8-12]. Furthermore, it was stated that VACTERL association would incorporate just the preaxial limb anomalies [13]. The inclusion of cardiac defects is still debated, however, as is the number of defects needed for the diagnosis of VACTERL association [13-19]. In 2001, other authors divided patients in an “upper” and a “lower” group, on the basis of the localization of their own anomalies as like as upper ones (cardiac, esophageal or limb deformities) and lower ones (caudal dysgenesis, renal, anorectal and lower vertebral anomalies) [16]. In a later study, patients with similar phenotypes were grouped in five peculiar clusters [20]. Since there is a wide margin of interpretation in diagnostic criteria, a thorough examination is mandatory in all patients with the suspicious of VACTERL association. In some studies the frequency was estimated between 1/10,000 and 1/40,000 infants [2]. Some studies have suggested that the condition is more common in males [21,22]. No specific distribution or geographic predominance in ethnic groups has been observed.
Because of the high clinical variability and sporadic occurrence, the etiology of VACTERL association is still unclear and a precise etiological cause has been identified just in a small fraction of patients so far (in example patients with hydrocephalus, VACTERL-H) [2].
In this study, data on 52 newborns with esophageal atresia (EA) with or without tracheoesophageal fistula (TEF) are shown in order to evaluate the contingent occurrence of typical VACTERL anomalies (VACTERL-type) and non typical VACTERL anomalies (non-VACTERL-type) such as single umbilical artery (SUA), duodenal atresia or stenosis, cleft lip and palate, genital anomalies, urinary tract abnormalities, hypothyroidism, anomalies of toes, lower limb anomalies and anomalies of respiratory, intestinal, vascular and nervous system.
Methods
We aim to evaluate data collected from our Neonatal Intensive Care Unit (NICU) in Palermo University, in which are referred most of newborns coming from all Western Sicily with congenital malformations requiring a surgical treatment. We enrolled in this retrospective analysis all newborns admitted between January 2003 and January 2013. We evaluated many variables such as sex, gestational age, body weight, length and cranial circumference, APGAR score, type of EA (according to Vogt-Roberts classification), VACTERL and non-VACTERL associated anomalies.
According to de Jong et al. [21], the inclusion criteria for the diagnosis of VATER/VACTERL association were the presence of EA with or without TEF and at least two of the following defects: vertebral anomalies (V), anal atresia (A), cardiovascular malformations (C), renal anomalies (R) and limb deformities (L). The diagnostic process included an ultrasound screening (cardiac, abdominal and cerebral ultrasonography), together with X-rays of spine and limbs. Karyotyping was performed in all children with multiple malformations, in order to identify patients with chromosomal abnormalities. In addition, in some patients, further molecular and cytogenetic analysis, like array-Comparative Genomic Hybridization (a-CGH) and FISH (Fluorescence In Situ Hybridization), were performed in order to assess potential microdeletions.
Results
Fifty-two patients have been enrolled in the study, 20 of wich (38,4%) had isolated EA and were therefore excluded from the subsequent analysis. Among 32 infants (61,5%) with EA and associated malformations, 22 patients showed a recognizable pattern of multiple congenital anomalies (associations, syndromes or sequences), in particular 7 (21,8%) with malformation syndromes or sequences (4 with Down syndrome, 2 with Nager syndrome and 1 with CHARGE syndrome), 15 with VACTERL associations (46,8%) and 10 with EA and other multiple malformations (31%; Figure 1). Feingold syndrome and other syndromes have been considered and excluded after accurate evaluation by an experienced clinical genetitian, who has personally addresses the clinical diagnosis and the eventual need of subsequent genetical investigations.
Figure 1.
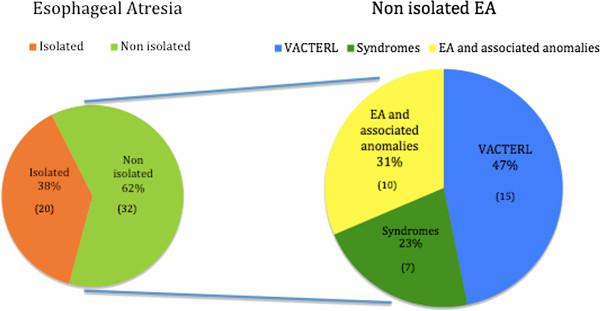
Patients with EA/TEF admitted in this study. Clustering of non-isolated-EA in syndromes, VACTERL association and EA with multiple malformations.
The 15 infants with VACTERL association, 8 males and 7 females, showed EA associated with at least two other malformations included in the classic phenotypical spectrum (Additional file 1). Only in 1 patient (6,6%) the full spectrum with six anomalies was observed, whereas 4 patients (26,6%) showed four defects and 10 patients (66,6%) three. Fourteen infants (93,3%) were preterm, namely 6 were born at < 34 weeks of gestation and 8 were late-preterm, born between the 34 and 37 weeks of gestation. One neonate was born full-term at 39,3 weeks of gestation. The birth weight ranged between 1490 and 2870 g, body length between 40 and 51 cm and cranial circumference between 28.5 and 33.5 cm. Eight infants were small for gestational age and seven were adequate for gestational age. The APGAR score was normal (between 8 and 10) for all patients, except 3 newborns who required intubation and/or intravenous adrenaline (Apgar score < 7). For all patients no parental consanguineity or familial recurrence for genetics disorders were found. All 15 infants with VACTERL association had a normal karyotype; three newborns also showed normal array-CGH analysis.
Table 1 displays the prevalence of the most frequent defects observed in our patients: vertebral anomalies were found in 10 patients (66,6%), 6 of which presented also rib abnormalities. Seven patients (46,6%) had anorectal malformations and specifically five anal atresia/stenosis and two anal anteriorization. Regarding congenital heart defects (11 patients, 73,3%), we observed 5 ventricular septal defects (VSD), 3 tetralogies of Fallot, 3 patent ductus arteriosus, 4 aortic arch defects and 2 atrial septal defects (ASD). Three patients (20%) showed renal malformations such as agenesis, dysgenesis and ectopia. Six patients (40%) had abnormalities of the upper limbs and more precisely 3 of them showed defects in forearm (radius/ulna) bones while the other 3 patients showed isolated thumb anomalies.
Table 1.
VACTERL-type defects observed in 15 newborns with esophageal atresia
| Defect | N. of patients | % |
|---|---|---|
| VERTEBRAL |
10 |
66,6 |
| - Vertebral anomalies |
8 |
53,3 |
| - Rib anomalies |
6 |
40 |
| ANAL |
7 |
46,6 |
| - Anal atresia/stenosis |
5 |
33,3 |
| - Abnormal placement of anus |
2 |
13,3 |
| CARDIOVASCULAR |
11 |
73,3 |
| - Atrial septum defect |
2 |
13,3 |
| - Ventricular septum defect |
5 |
33,3 |
| - Tetralogy of Fallot |
3 |
20 |
| - Aortic arch defects |
4 |
26,6 |
| - Patent ductus arteriosus |
3 |
20 |
| RENAL |
3 |
20 |
| - Renal agenesis |
1 |
6,6 |
| - Ectopic kidney |
1 |
6,6 |
| - Kidney malformation |
1 |
6,6 |
| LIMB |
6 |
40 |
| - Radius/ulna anomalies |
3 |
20 |
| - Thumb anomalies | 3 | 20 |
We also evaluated the presence of non-VACTERL-type malformations. In 9 VACTERL-patients we observed associated non-VACTERL-type anomalies and specifically 1 duodenal atresia, 2 digital anomalies, 1 venous anomalies (persistent left superior vena cava with dilatation of the coronary sinus), 3 single umbilical arteries, 2 genital abnormalities (undescended testis and micropenis), 2 developmental defects of the oropharyngeal region (cleft palate, microretrognathia and high arched palate), 1 respiratory system anomalies, 3 nervous system abnormalities (microphthalmia, coloboma, ventriculomegaly of the occipital horn).
The remaining 10 infants with EA did not have a fully expressed VACTERL phenotype: in 4 newborns the EA was exclusively associated with non-VACTERL-type defects, while in 6 patients EA was associated with only one of the major malformations typically required by the inclusion criteria. In these 6 infants (3 males and 3 females) not fully encountering the inclusion criteria, the most prevalent VACTERL-type defects were the cardiac ones, observed in 3 patients (one ASD and two VSD) and in 2 of them other malformations were not part of the phenotype (duodenal atresia, single umbilical artery, V finger clinodactyly and microcephaly). In the other 3 patients (out of six) we observed separately one with anal anteriorization, one with limb anomalies (supernumerary thumb) and venous abnormalities (portal-vein thrombosis), and one with vertebral anomalies and V finger clinodactyly. Instead in the other 4 babies with EA associated exclusively with non-VACTERL-type defects we observed one with duodenal atresia, another one with venous anomalies (azygos vein ecstasies) and two with congenital hypothyroidism (Additional file 1).
Discussion
In our study, the cardiovascular system was the most common affected district in association with EA, followed by spine, anal region, limbs and kidney. These results have been compared to those of previously published studies. Solomon et al. reported a higher prevalence of cardiac malformations (80%) followed by vertebral anomalies (78%), renal abnormalities (72%), anal atresia (55%), esophageal atresia (52%) and limb anomalies (47%) [20]. Cardiac anomalies were the most common defects and vertebral anomalies were frequent as well as in our study although, on the other hand, renal malformations were present in 72% of cases, according to other reports, compared to 20% in our population. Indeed, this study included all cases with any three or more of the six main defects, although not necessarily EA/TEF.
We enrolled only those patients with EA/TEF and at least two other typical defects within the spectrum of VACTERL association. There are only a few articles in the literature reporting similar data [15,21,23-25]. de Jong et al. used the same inclusion criteria and reported a higher prevalence of vertebral/rib anomalies (68,9%) and cardiovascular defects (65,6%), followed by anal atresia (42,2%), renal abnormalities (35,6%) and limb anomalies (32,2%) [21]. In this study, both vertebral anomalies and cardiac malformations were the most common VACTERL-type defects, while there are differences about other ones, especially anal atresia and kidney malformations.
The frequency of non-VACTERL-type anomalies varies in the literature: it has been reported a rate of 70% [22] versus 57% reported by other studies [15,22] and much lower percentages by other authors [22]. Anyway, once again it is difficult to compare the results, due to the high heterogeneity of the inclusion criteria chosen for patient selection in different studies. In our study 9 (60%) of 15 newborns with VACTERL association presented non-VACTERL-type anomalies (Table 2).
Table 2.
Frequency of non-VACTERL-type abnormalities in 15 patients with VACTERL association and in 10 patients with EA according to de Jong criteria[18]
| Non-VACTERL-type anomalies |
de Jong et al. VACTERL patients |
Palermo NICU VACTERL patients |
Palermo NICU EA patients |
|---|---|---|---|
| (≥ 3 anomalies) | (≥ 3 anomalies) | (< 3 anomalies) | |
| Number of patients |
90 |
15 |
10 |
| % cases with associated anomalies |
70% |
60% |
70% |
| Single umbilical artery |
20% |
20% |
20% |
| Duodenal atresia/stenosis |
8,90% |
6,6% |
20% |
| Cleft lip and palate |
4,40% |
13,3% |
0 |
| Genital anomalies |
7,80% |
13,3% |
0 |
| Respiratory system anomalies |
13,30% |
6,6% |
10% |
| Vascular anomalies |
7,70% |
6,6% |
20% |
| Nervous system anomalies |
11,10% |
20% |
10% |
| Anomalies of toes |
10% |
13,3% |
20% |
| Lower limb anomalies |
16,7% |
6,6% |
0% |
| Hypothyroidism | 0% | 0% | 20% |
Ten infants with EA and other multiple malformations did not met the criteria for VACTERL diagnosis. Seven of them (70%) showed non-VACTERL-type anomalies with similar percentages compared with our VACTERL population. The SUA malformation was the most common non-VACTERL-type anomaly found in our population, with a percentage of 20% in VACTERL infants and 20% in infants with EA and other malformations. Other studies reported frequency ranges from 8 to 70% [6,21,23,24], supporting the opportunity to extend the acronym with the letter S (VACTERLS), in spite of including it in the vascular anomalies (V) [2]. Another non-VACTERL-type anomaly which is frequently observed in VACTERL patients is duodenal atresia (8,9% [21], 8% [23,24]), which has been reported in 1 of our VACTERL-patients (6,6%) and 2 (20%) of our infants with EA and other malformations. Two VACTERL newborns (13,3%) presented cleft palate, comparing with 4,4% [21] and 13,2% [15] of other studies. Two newborns (13,3%) showed genital abnormalities (undescended testis and micropenis) against the 7,8% reported in literature [21]. Even the abnormalities of the fingers (ectrodactyly with duplication and/or aplasia of phalanges) are found in 20% of patients with EA associated with other non-VACTERL-type malformations and in the 13,3% of VACTERL infants. We have noticed the presence of venous malformations such as ectasies, dilatation or thrombosis, mainly involving the portal vein, vena cava or azygos vein in 20% of infants with EA and multiple malformations and in 6,6% of VACTERL-patients, percentages similar to those reported in the literature (7,7% [21]). We found abnormalities in the nervous system (such as ventriculomegaly, microcephaly, coloboma and microphthalmia) in 3 neonates (20%) with VACTERL association and 1 (10%) with EA and multiple malformations, according to other reports in the literature [21]. Hypothyroidism was present in 2 infants (20%) with EA and multiple malformations and respiratory system anomalies in 1 newborn (10%) with EA and multiple malformations and in 1 (6,6%) with VACTERL association. Finally, we found lower limb anomalies in 1 neonate (6,6%) with VACTERL association.
It is interesting to point out that between the two initially disjointed groups, VACTERL-patients with full phenotype and infants with EA and multiple malformations, there are several overlapping data, both for the most typical malformations associated with EA and for the non-VACTERL-type anomalies. On these bases, we can actually define three clusters of patients (Figure 2), identifying an additional subgroup with patients who do not show a full phenotype, therefore do not meet the classic diagnostic criteria but differ from infants having an EA associated only with non-VACTERL-type defects (Table 3, Figure 3). There 6 patients, presenting only two of the major components of the acronym VACTERL, can be considered at the extreme border of the VACTERL spectrum and can be defined as having a partial phenotype (Figure 4).
Figure 2.
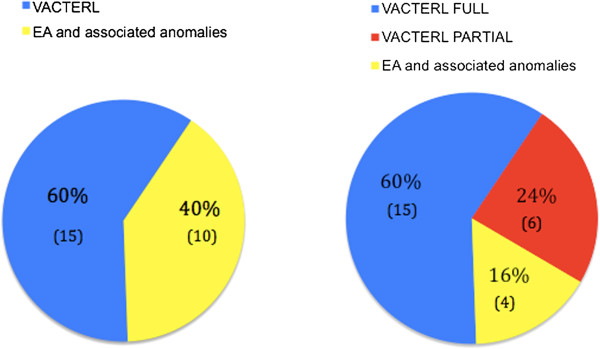
Partition of patients in three groups. Identification of three clusters of patients: VACTERL full phenotype, VACTERL partial phenotype and EA with only non-VACTERL-type defects.
Table 3.
VACTERL-type and non-VACTERL-type malformations in three clusters of our cohort
| Anomalies |
Full phenotype |
Partial phenotype |
Non-VACTERL associated anomalies |
|||
|---|---|---|---|---|---|---|
| (15) | (6) | (4) | ||||
| V |
10 |
66,6% |
1 |
16,6% |
0 |
0% |
| A |
7 |
50% |
1 |
16,6% |
0 |
0% |
| C |
10 |
71,4% |
3 |
50% |
0 |
0% |
| TE |
14 |
100% |
6 |
100% |
4 |
100% |
| R |
3 |
21,4% |
0 |
0% |
0 |
0% |
| L |
6 |
42,8% |
1 |
16,6% |
0 |
0% |
| SUA |
3 |
21,4% |
1 |
16,6% |
1 |
0% |
| Duodenal atresia |
1 |
7% |
1 |
16,6% |
1 |
25% |
| Cleft lip and palate |
2 |
14,2% |
0 |
0% |
0 |
0% |
| Genital anomalies |
2 |
14,2% |
0 |
0% |
0 |
0% |
| Hypothyroidism |
0 |
0% |
0 |
0% |
2 |
50% |
| Vascular anomalies |
1 |
7% |
1 |
16,6% |
1 |
25% |
| Nervous system anomalies |
3 |
21,4% |
1 |
16,6% |
0 |
0% |
| Anomalies of toes |
2 |
14,2% |
2 |
33,3% |
0 |
0% |
| Lower limb anomalies | 1 | 7% | 0 | 0% | 0 | 0% |
V vertebral anomalies, A anal atresia, C cardiovascular anomalies, TE tracheoesophageal fistula and/or esophageal atresia, R renal anomalies, L limb anomalies, SUA single umbilical artery.
Figure 3.
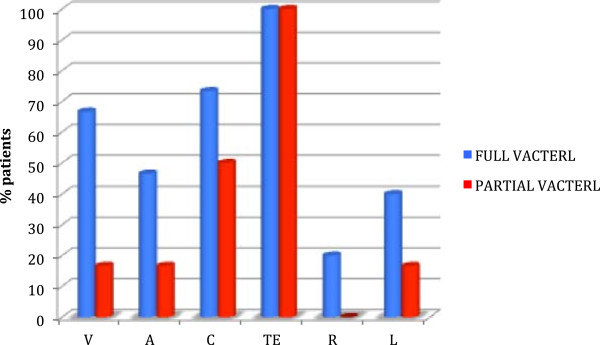
Comparison between patients with VACTERL full phenotype and VACTERL partial phenotype. There are several overlapping data for the typical VACTERL malformations. V, vertebral anomalies; A, anal atresia; C, cardiovascular anomalies; TE, tracheoesophageal fistula and/or esophageal atresia; R, renal anomalies; L, limb anomalies.
Figure 4.

Flow-chart showing patient group composition.
Given that the etiology of VACTERL association is still unknown, the diagnosis is only possible evaluating the phenotypical features, thanks to a good skilled physician. Consequently, these infants with partial phenotype may still belong to the VACTERL association, even though they present a limited clinical spectrum. Analyzing data showed in Additional file 1 it is possible to note the wide spectrum that characterizes patients from the most expressed phenotype to the most restricted one. It is questionable how to split these patients in groups with different prognosis and follow-up schedules. Waiting for more informative molecular-genetic data, it is necessary to avoid restriction of clinical criteria for recruitment, including SUA in the acronym (as already suggested by other authors [8]) and considering other structural abnormalities frequently associated, thus expanding the phenotypic continuum that characterizes the VACTERL association.
It is remarkable to consider that all defects observed in the VACTERL association spectrum, including EA, and most of the non typical associated anomalies take place during the blastogenetic period, the first 4 weeks after conception, in which the embryo is a single developmental field. Most of the associations depends on blastogenetic defects and may show a partial phenotypical overlapping affecting predominantly midline, independently from the etiological basis, due to the common timing and pathogenetic mechanism [11].
Conclusions
Many associated anomalies may be present in a patients with EA/TEF, which therefore need to be carefully investigated. These complex clinical pictures may represent a recognizable pattern of multiple malformations such as chromosomal or genetic syndromes or associations, mainly VACTERL association. Sometimes a recognizable malformation pattern is not present. The high frequency of non-VACTERL-type anomalies encountered in “full” and “partial” phenotype patients would suggest the need for an extension of the classical clinical criteria. All these patients, with high phenotypical variability, likely belong to a common wide clinical spectrum and is today very difficult to differentiate clearly distinct groups until more molecular genetic etiologic informations are available. More detailed clinical definition is important to start a proper short and long term follow-up, even in infants with EA/TEF without other VACTERL-type anomalies. The better definition of the extended phenotypical spectrum in VACTERL association allows a more complete diagnostic screening in order to detect even hidden or mild associated malformations, providing a more defined prognosis and follow-up program for the newborn. Future improvements in molecular genetic etiology should allow to assess a genotype-phenotype correlation for such an extended and variable clinical spectrum.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All the authors participated in the data collection. SLP and AG designed the study, carried out the data elaboration and coordinated the study. GC and MG revised the manuscript critically for important intellectual content. All authors read and approved the final manuscript.
Supplementary Material
Characteristics of 25 patients with esophageal atresia and associated anomalies.
Contributor Information
Simona La Placa, Email: simonalaplaca@libero.it.
Mario Giuffrè, Email: mario.giuffre@unipa.it.
Antonella Gangemi, Email: gangemiantonella@gmail.com.
Stefania Di Noto, Email: stefydinoto@tiscali.it.
Federico Matina, Email: fedematina@libero.it.
Federica Nociforo, Email: anglesite@libero.it.
Vincenzo Antona, Email: vincenzo.antona@unipa.it.
Maria Rita Di Pace, Email: mariarita.dipace@unipa.it.
Maria Piccione, Email: maria.piccione@unipa.it.
Giovanni Corsello, Email: giovanni.corsello@unipa.it.
References
- Corsello G, Giuffrè M. Congenital malformations. J Matern Fetal Neonatal Med. 2012;25(S1):25–29. doi: 10.3109/14767058.2012.664943. [DOI] [PubMed] [Google Scholar]
- Solomon BD. VACTERL/VATER association. Orphanet J Rare Dis. 2011;6:56. doi: 10.1186/1750-1172-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan L, Smith DW. In: The clinical delineation of birth defects. Volume XII. G.I. tract including liver and pancreas. Bergsma D, editor. Baltimore: The Williams and Wilkins company; 1972. The VATER association: vertebral defects, anal atresia, tracheoesophageal fistula with esophageal atresia, radial dysplasia; pp. 75–78. [Google Scholar]
- Quan L, Smith DW. The VATER association. Vertebral defects, Anal atresia, T-E fistula with esophageal atresia, Radial and Renal dysplasia: a spectrum of associated defects. J Pediatr. 1973;82:104–107. doi: 10.1016/S0022-3476(73)80024-1. [DOI] [PubMed] [Google Scholar]
- Hall BD. In: Management of genetic syndromes. Third. Suzanne B, Cassidy MD, Allanson JE, editor. Hoboken, New Jersey: Pubblished by John Wiley & sons Inc.; 2010. VATER/VACTERL association; pp. 871–878. [Google Scholar]
- Temtamy SA, Miller JD. Extending the scope of the VATER association: definition of the VATER syndrome. J Pediatr. 1974;85:345–349. doi: 10.1016/S0022-3476(74)80113-7. [DOI] [PubMed] [Google Scholar]
- Nora AH, Nora JJ. A syndrome of multiple congenital anomalies associated with teratogenic exposure. Arch Environ Health. 1975;30:17–21. doi: 10.1080/00039896.1975.10666626. [DOI] [PubMed] [Google Scholar]
- Kaufman RL. Birth defects and oral contraceptives. Lancet. 1973;1:1396. doi: 10.1016/s0140-6736(73)91731-5. [DOI] [PubMed] [Google Scholar]
- Apold J, Dahl E, Aarskog D. The Vater association: malformations of the male external genitalia. Acta Paediatr Scand. 1976;65:150–152. doi: 10.1111/j.1651-2227.1976.tb16528.x. [DOI] [PubMed] [Google Scholar]
- Solomon BD, Raam MS, Pineda-Alvarez DE. Analysis of genitourinary anomalies in patients with VACTERL (Vertebral anomalies, Anal atresia, Cardiac malformations, Tracheo-Esophageal fistula, Renal anomalies, Limb abnormalities) association. Congenit Anom (Kyoto) 2011;51:87–91. doi: 10.1111/j.1741-4520.2010.00303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsello G, Maresi E, Corrao AM, Dimita U, Lo Cascio M, Cammarata M, Giuffrè L. VATER/VACTERL association: clinical variability and expanding phenotype including laryngeal stenosis. Am J Med Genet. 1992;1;44(6):813–815. doi: 10.1002/ajmg.1320440619. [DOI] [PubMed] [Google Scholar]
- Ghirri P, Scaramuzzo RT, Bertelloni S, Pardi D, Celandroni A, Cocchi G, Danieli R, De Santis L, Di Stefano MC, Gerola O, Giuffrè M, Gragnani GS, Magnani C, Meossi C, Merusi I, Sabatino G, Tumini S, Corsello G, Boldrini A. Prevalence of hypospadias in Italy according to severity, gestational age and birthweight: an epidemiological study. Ital J Pediatr. 2009;35:18. doi: 10.1186/1824-7288-35-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botto LD, Khoury MJ, Mastroiacovo P, Castilla EE, Moore CA, Skjaerven R, Mutchinick OM, Borman B, Cocchi G, Czeizel AE, Goujard J, Irgens LM, Lancaster PA, Martínez-Frías ML, Merlob P, Ruusinen A, Stoll C, Sumiyoshi Y. The spectrum of congenital anomalies of the VATER association: an international study. Am J Med Genet. 1997;71:8–15. doi: 10.1002/(SICI)1096-8628(19970711)71:1<8::AID-AJMG2>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Czeizel A, Ludanyi I. An aetiological study of the VACTERL-association. Eur J Pediatr. 1985;144:331–337. doi: 10.1007/BF00441773. [DOI] [PubMed] [Google Scholar]
- Czeizel A, Telegdi L, Tusnàdy G. In: Multiple Congenital Anomalies. Akadémiai Kiadò, editor. Budapest: Czeizel, Telegdi, Tusnàdy; 1988. VACTERL-association; pp. 247–280. [Google Scholar]
- Kallen K, Mastroiacovo P, Castilla EE, Robert E, Kallen B. VATER non-random association of congenital malformations: study based on data from four malformation registers. Am J Med Genet. 2001;101:26–32. doi: 10.1002/ajmg.1201. [DOI] [PubMed] [Google Scholar]
- Khoury MJ, Cordero JF, Greenberg F, James LM, Erickson JD. A population study of the VACTERL association: evidence for its etiologic heterogeneity. Pediatrics. 1983;71:815–820. [PubMed] [Google Scholar]
- Martinez-Frias ML, Frias JL. VACTERL as primary, polytopic developmental field defects. Am J Med Genet. 1999;83:13–16. doi: 10.1002/(SICI)1096-8628(19990305)83:1<13::AID-AJMG4>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Rittler M, Paz JE, Castilla EE. VACTERL association, epidemiologic definition and delineation. Am J Med Genet. 1996;63:529–536. doi: 10.1002/(SICI)1096-8628(19960628)63:4<529::AID-AJMG4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Solomon BD, Pineda-Alvarez DE, Raam MS, Bous SM, Keaton AA, Vélez JI, Cummings DA. Analysis of component findings in 79 patients diagnosed with VACTERL association. Am J Med Genet A. 2010;152A:2236–2244. doi: 10.1002/ajmg.a.33572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong EM, Felix JF, Deurloo JA, van Dooren MF, Aronson DC, Torfs CP, Heij HA, Tibboel D. Non-VACTERL-type anomalies are frequent in patients with Esophageal Atresia/Tracheo-esophageal Fistula and full or partial VACTERL association. Birth Defects Res A Clin Mol Teratol. 2008;82:92–97. doi: 10.1002/bdra.20437. [DOI] [PubMed] [Google Scholar]
- Oral A, Caner I, Yigiter M, Kantarci M, Olgun H, Ceviz N, Salman AB. Clinical characteristics of neonates with VACTERL association. Pediatr Int. 2012;54(3):361–364. doi: 10.1111/j.1442-200X.2012.03566.x. [DOI] [PubMed] [Google Scholar]
- Chittmittrapap S, Spitz L, Kiely EM, Brereton MJ. Oesophageal atresia and associated anomalies. Arch Dis Child. 1989;64:364–368. doi: 10.1136/adc.64.3.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz L. Oesophageal atresia. Orphanet J Rare Dis. 2007;2:24. doi: 10.1186/1750-1172-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keckler SJ, St Peter SD, Valusek PA, Tsao K, Snyder CL, Holcomb GW 3rd, Ostlie DJ. VACTERL anomalies in patients with esophageal atresia: an updated delineation of the spectrum and review of the literature. Pediatr Surg Int. 2007;23:309–313. doi: 10.1007/s00383-007-1891-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characteristics of 25 patients with esophageal atresia and associated anomalies.