

Abstract
The Nrf2 (NF-E2 related factor 2)-ARE (antioxidant response element) pathway controls a powerful array of endogenous cellular antioxidant systems and is an important pathway in the detoxification of reactive oxygen species (ROS) in the brain. Using a combination of quantitative proteomics and siRNA screening, we have identified novel protective mechanisms of the Nrf2-ARE pathway against oxidative stress in astrocytes. Studies from our lab and others have shown Nrf2 overexpression protects astrocytes from oxidative stress. However, the exact mechanisms by which Nrf2 elicits these effects are unknown. In this study, we show that induction of Nrf2 reduces levels of reactive oxygen species (ROS) produced by various oxidative stressors and results in robust cytoprotection. To identify the enzymes responsible for these effects, we used stable isotope labeling by amino acids in cell culture (SILAC) and quantitative shotgun proteomics to identify 72 Nrf2-regulated proteins in astrocytes. We hypothesized a subset of these proteins might play a critical role in Nrf2 protection. In order to identify these critical proteins, we used bioinformatics to narrow our target list of proteins and then systematically screened each candidate with siRNA to assess the role of each in Nrf2 protection. We screened each target against H2O2, tert-butyl hydroperoxide, and 4-hydroxynonenal and subsequently identified three enzymes–catalase, prostaglandin reductase-1, and peroxiredoxin-6–that are critical for Nrf2-mediated protection in astrocytes.
Introduction
Oxidative stress has been implicated as a causative agent in a wide spectrum of neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis [1], [2], [3]. In the brain, the activation of the Nrf2-ARE pathway is protective against various stressors, including glutathione depletion, peroxides, excitotoxins, mitochondrial toxins, and intracellular calcium overload [4], [5], [6], [7], [8], [9], [10]. The pathway is composed of an enhancer element, the antioxidant response element (ARE), and its transcription factor, NF-E2-related factor-2 (Nrf2) [4], [11], [12], [13]. Nrf2 is regulated by its binding partner Kelch ECH associating protein 1 (Keap1) [14], [15], [16] and under normal conditions is sequestered by Keap1 in the cytoplasm; however, under conditions of oxidative stress, Nrf2 is released from Keap1 and translocates to the nucleus where it binds the ARE and drives gene expression [17]. Genes under the putatiave control of the Nrf2-ARE pathway are direct antioxidants, including glutathione [18], [19]; enzymes that inactivate ROS, including superoxide dismutase and catalase [20], [21]; reductive co-factors, including NADPH, [22]; and enzymes involved in protein turnover and homeostasis [23].
In vitro studies have indicated that the Nrf2 pathway is relatively unresponsive in neurons but highly inducible in astrocytes. Furthermore, astrocytic-specific Nrf2 activation confers protection against ROS to co-cultured neurons [6], [7], [9]. Astrocyte-specific overexpression in vivo has been shown to mitigate disease pathogenesis in animal models of Huntington’s disease, amyotrophic lateral sclerosis, Parkinson’s disease, and Alexander’s disease [9], [24], [25], [26].
In primary astrocyte cultures, gene expression profiling has revealed a number of Nrf2-regulated cellular defense pathways including those involved in the production and utilization of glutathione [4], [6], [7]. Additional studies have demonstrated the central importance of glutathione in Nrf2-mediated neuroprotection [6], [27], [28]. However, the exact mechanisms of Nrf2-mediated protection in astrocytes has yet to be identified. In light of this, we have undertaken the current study using a combination of quantitative proteomics and siRNA screening to elucidate the specific mechanisms of astrocytic Nrf2 protection against oxidative stress.
Results
Nrf2 Activation Protects Astrocytes from Hydrogen Peroxide
Although Nrf2 induction has previously been shown to protect primary astrocyte cultures from H2O2 toxicity, we sought to establish a toxicity assay to identify specific mechanisms of Nrf2 protection [5], [6], [7]. In order to achieve this, we generated H2O2 toxicity curves and then assessed cell viability by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H tetrazolium (MTS) and lactate dehydrogenase release (LDH) assays. Nrf2 induction with tBHQ confers robust protection against H2O2 toxicity ( Figure 1A ). Furthermore, this protection is reversed in Nrf2 knockout astrocytes, demonstrating that Nrf2 is required for tBHQ protection ( Figure 1B ). In addition to tBHQ treatment, adenoviral overexpression of Nrf2 was also able to confer robust protection (Figure S1A). To validate the results of the MTS assay, we also assessed toxicity by LDH assay, the results of which closely mirror that of the MTS assay (Figure S1B). These data show a robust Nrf2-dependent protection against H2O2 toxicity in primary astrocytes.
Figure 1. Effects of Nrf2 induction on H2O2 toxicity.
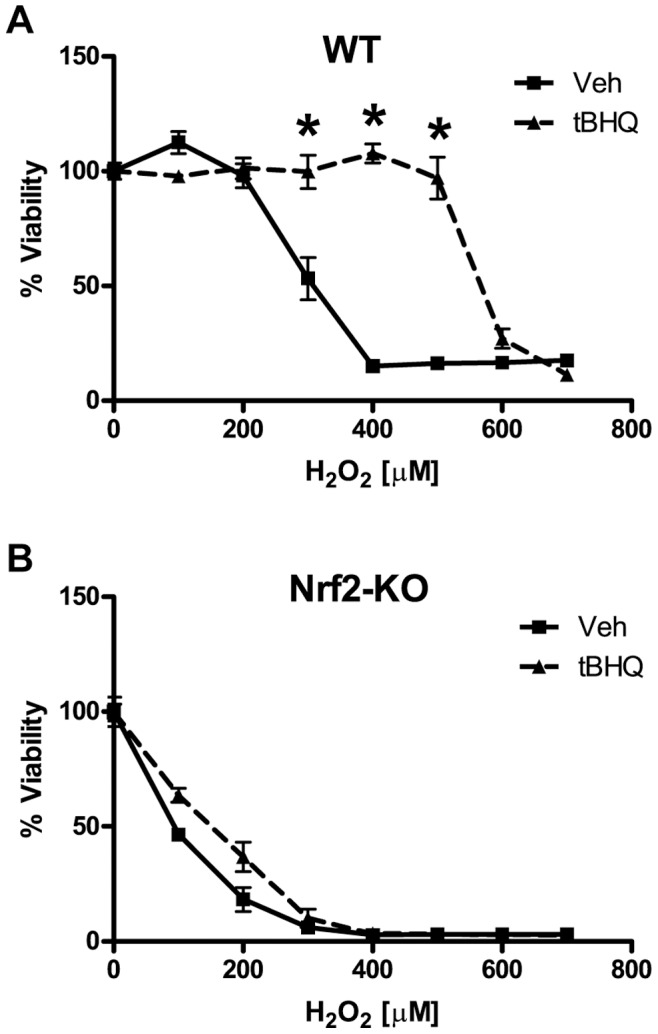
A) Wild-type (WT) or B) Nrf2-knockout (Nrf2-KO) astrocytes were pretreated with vehicle or tBHQ and then treated with H2O2 as indicated. Cell viability was determined by MTS. Statistics were performed using 2-way ANOVA, * indicates p<0.01.
Nrf2 Reduces ROS Induced by H2O2 Treatment and Enhances H2O2 Clearance
We hypothesized that Nrf2 protects astrocytes from H2O2 toxicity by reducing the levels of reactive oxidative species (ROS) produced during H2O2 treatment. In order to assess this hypothesis, we monitored ROS levels by 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA), which is a general ROS indicator, and diphenyl-1-prenylphosphine (DPPP), which is an indicator of lipid oxidation [29]. In vehicle treated astrocytes, the levels of ROS (as indicated by DCFDA fluorescence) and lipid oxidation (as indicated by DPPP fluorescence) increased with higher concentrations of H2O2 ( Figure 2A, B ). Nrf2 induction reduced levels of ROS and lipid oxidation in comparison to vehicle treatment ( Figure 2A, B ). This effect was particularly dramatic at the highest concentrations of H2O2. To further investigate how Nrf2 activation reduces the levels of ROS, we assessed the effect of Nrf2 activation on the clearance of extracellular H2O2. We added H2O2 directly to primary astrocyte cultures and then monitored the clearance of H2O2 from the culture medium over time. As has been previously shown, astrocytes exhibit a robust capacity to clear extracellular hydrogen peroxide [30], [31], [32], [33]. In wild-type astrocytes, Nrf2 activation resulted in an almost 2-fold increase in H2O2 clearance over the vehicle treated cells ( Figure 2C and Table 1 ). In Nrf2 knockout (Nrf2-KO) astrocytes, the H2O2 clearance rate was unchanged after tBHQ treatment, demonstrating the dependence of this effect on Nrf2 ( Figure 2D and Table 1 ). As these data demonstrate, Nrf2 activation significantly enhances the antioxidant capacity of astrocytes, resulting in a more robust clearance of H2O2 and a reduction of both the ROS and lipid oxidation levels produced by H2O2 treatment.
Figure 2. Effects of Nrf2 induction on ROS generation and extracellular H2O2 clearance.

Wild-type astrocytes were pretreated with vehicle or tBHQ. H2O2 was added as indicated and after 4 hours the generation of ROS was monitored by either A) DCFDA or B) DPPP. Astrocytes were pretreated with vehicle or tBHQ and the rate of H2O2 clearance from the extracellular medium was measured over time for C) Wild-type (WT) or D) Nrf2-knockout (Nrf2-KO) cells. Statistics were performed using 2-way ANOVA, * indicates p<0.01.
Table 1. Extracellular Peroxide Clearance.
| 1/2 Time min | Clearance µmole/(min×mg) | Clearance % Veh | ||
| WT | Veh | 3.3 | 4.8 | |
| tBHQ | 1.7 | 9.5 | 190 | |
| Nrf2-KO | Veh | 3.0 | 5.4 | |
| tBHQ | 2.6 | 6.2 | 113 |
Nrf2 Protection against tBOOH and 4-HNE
To examine the role of the Nrf2-ARE pathway in protecting astrocytes from other oxidative stressors, we chose the organic peroxide tert-butyl hydroperoxide (tBOOH) and 4-hydroxynonenal (4-HNE) both of which are produced during conditions of oxidative stress. Organic peroxides, such as tBOOH, are produced physiologically during eicosanoid metabolism. Lipid peroxidation products, such as 4-HNE, are produced after the reaction of free radicals with cellular lipids [1]. Nrf2 activation via tBHQ produced robust protection against both tBOOH and 4-HNE ( Figure 3A, B ). This protection was reversed in Nrf2-KO astrocytes, demonstrating the requirement of Nrf2 for tBHQ protection ( Figure 3C, D ).
Figure 3. Effects of Nrf2 induction on tBOOH and 4-HNE toxicity.

Wild-type (WT) or Nrf2-knockout (Nrf2-KO) astrocytes were pretreated with vehicle or tBHQ (40 µM) for 48 hours and then treated with tBOOH, A) WT and B) Nrf2-KO, or with 4-HNE, C) WT and D) Nrf2-KO. Cell viability was determined by MTS. Statistics were performed using 2-way ANOVA, * indicates p<0.01.
Identification of Nrf2 Regulated Proteins by Quantitative Proteomics
In order to identify protein targets of the Nrf2-ARE pathway, we performed quantitative proteomics on primary astrocytes isolated from mice that overexpress Nrf2 under the control of the astrocyte-specific promoter glial fibrillary acid protein (GFAP-Nrf2) [26]. We used stable isotope labeling by amino acids in cell culture (SILAC) and quantitative shotgun proteomics to quantify changes in protein expression between wild-type and GFAP-Nrf2 astrocytes (see Figure 4 for an overview). We filtered the results of these experiments based on a fold-change cutoff and a requirement that a protein be identified and quantified in all three experimental replicates. From these experiments, we identified 72 differentially regulated proteins (Table S3). In order to further validate the proteomics results and distill our target genes/proteins into an experimentally manageable number, we compared the list of differentially expressed proteins with differentially regulated mRNA transcripts that were previously identified by microarray in our laboratory [5], [7]. After filtering each microarray dataset according to a relative fold-change and rank analysis threshold, we compared the identified mRNA transcripts against the proteins identified by quantitative proteomics. We required each putative Nrf2-regulated gene/protein to be differentially regulated in at least two out of the three datasets. From this analysis, we identified 29 differentially regulated genes/proteins ( Table 2 ). In order to identify those enzymes that might be critical for the antioxidant and protective capacities of the Nrf2-ARE pathway, we performed a gene ontology functional annotation analysis using the DAVID Bioinformatic Resources. According to this analysis, a number of functional categories related to oxidative stress and/or cellular redox status were significantly enriched (Table S4). We combined those genes/proteins identified in the top three gene ontology terms related to oxidative stress (GO:0006979, 0055114, and 0045454) to generate a target list of 15 genes/proteins, including genes associated with the production and utilization of NADPH and glutathione as well as enzymes associated with the direct enzymatic detoxification of reactive oxygen species (Table S5). After an extensive review of the literature, we further refined our target list to a core set of eight enzymes that we hypothesized might be necessary for Nrf2-mediated protection against oxidative stressors, including: NAD(P)H dehydrogenase (quinone)-1 (NQO1), heme oxygenase-1 (HO-1), catalase (CAT), glutathione S-transferase A4 (GSTA4), prostaglandin reductase-1 (PTGR1), glutamate-cysteine ligase modifier subunit (GCLM), and peroxiredoxin-1 and -6 (Prdx1 and Prdx6).
Figure 4. Overview of SILAC workflow.

Astrocytes were grown in “light” or “heavy” amino acid containing media. Proteins from the wild-type (“light” labeled) and GFAP-Nrf2 (“heavy” labeled) cells were mixed and digested using trypsin. Tryptic peptides were then separated offline by high pH reverse phase HPLC. Fractions were collected and then individually analyzed by LC-MS/MS. The differences in expression were quantified by calculating the area under the curve for the “light” and “heavy” peptide pairs for each protein.
Table 2. Comparison of Quantitative Proteomics and Microarray Datasets.
| SwissProt ID | DAVID Description | SILAC | Microarray 1 | Microrray 2 |
| O08709 | peroxiredoxin 6 | 2.11 | 1.70 | 2.43 |
| O08739 | adenosine monophosphate deaminase 3 | NA | 2.33 | 2.14 |
| O54754 | aldehyde oxidase 1 | NA | 2.62 | 7.30 |
| P06801 | malic enzyme 1 | NA | 1.74 | 3.45 |
| P10649 | glutathione S-transferase, mu 1 | 2.14 | 1.54 | 2.19 |
| P14901 | heme oxygenase (decycling) 1 | 10.41 | 2.65 | NA |
| P17809 | solute carrier family 2, member 1 | NA | 1.49 | 5.15 |
| P19639 | glutathione S-transferase, mu 3 | NA | 1.77 | 1.67 |
| P24270 | catalase | NA | 1.93 | 1.61 |
| P24472 | glutathione S-transferase, alpha 4 | 4.85 | 3.95 | 9.53 |
| P35700 | peroxiredoxin 1 | 2.74 | 1.60 | 1.68 |
| P19157 | glutathione S-transferase, pi 1 | 2.71 | 2.34 | 2.72 |
| P47791 | glutathione reductase | 14.95 | 2.60 | NA |
| P52760 | heat-responsive protein 12 | 3.01 | 2.58 | 4.88 |
| P56395 | cytochrome b-5 | NA | 1.63 | 2.10 |
| P97494 | glutamate-cysteine ligase, catalytic subunit | NA | 2.24 | 1.57 |
| Q00612 | glucose-6-phosphate dehydrogenase | NA | 2.15 | 3.24 |
| Q60963 | phospholipase A2, group VII | NA | 2.70 | 2.44 |
| Q64337 | p62 | NA | 1.61 | 1.72 |
| Q64669 | NAD(P)H dehydrogenase, quinone 1 | 3.80 | 5.10 | NA |
| Q91YR9 | prostaglandin reductase 1 | NA | 2.94 | 4.30 |
| Q93092 | transaldolase 1 | 2.42 | 2.94 | 3.32 |
| Q9D379 | epoxide hydrolase 1, microsomal | NA | 1.98 | 3.32 |
| Q9D6Y9 | glucan (1,4-alpha-), branching enzyme 1 | NA | 2.01 | 22.51 |
| Q9D975 | sulfiredoxin 1 | NA | 10.07 | 10.73 |
| Q9DCD0 | phosphogluconate dehydrogenase | 3.54 | 1.97 | 2.01 |
| Q9JMH6 | thioredoxin reductase 1 | 2.77 | 3.87 | 2.93 |
| Q9R0P3 | esterase D/formylglutathione hydrolase | NA | 2.42 | 2.34 |
| Q9R0P9 | ubiquitin carboxy-terminal hydrolase L1 | 1.50 | 1.48 | NA |
Nrf2 Modulated Genes
In order to examine transcript level changes of our core enzymes, we performed quantitative PCR (qPCR). All of eight target genes were robustly activated by tBHQ treatment ( Figure 5A ). In addition, the mRNA levels of these same genes were strongly upregulated in astrocytes infected with an Nrf2 adenoviral construct as determined by qPCR (data not shown). To identify the contribution of these individual genes to the Nrf2 protection, we used siRNA constructs to knockdown each of the eight genes. The siRNA knockdown was validated by qPCR and/or western blot ( Figure 5B, C ).
Figure 5. Validation of Nrf2 targets and siRNA knockdown by quantitative PCR and western blot.

A) Wild-type (WT) astrocytes were treated with vehicle or tBHQ. Differences in target gene expression were analyzed by qPCR. Statistics were performed on vehicle versus tBHQ for each gene using a paired t-test, * indicates p<0.05. WT astrocytes were pretreated with siRNA constructs against the indicated genes and a non-targeting siRNA control (siNT) prior to vehicle or tBHQ treatment. RNA or protein extracts were subjected to B) qPCR and/or C) western blot to validate knockdown. Statistics were performed on siNT plus tBHQ versus targeted siRNA plus tBHQ for each gene or protein using a paired t-test, * indicates p<0.05.
The Role of Catalase in Nrf2 Protection against H2O2
After the knockdown of all each individual gene by siRNA, astrocytes were treated with tBHQ to activate Nrf2 and then treated with H2O2 to assess the effects of siRNA knockdown on Nrf2-mediated protection. The relative protection afforded by tBHQ treatment was robustly reduced in the cells treated with siRNA against catalase (siCAT) when compared to the non-targeting siRNA (siNT) treated cells ( Figure 6A, C ). To validate the siRNA data, we treated astrocytes with the specific catalase inhibitor 3-aminotriazole (3AT). Chemical inhibition by 3AT also reversed Nrf2 protection to a similar degree as siRNA treatment ( Figure 6B, D ). Knockdown of other candidate genes did not substantially modulate Nrf2 protection against H2O2 (data not shown).
Figure 6. The effect of catalase siRNA knockdown on Nrf2 protection against H2O2.

Vehicle or tBHQ treated astrocytes were pretreated with A) non-targeting siRNA (siNT) or B) catalase siRNA (siCAT) or C) a vehicle control (Veh) or D) a catalase inhibitor, 3-aminotriazole (3AT). H2O2 toxicity curves were performed as indicated. Cell viability was determined by MTS. Statistics were performed using 2-way ANOVA, * indicates p<0.01.
The Role of Prdx6 in Nrf2 Protection against tBOOH
To evaluate the relative contribution of individual genes to Nrf2 protection against tBOOH, we used siRNA constructs to knockdown each candidate gene before treatment with tBOOH. Nrf2 protection was considerably reduced after knockdown of Prdx6 ( Figure 7A, C ). In order to validate the siRNA data, we treated astrocytes with the Prdx6 inhibitor mercaptosuccinate (MS) before treatment with tBOOH. In concordance with the siRNA data, mercaptosuccinate reversed the Nrf2-mediated protection against tBOOH ( Figure 7B, D ). Knockdown of other candidate genes did not substantially modulate Nrf2 protection against tBOOH (data not shown).
Figure 7. The effect of Prdx6 siRNA knockdown on Nrf2 protection against tBOOH.

Vehicle or tBHQ treated astrocytes were pretreated with A) non-targeting siRNA (siNT) or B) peroxiredoxin-6 siRNA (siPrdx6) or with either C) a vehicle control (Veh) or D) a peroxiredoxin-6 inhibitor, mercaptosuccinate (MS). tBOOH toxicity curves were performed as indicated. Cell viability was determined by MTS. Statistics were performed using 2-way ANOVA, * indicates p<0.01.
The Role of PTGR-1 in Nrf2 Protection against 4-Hydroxynonenal
In addition to modulating enzymes directly involved in the processing of ROS, Nrf2 also modulates the expression of genes responsible for the detoxification of lipid oxidation products, including glutathione S-transferase-A4 (GSTA4) and prostaglandin reductase-1 (PTGR1). In order to examine the role of individual enzymes in Nrf2 protection against 4-HNE, we used siRNA to knockdown each candidate gene before treatment with 4-HNE. The knockdown of PTGR-1 considerably reduced Nrf2-mediated protection against 4-HNE ( Figure 8A, B ). Knockdown of the other candidate genes did not substantially modulate Nrf2 protection against 4-HNE (data not shown).
Figure 8. The effect of PTGR1 siRNA knockdown on Nrf2 protection against 4-HNE.
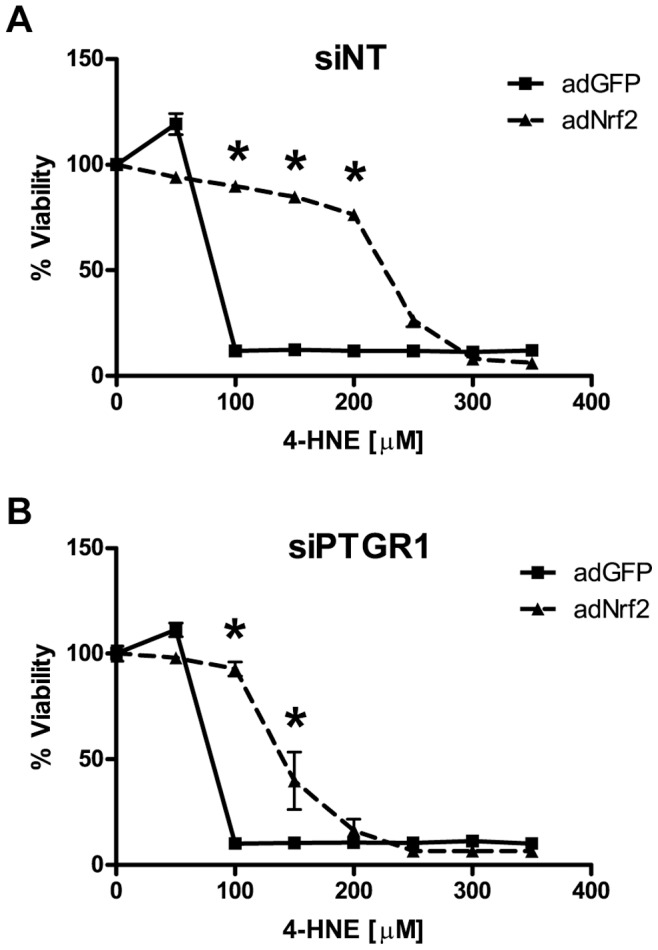
Vehicle or tBHQ treated astrocytes were pretreated with A) non-targeting siRNA (siNT) or B) prostaglandin reductase-1 siRNA (siPTGR1). 4-HNE toxicity curves were performed as indicated. Cell viability was determined by MTS. Statistics were performed using 2-way ANOVA, * indicates p<0.01.
Nrf2 Protection and Glutathione
In order to evaluate the contribution of glutathione to the protective effects of Nrf2, we induced Nrf2 expression in primary astrocytes isolated from mice that lack the glutamate-cysteine ligase modifier subunit (GCLM-KO) and then treated them with H2O2 or tBOOH [34]. Nrf2 activation conferred robust protection against both H2O2 and tBOOH in GCLM-KO astrocytes ( Figure 9C, D ). In addition, siRNA knockdown of GCLM had no effect on Nrf2 protection against 4-HNE (Figure S2).
Figure 9. The effects of glutathione deficiency on Nrf2 protection.

Wild-type (WT) or glutamate-cysteine ligase, modifier subunit knockout (GCLM-KO) astrocytes were infected with adGFP or adNrf2 virus prior to stressor treatment. A) WT cells or B) GLCM-KO cells were treated with H2O2 as indicated. C) WT cells or D) GLCM-KO cells were treated with tBOOH as indicated. Cell viability was determined by MTS. Statistics were performed using 2-way ANOVA, * indicates p<0.01.
Discussion
In order to elucidate mechanisms of astrocytic Nrf2 protection, we used molecular, biochemical, and proteomics approaches to identify specific enzymes responsible for Nrf2 protection against oxidative stress. One of the oxidative stressors used in this study, H2O2, is relatively unreactive towards biomolecules; however, it rapidly reacts with physiological iron via the Fenton reaction to produce the highly reactive and damaging hydroxyl radical [1]. The hydroxyl radical can damage DNA, protein, lipids, and organelles. In order to examine changes in ROS and lipid oxidation as a result of H2O2 treatment, we employed the fluorescent ROS probes DCFDA and DPPP ( Figure 2A, B ). These assays were performed 4 hours after H2O2 treatment and were an assessment of changes in ROS and lipid oxidation levels which occur as a secondary consequence of H2O2 treatment [30], [31], [32], [33]. This secondary production of ROS and lipid oxidation is most likely due to damage to the mitochondrial electron transport chain and/or the induction of NADPH oxidases. H2O2 has been shown to damage mitochondrial DNA and lipids as well as disrupt the electron transport chain, causing an increase in mitochondrial superoxide production [35], [36]. In addition, recent evidence has emerged implicating H2O2 as a signaling molecule capable of stimulating ROS production via NADPH oxidases [37], [38]. In this study, Nrf2 induction produced a robust increase in cellular antioxidant capacity that reduced ROS and lipid oxidation as well as enhanced extracellular H2O2 clearance ( Figure 2A, B ).
In order to identify the Nrf2-regulated enzymes responsible for these changes, we used quantitative proteomics in combination with stable isotope labeling by amino acids in cell culture (SILAC) to identify differentially expressed proteins in GFAP-Nrf2 astrocytes ( Figure 4 ). Typically in a SILAC experiment, cells from one experimental condition are grown in a medium that contains stable isotopologues of essential amino acids while cells from the other condition are grown in a medium with natural amino acids. The amino acids from the medium are incorporated into the cellular proteome as the cells grow and proliferate, producing differentially labeled “light” and “heavy” protein samples. Protein extracts from the two experimental samples are then harvested, mixed, digested, and analyzed by mass spectrometry (MS). The relative protein expression changes between the two samples are then quantified by comparing relative MS signal intensity between the “light” and “heavy” peptide peaks. Using this technique, we were able to identify 72 differentially expressed proteins in GFAP-Nrf2 versus wild-type (WT) astrocytes–25 proteins were increased and 47 proteins were decreased relative to the WT control (Table S3).
In order to identify genes/proteins that are important for Nrf2-mediated protection, we used an iterative approach to refine our dataset. First, we compared the proteins identified in the proteomics screen to two previously published mRNA microarray data sets [5], [7]. By doing this we significantly reduced the number of putative Nrf2-regulated genes to a total of 29 ( Table 2 ). There is a high degree of concordance between the microarray and proteomics datasets. However, the proteomics analysis also revealed an additional 11 upregulated proteins that were not identified in the microarray datasets (Table S3). It is interesting to note that over a third of these proteins are ubiquitin-conjugating enzymes, indicating the possible importance of the Nrf2 pathway in the regulating ubiquitin conjugation under conditions of oxidative stress. Kopito and co-workers have shown the importance of Nrf2 in the ubiquitin-autophagy pathway via its regulation of p62 [39]. There were no commonly downregulated genes among the datasets (data not shown). It is clear that analyzing both microarray and proteomics data in parallel is a powerful approach that can reduce spurious results as well as reveal subtle differences in transcript- and protein-level regulation of protein expression.
From these data, catalase was identified as a potentially important contributor to Nrf2-mediated protection against ROS. Catalase is one of the earliest antioxidant enzymes discovered and plays a central role in cellular protection against ROS by catalyzing the decomposition of H2O2 into water and oxygen [40]. The importance of catalase for the clearance and protection of astrocytes from H2O2 toxicity is well established [31], [41], [42], [43]. However, the importance of catalase in Nrf2-mediated protection in astrocytes was not known before this study. After siRNA knockdown or chemical inhibition of catalase, Nrf2 protection against H2O2 was almost completely ablated ( Figure 6 ). These data indicate the central importance of catalase in Nrf2-mediated protection against H2O2.
Two enzymes that are thought to be important in oxidative signaling, peroxiredoxin-1 (Prdx1) and peroxiredoxin-6 (Prdx6), were both strongly induced by Nrf2 ( Figure 5A ). There are six known mammalian members of the peroxiredoxin family. Peroxiredoxin-1 through -5 are thioredoxin-dependent enzymes while Prdx6 is glutathione-dependent [44]. Peroxiredoxins are primarily known as peroxidases; however, they possess important functions that are distinct from their peroxidase activity [45], [46], [47], [48]. In the case of Prdx6, the oxidation of the specific reactive cysteines changes it from a peroxidase to a phospholipase [46]. In the case of Prdx1, cysteine oxidation results in the oligomerization of Prdx1 into a protein chaperone [45]. These changes are thought to be important in modulating oxidative signaling and inducing cell survival pathways [48].
In regards to Nrf2 protection, Prdx6 appears to play a substantial role in Nrf2 protection against tBOOH. Prdx6 knockdown by siRNA and chemical inhibition by mercaptosuccinate (MS) dramatically reduced Nrf2 protection against tBOOH ( Figure 7 ). While Prdx6 is catalytically less efficient than glutathione peroxidase, it is significantly more important in protecting cells against tBOOH than glutathione peroxidase-1 [49]. Additionally, Prdx6 is upregulated in patients with Alzheimer’s disease and ALS, suggesting a potential role in neurodegenerative disease [50], [51], [52].
Sulfiredoxin is another target of Nrf2 and maintains Prdx1 activity by preventing its catalytic cysteine from becoming hyperoxidized. Sulfiredoxin has also been shown to be protective against oxidative stress [39]. However, in the context of the experiments presented here, knockdown by siRNA produced an increase of both basal resistance and Nrf2-mediated protection against H2O2 (Figure S3). We have no explanation for this unexpected result. However, it is clear that sulfiredoxin is vitally important to redox signaling and cellular responses to oxidative stress and its regulation by Nrf2, as well as that of Prdx1 and Prdx6, could represent a central control point for redox signaling via the Nrf2-ARE pathway.
One of the most studied toxic byproducts of lipid oxidation is 4-hydroxynonenal (4-HNE). 4-HNE is formed when polyunsaturated fatty acids undergo free radical reactions during conditions of oxidative stress. 4-HNE readily reacts with lysine, cysteine, and histidine to form protein adducts [53]. These 4-HNE adducts have been implicated in the etiology of Alzheimer’s disease and Parkinson’s disease [54], [55]. Nrf2 activation robustly protects astrocytes from 4-HNE toxicity ( Figure 3 ) and regulates genes responsible for detoxifying 4-HNE, including both glutathione S-transferase A4 (GSTA4) and prostaglandin reductase-1 (PTGR1) ( Figure 5 ) [5], [7], [56], [57], [58]. GSTA4 detoxifies 4-HNE via direct conjugation while PTGR1 catalyzes the reduction of the highly reactive α,β-carbon double bond of 4-HNE to a non-reactive single carbon bond [58]. The knockdown of PTGR1 had a dramatic effect on the ability of Nrf2 to protect against 4-HNE toxicity ( Figure 8 ). However, siRNA knockdown of GSTA4 had no effect on Nrf2-mediated resistance to 4-HNE (data not shown). These data indicate the importance of PTGR1 in Nrf2 protection against 4-HNE.
In astrocytes, Nrf2 has been shown to modulate enzymes responsible for the production and processing of glutathione as well as enzymes which utilize glutathione for cellular detoxification processes [5], [6], [7], [27]. A number of studies have demonstrated the importance of glutathione in Nrf2-mediated neuroprotection [6], [27], [28]. The rate-limiting enzyme in the glutathione biosynthetic pathway is γ-glutamylcysteine ligase (GCL). This enzyme is composed of a catalytic subunit (GCLC) and a modifier subunit (GCLM). GCLM expression controls the rate of glutathione biosynthesis by modulating the catalytic efficiency of GCLC [34]. GCLM is highly induced by Nrf2 ( Figure 5 ) and knockdown of GCLM by siRNA completely inhibits the ability of Nrf2 to increase total glutathione (data not shown). To study the role of glutathione in Nrf2 protection, we employed primary cortical astrocytes from GCLM knockout (GCLM-KO) mice that contain 80% less total glutathione than wild-type astrocytes. It has been shown that Nrf2 activation in GCLM-KO astrocytes fails to increase cellular levels of glutathione [59]. Surprisingly, we found that GCLM-KO astrocytes still exhibit a robust Nrf2 protection against H2O2 and tBOOH even without the ability to upregulate glutathione synthesis ( Figure 9 ). In addition, siRNA knockdown of GCLM does not effect Nrf2 protection against 4-HNE (Figure S2). While these data are somewhat unexpected, other studies have shown that Nrf2 does not require de novo glutathione synthesis for its protective effects. For example, in mouse embryo fibroblasts isolated from GCLM-KO mice Nrf2 induction results in protection from arsenite, a compound known to induce ROS [60]. In addition, in vivo astrocyte-specific Nrf2 overexpression reversed many of the pathological hallmarks of Alexander’s disease while glutathione deficiency had no affect on pathology [24]. These data provide strong evidence that Nrf2 protection in astrocytes is largely independent of de novo glutathione synthesis.
Both NQO1 and HO-1 are widely recognized as canonical Nrf2 genes and both have been shown to possess potent cytoprotective activity. HO-1 catalyzes the degradation of heme to produce biliverdin that is subsequently converted to bilirubin, a powerful radical scavenger [61]. NQO1 has also been shown to be neuroprotective against oxidative damage by reducing highly reactive quinones to less reactive hydroquinones [62]. In the brain, HO-1 has been shown to be cytoprotective in models of stroke, excitotoxicity, Parkinson’s disease, and Alzheimer’s disease while NQO1 has been shown to be protective against a model of Parkinson’s disease [63], [64], [65], [66], [67], [68]. More recently, both HO-1 and NQO1 have been shown to be responsible for the neuroprotective properties of Nrf2 against H2O2 in astrocytes [69]. However, our data indicates no dependence of Nrf2 protection on either NQO1 or HO-1 for any of the stressors tested, including H2O2 (data not shown). It is unclear why our results differ from the study by Park and co-workers; however, in that study, chemical inhibitors of both HO-1 and NQO1 were used instead of the more specific siRNA knockdown used in our study.
In summary, we have shown the importance of the Nrf2-ARE pathway in protecting astrocytes against oxidative stress. This protection appears to require discrete enzymes working synergistically to detoxify specific oxidative stressors, i.e. catalase is required for Nrf2 protection against H2O2, Prdx6 is required for protection against tBOOH, and PTGR1 is required for protection against 4-HNE (summarized in Figure 10 ). Finally, while glutathione is certainly an extremely important physiological antioxidant and is essential for cellular survival, it does not appear to be critical for Nrf2 protection against the H2O2, tBOOH, or 4-HNE in astrocytes. These data indicate the complex mechanisms of Nrf2 protection and the requirement of multiple enzymes to execute the powerful protective effects of the Nrf2-ARE pathway.
Figure 10. Overview of results.
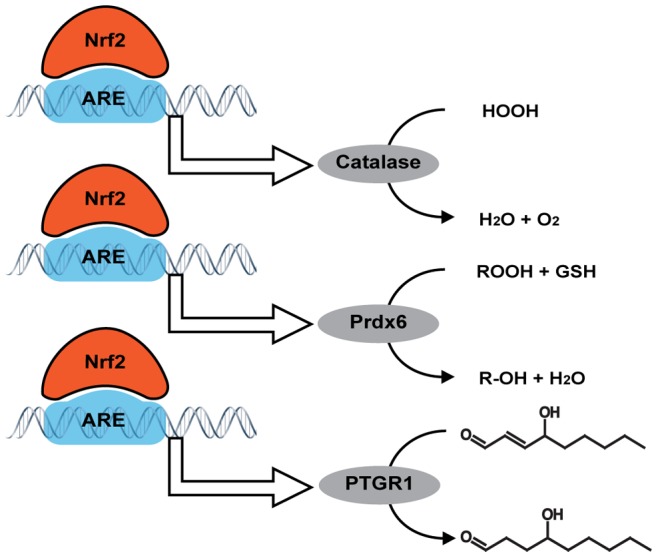
Binding of Nrf2 to the antioxidant response element (ARE) induces the expression of catalase, peroxiredoxin-6 (Prdx6), and prostaglandin reductase-1 (PTGR1). Each of these enzymes has a specific role in detoxifying the oxidative stressors H2O2, tBOOH, and 4-HNE.
Materials and Methods
Animals
Transgenic mice overexpressing Nrf2 under the astrocyte-specific glial fibrillary acid protein promoter (GFAP-Nrf2) and transgenic mice lacking the gene for glutamate-cysteine ligase modifier subunit (GCLM-KO) were utilized as indicated [26], [70]. This study was approved by the University of Wisconsin-Madison Institutional Animal Care and Use Committee (IACUC) and all animal procedures were performed in accordance with the requirements of the IACUC. The UW-Madison USDA Research Registration number is 35-R-1 and the Laboratory Animal Welfare (OLAW) Public Health Service (PHS) Assurance Number is A3368-01.
Materials
The Hank’s balanced salt solution (HBSS), Dulbecco’s modified Eagle’s medium (DMEM), Opti-MEM, fetal bovine serum (FBS), penicillin/streptomycin, TRIzol, Lipofectamine, and 2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA) were obtained from Life Technologies (Carlsbad, CA). 4-hydroxynonenal (4-HNE) and diphenyl-1-prenylphosphine (DPPP) were purchased from Cayman Chemical (Ann Arbor, MI). 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS), lactate dehydrogenase (LDH) release, and reverse transcriptase kits were purchased from Promega (Madison, WI). Heavy isotopes for SILAC labeling were purchased from Cambridge Isotopes (Andover, MA). All siRNA constructs and primers were obtained from Integrated DNA Technologies (Coralville, IA). Cycler480 SYBR Green I Master mix was purchased from Roche Applied Sciences (Indianapolis, IN). All other reagents were obtained from Sigma (St. Louis, MO).
Primary Astrocyte Culture
Primary astrocyte cultures were prepared from postnatal day 1 (P1) mice. The cerebral cortices from individual P1 pups were removed, placed in ice-cold HBSS, minced with a scalpel blade, and then transferred to a tube with 10 ml of 0.25% trypsin at 37°C. After 25 minutes, 10 ml of DMEM+FBS (1.2 mg/ml sodium bicarbonate, 3.6 mg/ml HEPES, 10% fetal bovine serum (FBS), penicillin/streptomyocin at 100 IU/ml and 100 µg/ml) was added to deactivate the trypsin. DNase was added to a final concentration of 0.05 mg/ml and then the cells were pelleted at 400 g for 3 minutes. The supernatant was removed and the tissue pellet was re-suspended in 5 ml of fresh DMEM+FBS. After triturating 15–20 times with a 5 ml pipette, the disassociated cells passed through a 70 µm cell strainer. Cells from the cortices of three pups were plated onto a uncoated T75 flask. The culture medium was changed after the first day and then every 3 days thereafter. After the astrocytes reached confluency (7–8 days), the flasks were placed on a rotary shaker (200 rpm). After 18 hours, the cells were lifted with trypsin+EDTA and then re-plated at 35,000 cells/cm2. The astrocytes were allowed to reach confluency (5–7 days) before use. For the SILAC experiments, the DMEM was supplemented with either the natural isotopes of lysine and arginine for the “light” SILAC medium or “heavy” lysine (U-13C6, 99%; U-15N2, 99%, Cat# CNLM-291-H-0.1) and “heavy” arginine (U-13C6, 99%, Cat# M-2265-H-0.1) from Cambridge Isotopes (Andover, MA) for the “heavy” SILAC medium. Briefly, cells from the GFAP-Nrf2 pups were grown and maintained throughout the culturing period with “heavy” DMEM+FBS while the wild-type mice were grown and maintained with “light” DMEM+FBS. Please refer to Figure 4 for an overview. Littermate controls were used for all experiments.
Toxicity Curves
Astrocytes were treated with either adenovirus overexpressing GFP (control) and Nrf2 (50 MOI) or a vehicle control and a 40 µM tert-butyl hydroquinone (tBHQ), as previously published [7]. After 48 hours, toxicity curves were performed using hydrogen peroxide, tert-butyl-hydrogen peroxide, or 4-hydroxynonenal at the concentrations indicated. Each concentration of stressor was used to treat four replicates per condition. Additionally, 3-aminotriazol (a catalase inhibitor) at 10 mM, or mercaptosuccinate (a peroxiredoxin-6 inhibitor) at 0.5 mM, was added to cell cultures as indicated two hours prior to treatment. After 24 hours of treatment, cell viability was measured by either MTS or LDH assay, according to the manufacturer’s instructions. Viability was reported as a percentage of the untreated vehicle control.
Measurement of ROS
DPPP (100 µM) or carboxy-H2DCFDA (10 µM) in DMEM without FBS was added to confluent astrocytes in a 96-well plate. After 30 minutes, the solution was replaced with DMEM+FBS without phenol red. The cells were treated with hydrogen peroxide at various concentrations and the fluorescence (DPPP at Excitation: 351 nm, Emission: 380 nm or DCFDA at Excitation: 485 nm, Emission: 540 nm) was measured on a SpectraMax M3 plate reader from Molecular Devices (Sunnyvale, CA) after 4 hours.
Peroxide Clearance
The rate of peroxide clearance was determined as described by Dringen and co-workers [31]. Briefly, the standard culture medium on a confluent astrocyte culture was replaced with incubation buffer (20 mM HEPES, 145 mM NaCl, 1.8 mM CaCl2, 5.4 mM KCl, 1 mM MgCl2, 0.8 mM Na2HPO4, and 5 mM glucose, pH 7.4). The cells were kept at 37°C and hydrogen peroxide was added to a final concentration of 200 µM. Aliquots of media (10 µl) were taken every minute for 6 minutes and added directly to 25 mM sulfuric acid in a 96-well plate. To these aliquots, 190 µl of reaction mixture (0.5 mM (NH4)2Fe(SO4)2, 200 µM xylenol orange and 200 mM sorbitol in 25 mM sulfuric acid) was added to each well. After incubating for 45 minutes, the hydrogen peroxide concentration was determined by comparing the absorbance (540 nm) of the samples versus a hydrogen peroxide concentration curve using a SpectraMax M3 plate reader.
Protein Extraction for Quantitative Proteomics
After reaching confluence, the astrocytes were washed with phosphate buffered saline (PBS). Hypotonic lysis buffer (50 mM Tris-HCl at pH 7.4) was added to each flask and the cells were detached with a cell scraper and the resulting lysate was flash frozen in liquid nitrogen. The cell lysate was thawed and spun at 15,000 g for 3 minutes in a refrigerated centrifuge (4°C). The supernatant was removed and subjected to bicinchoninic acid (BCA) to determine protein concentration and each “light” labeled wild-type sample was combined with a “heavy” GFAP-Nrf2 sample at equal total protein amounts (n = 3 total).
Protein Digestion for Quantitative Proteomics
Protein samples were diluted into 6 M urea/50 mM ammonium bicarbonate (pH 8). Cysteinyl disulfides were reduced via the addition of 2 mM Tris[2-carboxyethyl] phosphine (TCEP) for 30 minutes at 37°C. Reduced disulfides were then alkylated by the addition of 10 mM iodoacetamide (IAA) for 30 minutes in the dark. The urea was then diluted to <1 M with 50 mM ammonium bicarbonate, the sample concentrated via ultrafiltration (10 kDa cut-off), and the pH was adjusted to pH 8 and acetonitrile was added to 20%. Trypsin was added at a 1∶20 weight-to-weight ratio and incubated for 18 hours at 37°C. After digestion, the sample was dried by vacuum centrifugation.
Offline High pH Reverse Phase Separation for 2D LC-MS/MS
100 µg of protein digest was reconstituted in 100 µl of 50 mM ammonium formate at pH 10. Peptides were separated by off-line high pH reverse phase using a Gemini C18 RP column (2×150 mm, 3 µM, 110 Å) from Phenomenex (Torrance, CA) with a 50 mM ammonium formate (pH 10) and acetonitrile mobile phase. Peptides were eluted with a linear gradient of acetonitrile from 5 to 35% over 60 minutes. Fractions were collected every 6 minutes for a total of 10 fractions. Each fraction was vacuum-centrifuged to dryness and then reconstituted in 30 µl of 0.1% formic acid and analyzed by LC-MS/MS.
LC-MS/MS
Using a Waters NanoAcquity HPLC, tryptic peptides were separated with a 5–40% linear gradient of 0.1% formic acid in acetonitrile at a flow rate of 300 nl/min over 90 minutes. The eluted peptides were analyzed by a nanoelectrospray ionization (nESI) ion trap mass spectrometer (amaZon ETD) from Bruker Daltonics. The MS survey scan was performed in positive ion mode from m/z 400 to 2000, followed by data-dependent MS/MS using the Stable Isotope Pairs (SILE) acquisition method. The signal threshold for switching from the survey scan to MS/MS was set at 3000. Normalized collision energy was set at 35; capillary voltage, 3000 V; capillary temperature, 200°C. Dynamic exclusion was activated with the following parameters: repeat count was 1, repeat duration was 60 s, and the exclusion duration was 60 s.
Database Search for Quantitative Proteomics
The result files (.yep) from each sample were searched using the Mascot Server (version 2.1.1) from Matrix Science. Using Mascot, the files were searched against the SwissProt database (UniProt) with the following parameters: taxonomy was limited to mus musculus, parent mass tolerance was 2.0 Da, fragment mass tolerance was 0.8 Da, and a maximum of two missed cleavages was allowed. Carbamidomethylation at cysteine residues was set as a fixed modification and oxidation of methionine was set as a variable modification. The false discovery rate (FDR) was determined to be less than 1% via the Mascot Search Engine.
SILAC Quantitation, Functional Analysis, and Microarray Comparison
Expression differences between “heavy” (GFAP-Nrf2) and “light” (Wild-type) peptide pairs were quantified using Mascot Distiller from Matrix Science. Fold changes were exported to Microsoft Excel. Fold changes were converted to a log2 scale and then checked for a normal distribution via a histogram plot (Figure S5). Peptide pairs were required to be identified in all three replicates and were only reported if they exhibited an average fold change of greater than +1.35 or less than −1.35. Only peptide pairs identified in all three SILAC replicates were included in the results. Functional analysis of the differentially expressed proteins was performed with the online tool DAVID Bioinformatics Resources 6.7 [71], [72]. Comparison against two previously published microarray data sets was performed [5], [7]. For the microarray data, each gene was required to have a fold change greater than 1.5 fold, a rank analysis score of at least 5, and a coefficient of variation of less than 1.
siRNA Treatment
Confluent astrocytes were transfected with siRNA constructs with RNAiMAX Lipofectamine according to the manufacturer’s instructions (see Table S1 for siRNA sequences). Briefly, each siRNA construct was diluted in Opti-MEM and then mixed with an equal volume of Lipofectamine diluted in Opti-MEM (1∶1 ratio Lipofectamine to siRNA) and allowed to incubate for 10 minutes. After incubation, the siRNA/Lipofectamine solution was added directly to cells without removing the original cell media. The final siRNA concentration was 25 nM. After 18 hours, the siRNA was removed and cells were infected with adenovirus or treated with tBHQ or vehicle.
Quantitative PCR
After 24 hours of treatment vehicle control or tBHQ, astrocytes were rinsed once with PBS and total RNA was isolated from cells using TRIzol reagent according to manufacturer’s instructions. RNA quality was assessed with a Bioanalyzer 2100 from Agilent Technologies (Santa Clara, CA). The RNA Integrity Number (RIN) was required to be 8.5 or above. Reverse transcriptase was used to make cDNA using Oligo(dT)15 primers at 42°C for 1 hour. The quantitative PCR was performed with a LightCycler480 Real-Time PCR System from Roche. Briefly, Cycler480 SYBR Green I Master mix containing 1 µl of cDNA for each sample and 50 pmol of each primer were amplified (see Table S2 for primer sequences).
Western Blot Analysis
After 48 hours of treatment with adenovirus (adGFP or adNrf2) or chemical treatment (vehicle or tBHQ), astrocytes were rinsed once with PBS and then harvested in RIPA buffer. Protein concentration was assayed by bicinchoninic acid assay (BCA). Protein samples (10 µg) were resolved on a 12% SDS-PAGE gel and transferred to a Hybond-P membrane. The membrane was blocked with 5% non-fat milk in TBS with 0.1% Tween (TBS-T) for 1 hour before overnight incubation at 4°C with primary antibody. The following antibodies were used: polyclonal anti-GCLM (1∶10,000) a kind gift from Dr. Terrence Kavanaugh, rabbit monoclonal anti-Prdx6 (1∶2,000) from Epitomics (Burlingame, CA), rabbit monoclonal anti-Prdx1 (1∶10,000) from Epitomics, and mouse monoclonal anti-β-tubulin (1∶1,000) from the University of Iowa Developmental Studies Hybridoma Bank (Iowa City, IA). The membrane was incubated for 1 hour at room temperature with a horseradish peroxidase conjugated secondary antibody (1∶4000). The membrane was developed using an enhanced chemiluminescence (ECL) kit and then imaged and quantified using the G:Box imaging system from Syngene (Frederick, MD).
Statistical Analysis
All data presented is reported as mean +/− standard deviation unless otherwise stated. All statistical computations were performed using GraphPad Prism 4.0 from GraphPad Software. Statistical significance was determined by using a Student’s t-test (p<0.05) or ANOVA (p<0.05) followed by a Newman-Keuls posthoc analysis to determine statistically significant paired comparisons (p<0.05).
Supporting Information
Adenoviral overexpression of Nrf2 and its effects on H2O2 toxicity. Wild-type (WT) astrocytes were infected with adGFP or adNrf2 adenovirus and then treated with H2O2 as indicated. Cell viability was determined by A) MTS or B) LDH. Statistics were performed using 2-way ANOVA, * indicates p<0.01.
(TIF)
The effect of GCLM siRNA knockdown on Nrf2 protection against 4-HNE. adGFP or adNrf2 infected astrocytes were pretreated with A) non-targeting siRNA (siNT) or B) glutamate-cysteine ligase, modifier subunit siRNA (siGCLM). 4-HNE toxicity curves were performed as indicated. Cell viability was determined by MTS. Statistics were performed using 2-way ANOVA, * indicates p<0.01.
(TIF)
The effect of sulfiredoxin-1 knockdown on cellular sensitivity to toxins and Nrf2 mediated protection. Vehicle/tBHQ or adGFP/adNrf2 treated astrocytes were pretreated with either non-targeting (siNT) or sulfiredoxin-1 (siSrdxn) siRNA. Toxicity curves were performed as indicated: A) H2O2, B) tBOOH, or C) 4-HNE. Cell viability was determined by MTS. Statistics were performed using 2-way ANOVA, * indicates p<0.01.
(TIF)
The effect of glutathione deficiency on extracellular H2O2 clearance. Wild-type (WT) or glutamate-cysteine ligase, modifier subunit knockout (GCLM-KO) astrocytes were infected with adGFP or adNrf2 virus. The rate of H2O2 clearance from the extracellular medium was measured over time for wild-type (WT) or GCLM-knockout (GCLM-KO) cells. Statistics were performed using 2-way ANOVA, * indicates p<0.01.
(TIF)
Log2 Histogram of SILAC Fold Change. To check for a normal distribution of expression changes, raw fold changes for each protein were log2 converted, binned, and plotted.
(TIF)
siRNA Sequences
(XLSX)
Primer Sequences for qPCR
(XLSX)
(XLSX)
Gene Ontologhy Analysis
(XLSX)
Oxidative Stress Related Proteins
(XLSX)
Funding Statement
National Institute of Environmental Health Sciences (NIEHS ES08089). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Halliwell B (1992) Reactive oxygen species and the central nervous system. J Neurochem 59: 1609–1623. [DOI] [PubMed] [Google Scholar]
- 2. Coyle JT, Puttfarcken P (1993) Oxidative stress, glutamate, and neurodegenerative disorders. Science 262: 689–695. [DOI] [PubMed] [Google Scholar]
- 3. Olanow CW (1993) A radical hypothesis for neurodegeneration. Trends Neurosci 16: 439–444. [DOI] [PubMed] [Google Scholar]
- 4. Li J, Lee JM, Johnson JA (2002) Microarray analysis reveals an antioxidant responsive element-driven gene set involved in conferring protection from an oxidative stress-induced apoptosis in IMR-32 cells. Journal of Biological Chemistry 277: 388–394. [DOI] [PubMed] [Google Scholar]
- 5. Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA (2003) Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. Journal of Biological Chemistry 278: 12029–12038. [DOI] [PubMed] [Google Scholar]
- 6. Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, et al. (2003) Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. The Journal of neuroscience : the official journal of the Society for Neuroscience 23: 3394–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kraft AD, Johnson DA, Johnson JA (2004) Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. The Journal of neuroscience : the official journal of the Society for Neuroscience 24: 1101–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jakel RJ, Townsend JA, Kraft AD, Johnson JA (2007) Nrf2-mediated protection against 6-hydroxydopamine. Brain Research 1144: 192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Calkins MJ, Jakel RJ, Johnson DA, Chan K, Kan YW, et al. (2005) Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription. Proceedings of the National Academy of Sciences of the United States of America 102: 244–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee JM, Shih AY, Murphy TH, Johnson JA (2003) NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J Biol Chem 278: 37948–37956. [DOI] [PubMed] [Google Scholar]
- 11. Rushmore TH, King RG, Paulson KE, Pickett CB (1990) Regulation of glutathione S-transferase Ya subunit gene expression: identification of a unique xenobiotic-responsive element controlling inducible expression by planar aromatic compounds. Proceedings of the National Academy of Sciences of the United States of America 87: 3826–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnson JA, Johnson DA, Kraft AD, Calkins MJ, Jakel RJ, et al. (2008) The Nrf2-ARE pathway: an indicator and modulator of oxidative stress in neurodegeneration. Annals of the New York Academy of Sciences 1147: 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moi P, Chan K, Asunis I, Cao A, Kan YW (1994) Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proceedings of the National Academy of Sciences of the United States of America 91: 9926–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Itoh K, Tong KI, Yamamoto M (2004) Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free radical biology & medicine 36: 1208–1213. [DOI] [PubMed] [Google Scholar]
- 15. Zhang DD, Lo SC, Sun Z, Habib GM, Lieberman MW, et al. (2005) Ubiquitination of Keap1, a BTB-Kelch substrate adaptor protein for Cul3, targets Keap1 for degradation by a proteasome-independent pathway. J Biol Chem 280: 30091–30099. [DOI] [PubMed] [Google Scholar]
- 16. Sekhar KR, Yan XX, Freeman ML (2002) Nrf2 degradation by the ubiquitin proteasome pathway is inhibited by KIAA0132, the human homolog to INrf2. Oncogene 21: 6829–6834. [DOI] [PubMed] [Google Scholar]
- 17. Zhang DD, Hannink M (2003) Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol 23: 8137–8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Prestera T, Talalay P, Alam J, Ahn YI, Lee PJ, et al. (1995) Parallel induction of heme oxygenase-1 and chemoprotective phase 2 enzymes by electrophiles and antioxidants: regulation by upstream antioxidant-responsive elements (ARE). Mol Med 1: 827–837. [PMC free article] [PubMed] [Google Scholar]
- 19. Primiano T, Kensler TW, Kuppusamy P, Zweier JL, Sutter TR (1996) Induction of hepatic heme oxygenase-1 and ferritin in rats by cancer chemopreventive dithiolethiones. Carcinogenesis 17: 2291–2296. [DOI] [PubMed] [Google Scholar]
- 20. Singh A, Rangasamy T, Thimmulappa RK, Lee H, Osburn WO, et al. (2006) Glutathione peroxidase 2, the major cigarette smoke-inducible isoform of GPX in lungs, is regulated by Nrf2. Am J Respir Cell Mol Biol 35: 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhu H, Itoh K, Yamamoto M, Zweier JL, Li Y (2005) Role of Nrf2 signaling in regulation of antioxidants and phase 2 enzymes in cardiac fibroblasts: protection against reactive oxygen and nitrogen species-induced cell injury. FEBS Lett 579: 3029–3036. [DOI] [PubMed] [Google Scholar]
- 22. Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, et al. (2002) Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res 62: 5196–5203. [PubMed] [Google Scholar]
- 23. Kwak MK, Wakabayashi N, Greenlaw JL, Yamamoto M, Kensler TW (2003) Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol Cell Biol 23: 8786–8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lapash Daniels CM, Austin EV, Rockney DE, Jacka EM, Hagemann TL, et al. (2012) Beneficial effects of nrf2 overexpression in a mouse model of alexander disease. J Neurosci 32: 10507–10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen PC, Vargas MR, Pani AK, Smeyne RJ, Johnson DA, et al. (2009) Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proceedings of the National Academy of Sciences of the United States of America 106: 2933–2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA (2008) Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. The Journal of neuroscience : the official journal of the Society for Neuroscience 28: 13574–13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vargas MR, Pehar M, Cassina P, Beckman JS, Barbeito L (2006) Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR-dependent motor neuron apoptosis. J Neurochem 97: 687–696. [DOI] [PubMed] [Google Scholar]
- 28. Bell KF, Fowler JH, Al-Mubarak B, Horsburgh K, Hardingham GE (2011) Activation of Nrf2-regulated glutathione pathway genes by ischemic preconditioning. Oxid Med Cell Longev 2011: 689524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Okimoto Y, Warabi E, Wada Y, Niki E, Kodama T, et al. (2003) A novel method of following oxidation of low-density lipoprotein using a sensitive fluorescent probe, diphenyl-1-pyrenylphosphine. Free Radic Biol Med 35: 576–585. [DOI] [PubMed] [Google Scholar]
- 30. Desagher S, Glowinski J, Premont J (1996) Astrocytes protect neurons from hydrogen peroxide toxicity. J Neurosci 16: 2553–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dringen R, Hamprecht B (1997) Involvement of glutathione peroxidase and catalase in the disposal of exogenous hydrogen peroxide by cultured astroglial cells. Brain Res 759: 67–75. [DOI] [PubMed] [Google Scholar]
- 32. Dringen R, Kussmaul L, Gutterer JM, Hirrlinger J, Hamprecht B (1999) The glutathione system of peroxide detoxification is less efficient in neurons than in astroglial cells. Journal of Neurochemistry 72: 2523–2530. [DOI] [PubMed] [Google Scholar]
- 33. Dringen R, Kussmaul L, Hamprecht B (1998) Detoxification of exogenous hydrogen peroxide and organic hydroperoxides by cultured astroglial cells assessed by microtiter plate assay. Brain Res Brain Res Protoc 2: 223–228. [DOI] [PubMed] [Google Scholar]
- 34. Yang Y, Dieter MZ, Chen Y, Shertzer HG, Nebert DW, et al. (2002) Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm(−/−) knockout mouse. Novel model system for a severely compromised oxidative stress response. J Biol Chem 277: 49446–49452. [DOI] [PubMed] [Google Scholar]
- 35. Turrens JF (2003) Mitochondrial formation of reactive oxygen species. J Physiol 552: 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ricci C, Pastukh V, Leonard J, Turrens J, Wilson G, et al. (2008) Mitochondrial DNA damage triggers mitochondrial-superoxide generation and apoptosis. Am J Physiol Cell Physiol 294: C413–422. [DOI] [PubMed] [Google Scholar]
- 37. El Jamali A, Valente AJ, Clark RA (2010) Regulation of phagocyte NADPH oxidase by hydrogen peroxide through a Ca(2+)/c-Abl signaling pathway. Free Radic Biol Med 48: 798–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krause KH (2007) Aging: a revisited theory based on free radicals generated by NOX family NADPH oxidases. Exp Gerontol 42: 256–262. [DOI] [PubMed] [Google Scholar]
- 39. Riley BE, Kaiser SE, Shaler TA, Ng AC, Hara T, et al. (2010) Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: a potential role for protein aggregation in autophagic substrate selection. The Journal of cell biology 191: 537–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sumner JB, Dounce AL (1937) Crystalline Catalase. Science 85: 366–367. [DOI] [PubMed] [Google Scholar]
- 41. Liddell JR, Robinson SR, Dringen R (2004) Endogenous glutathione and catalase protect cultured rat astrocytes from the iron-mediated toxicity of hydrogen peroxide. Neurosci Lett 364: 164–167. [DOI] [PubMed] [Google Scholar]
- 42. Sokolova T, Gutterer JM, Hirrlinger J, Hamprecht B, Dringen R (2001) Catalase in astroglia-rich primary cultures from rat brain: immunocytochemical localization and inactivation during the disposal of hydrogen peroxide. Neurosci Lett 297: 129–132. [DOI] [PubMed] [Google Scholar]
- 43. Hirrlinger J, Resch A, Gutterer JM, Dringen R (2002) Oligodendroglial cells in culture effectively dispose of exogenous hydrogen peroxide: comparison with cultured neurones, astroglial and microglial cells. J Neurochem 82: 635–644. [DOI] [PubMed] [Google Scholar]
- 44. Neumann CA, Cao J, Manevich Y (2009) Peroxiredoxin 1 and its role in cell signaling. Cell Cycle 8: 4072–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jang HH, Lee KO, Chi YH, Jung BG, Park SK, et al. (2004) Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell 117: 625–635. [DOI] [PubMed] [Google Scholar]
- 46. Chen JW, Dodia C, Feinstein SI, Jain MK, Fisher AB (2000) 1-Cys peroxiredoxin, a bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. J Biol Chem 275: 28421–28427. [DOI] [PubMed] [Google Scholar]
- 47. Lee YM, Park SH, Shin DI, Hwang JY, Park B, et al. (2008) Oxidative modification of peroxiredoxin is associated with drug-induced apoptotic signaling in experimental models of Parkinson disease. J Biol Chem 283: 9986–9998. [DOI] [PubMed] [Google Scholar]
- 48. Rhee SG, Woo HA, Kil IS, Bae SH (2012) Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J Biol Chem 287: 4403–4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu G, Feinstein SI, Wang Y, Dodia C, Fisher D, et al. (2010) Comparison of glutathione peroxidase 1 and peroxiredoxin 6 in protection against oxidative stress in the mouse lung. Free Radic Biol Med 49: 1172–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chowdhury I, Mo Y, Gao L, Kazi A, Fisher AB, et al. (2009) Oxidant stress stimulates expression of the human peroxiredoxin 6 gene by a transcriptional mechanism involving an antioxidant response element. Free Radic Biol Med 46: 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Strey CW, Spellman D, Stieber A, Gonatas JO, Wang X, et al. (2004) Dysregulation of stathmin, a microtubule-destabilizing protein, and up-regulation of Hsp25, Hsp27, and the antioxidant peroxiredoxin 6 in a mouse model of familial amyotrophic lateral sclerosis. Am J Pathol 165: 1701–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Power JH, Asad S, Chataway TK, Chegini F, Manavis J, et al. (2008) Peroxiredoxin 6 in human brain: molecular forms, cellular distribution and association with Alzheimer’s disease pathology. Acta Neuropathol 115: 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Esterbauer H, Schaur RJ, Zollner H (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 11: 81–128. [DOI] [PubMed] [Google Scholar]
- 54. Sompol P, Ittarat W, Tangpong J, Chen Y, Doubinskaia I, et al. (2008) A neuronal model of Alzheimer’s disease: an insight into the mechanisms of oxidative stress-mediated mitochondrial injury. Neuroscience 153: 120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wey MC, Fernandez E, Martinez PA, Sullivan P, Goldstein DS, et al. (2012) Neurodegeneration and motor dysfunction in mice lacking cytosolic and mitochondrial aldehyde dehydrogenases: implications for Parkinson’s disease. PLoS One 7: e31522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gallagher EP, Huisden CM, Gardner JL (2007) Transfection of HepG2 cells with hGSTA4 provides protection against 4-hydroxynonenal-mediated oxidative injury. Toxicol In Vitro 21: 1365–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dick RA, Kensler TW (2004) The catalytic and kinetic mechanisms of NADPH-dependent alkenal/one oxidoreductase. J Biol Chem 279: 17269–17277. [DOI] [PubMed] [Google Scholar]
- 58. Dick RA, Kwak MK, Sutter TR, Kensler TW (2001) Antioxidative function and substrate specificity of NAD(P)H-dependent alkenal/one oxidoreductase. A new role for leukotriene B4 12-hydroxydehydrogenase/15-oxoprostaglandin 13-reductase. J Biol Chem 276: 40803–40810. [DOI] [PubMed] [Google Scholar]
- 59. Lavoie S, Chen Y, Dalton TP, Gysin R, Cuenod M, et al. (2009) Curcumin, quercetin, and tBHQ modulate glutathione levels in astrocytes and neurons: importance of the glutamate cysteine ligase modifier subunit. J Neurochem 108: 1410–1422. [DOI] [PubMed] [Google Scholar]
- 60. Kann S, Estes C, Reichard JF, Huang MY, Sartor MA, et al. (2005) Butylhydroquinone protects cells genetically deficient in glutathione biosynthesis from arsenite-induced apoptosis without significantly changing their prooxidant status. Toxicol Sci 87: 365–384. [DOI] [PubMed] [Google Scholar]
- 61. Maines MD (1988) Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J 2: 2557–2568. [PubMed] [Google Scholar]
- 62. Talalay P, Dinkova-Kostova AT (2004) Role of nicotinamide quinone oxidoreductase 1 (NQO1) in protection against toxicity of electrophiles and reactive oxygen intermediates. Methods Enzymol 382: 355–364. [DOI] [PubMed] [Google Scholar]
- 63. Wang HQ, Xu YX, Zhu CQ (2012) Upregulation of heme oxygenase-1 by acteoside through ERK and PI3 K/Akt pathway confer neuroprotection against beta-amyloid-induced neurotoxicity. Neurotox Res 21: 368–378. [DOI] [PubMed] [Google Scholar]
- 64. Shah ZA, Nada SE, Dore S (2011) Heme oxygenase 1, beneficial role in permanent ischemic stroke and in Gingko biloba (EGb 761) neuroprotection. Neuroscience 180: 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hung SY, Liou HC, Kang KH, Wu RM, Wen CC, et al. (2008) Overexpression of heme oxygenase-1 protects dopaminergic neurons against 1-methyl-4-phenylpyridinium-induced neurotoxicity. Mol Pharmacol 74: 1564–1575. [DOI] [PubMed] [Google Scholar]
- 66. Ahmad AS, Zhuang H, Dore S (2006) Heme oxygenase-1 protects brain from acute excitotoxicity. Neuroscience 141: 1703–1708. [DOI] [PubMed] [Google Scholar]
- 67. Panahian N, Yoshiura M, Maines MD (1999) Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J Neurochem 72: 1187–1203. [DOI] [PubMed] [Google Scholar]
- 68. Xiao H, Lv F, Xu W, Zhang L, Jing P, et al. (2011) Deprenyl prevents MPP(+)-induced oxidative damage in PC12 cells by the upregulation of Nrf2-mediated NQO1 expression through the activation of PI3K/Akt and Erk. Toxicology 290: 286–294. [DOI] [PubMed] [Google Scholar]
- 69. Park JS, Jung JS, Jeong YH, Hyun JW, Le TK, et al. (2011) Antioxidant mechanism of isoflavone metabolites in hydrogen peroxide-stimulated rat primary astrocytes: critical role of hemeoxygenase-1 and NQO1 expression. J Neurochem 119: 909–919. [DOI] [PubMed] [Google Scholar]
- 70. Yang Y, Dieter MZ, Chen Y, Shertzer HG, Nebert DW, et al. (2002) Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm(−/−) knockout mouse. Novel model system for a severely compromised oxidative stress response. The Journal of biological chemistry 277: 49446–49452. [DOI] [PubMed] [Google Scholar]
- 71. Huang da W, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 4: 44–57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Adenoviral overexpression of Nrf2 and its effects on H2O2 toxicity. Wild-type (WT) astrocytes were infected with adGFP or adNrf2 adenovirus and then treated with H2O2 as indicated. Cell viability was determined by A) MTS or B) LDH. Statistics were performed using 2-way ANOVA, * indicates p<0.01.
(TIF)
The effect of GCLM siRNA knockdown on Nrf2 protection against 4-HNE. adGFP or adNrf2 infected astrocytes were pretreated with A) non-targeting siRNA (siNT) or B) glutamate-cysteine ligase, modifier subunit siRNA (siGCLM). 4-HNE toxicity curves were performed as indicated. Cell viability was determined by MTS. Statistics were performed using 2-way ANOVA, * indicates p<0.01.
(TIF)
The effect of sulfiredoxin-1 knockdown on cellular sensitivity to toxins and Nrf2 mediated protection. Vehicle/tBHQ or adGFP/adNrf2 treated astrocytes were pretreated with either non-targeting (siNT) or sulfiredoxin-1 (siSrdxn) siRNA. Toxicity curves were performed as indicated: A) H2O2, B) tBOOH, or C) 4-HNE. Cell viability was determined by MTS. Statistics were performed using 2-way ANOVA, * indicates p<0.01.
(TIF)
The effect of glutathione deficiency on extracellular H2O2 clearance. Wild-type (WT) or glutamate-cysteine ligase, modifier subunit knockout (GCLM-KO) astrocytes were infected with adGFP or adNrf2 virus. The rate of H2O2 clearance from the extracellular medium was measured over time for wild-type (WT) or GCLM-knockout (GCLM-KO) cells. Statistics were performed using 2-way ANOVA, * indicates p<0.01.
(TIF)
Log2 Histogram of SILAC Fold Change. To check for a normal distribution of expression changes, raw fold changes for each protein were log2 converted, binned, and plotted.
(TIF)
siRNA Sequences
(XLSX)
Primer Sequences for qPCR
(XLSX)
(XLSX)
Gene Ontologhy Analysis
(XLSX)
Oxidative Stress Related Proteins
(XLSX)