

Abstract
The gene encoding type II DNA topoisomerase from the kinetoplastid hemoflagellated protozoan parasite Leishmania donovani (LdTOP2) was isolated from a genomic DNA library of this parasite. DNA sequence analysis revealed an ORF of 3711 bp encoding a putative protein of 1236 amino acids with no introns. The deduced amino acid sequence of LdTOP2 showed strong homologies to TOP2 sequences from other kinetoplastids, namely Crithidia and Trypanosoma spp. with estimated identities of 86 and 68%, respectively. LdTOP2 shares a much lower identity of 32% with its human homologue. LdTOP2 is located as a single copy on a chromosome in the 0.7 Mb region in the L.donovani genome and is expressed as a 5 kb transcript. 5′-Mapping studies indicate that the LdTOP2 gene transcript is matured post-transcriptionally with the trans-splicing of the mini-exon occurring at –639 from the predicted initiation site. Antiserum raised in rabbit against glutathione S-transferase fusion protein containing the major catalytic portion of the recombinant L.donovani topoisomerase II protein could detect a band on western blots at ∼132 kDa, the expected size of the entire protein. Use of the same antiserum for immunolocalisation analysis led to the identification of nuclear, as well as kinetoplast, antigens for L.donovani topoisomerase II. The in vitro biochemical properties of the full-length recombinant LdTOP2 when overexpressed in E.coli were similar to the Mg(II) and ATP-dependent activity found in cell extracts of L.donovani.
INTRODUCTION
The biological importance of DNA topoisomerases is well recognised (1,2). These enzymes, classified as type I topoisomerases if they cut a single strand of the DNA duplex or as type II topoisomerases if both strands are cleaved, participate in nearly all events relating to DNA metabolism including replication, transcription and recombination. Eukaryotic type II topoisomerases are also important because of their essential role in chromosome segregation (3,4) and maintenance of chromosome structure (5,6). In addition, DNA topoisomerases have emerged as proven targets for clinically useful antitumour, antibacterial (7) and antiparasitic drugs (8). Work on DNA topoisomerases from the kinetoplastid protozoan parasites has been a major focus of interest, since these enzymes are believed to play an important role in the replication of the unusual kinetoplast DNA (kDNA) harboured in the mitochondrion (kinetoplast) of these pathogens.
Type II topoisomerases have been isolated from several kinetoplastid protozoans and the genes encoding these enzymes have been cloned and sequenced in Trypanosoma brucei (9), Trypanosoma cruzi (10) and Crithidia fasciculata (11). The proteins are dimers, usually 137–160 kDa in size. We have previously reported type II topoisomerase activities from cell extracts of the kinetoplastid hemoflagellated protozoan parasite Leishmania donovani, which has ATP-dependent and -independent decatenating properties (12,13). However, it is uncertain whether the L.donovani enzyme preparations contained primarily one and the same enzyme or whether they contained distinct activities.
To elucidate this, as well as to delineate features differentiating parasite topoisomerase II enzyme from its counterpart in higher eukaryotes, so that drug selectivity between the host and parasite replication apparatus can be exploited for antileishmanial therapy, we undertook isolation and characterisation of the TOP2 gene from L.donovani (LdTOP2). To the best of our knowledge, this is the first report of a gene encoding type II DNA topoisomerase from Leishmania.
MATERIALS AND METHODS
Parasite and culture condition
Leishmania donovani D1700 parasites were maintained as promastigotes at 22°C in M199 media containing 10% foetal bovine serum (BRL Life Technologies), penicillin (50 U/ml) and streptomycin (50 µg/ml). Cells were harvested in late log phase (1 × 107 parasites/ml) by centrifugation at 1200 g at 4°C for 10 min, and washed with phosphate-buffered saline (PBS) pH 7. The cell pellet was used at this stage or stored at –70°C. Amastigotes were isolated from spleen of L.donovani-infected golden hamsters as described (14).
Nucleic acid isolation, pulsed field gradient gel electrophoresis (PFGE) and hybridisation analysis
Genomic DNA was isolated from ∼2 × 109 L.donovani D1700 promastigote cells by standard procedures (15). Total RNA was isolated from 109 cells using an RNA isolation kit (Boehringer Mannheim). λ DNAs were prepared by the Qiagen lambda isolation kit. Plasmid DNAs were isolated by the alkaline lysis procedure (15). Leishmania chromosomes were separated by PFGE in which low melting agarose blocks containing embedded cells (107/ml log phase promastigotes) were electrophoresed in a contour clamped homogenous electric field apparatus (CHEF DRIII, Bio-Rad) in 0.5× TBE, with buffer circulation at a constant temperature of 14°C. Saccharomyces cerevisiae chromosomes were used as markers. DNA, RNA and chromosome blots were hybridised with random primed α-32P-labeled DNA probes. All post-hybridisation washes were to a final stringency of 0.1× SSC, 0.1% SDS at 65°C.
Polymerase chain reaction (PCR), library screening and sequence analysis
PCR was performed in 50 µl reaction volume containing 100 ng of genomic DNA/plasmid DNA, 100 pmol each of either degenerate or gene-specific primer sets, 200 µM of each dNTP and 2.5 U Taq DNA polymerase (Boehringer Mannheim). For amplifying products >1 kb, the Expand high fidelity Taq DNA polymerase (Boehringer Mannheim) was used.
Leishmania donovani (D1700, a clone of the 1S Sudanese strain) genomic DNA library (a kind gift from Prof. Buddy Ullman, Oregon Health Services University, OR) was constructed by the insertion of partial Sau3AI digest (16–24 kb fragments) of L.donovani strain D1700 DNA into the BamHI site of phage λGEM11 (Promega). 5 × 104 p.f.u. were plated and screened with the random-labeled 800 bp PCR-derived probe. Positive plaques were selected and purified through tertiary screening. The isolated DNA from λ recombinant clones was digested with restriction enzymes and relevant restriction fragments that mapped to the PCR-derived probe were inserted into pBluescript (SK+) for automated sequencing.
DNA sequence analyses were performed by DNASIS (v. 3.0). Comparisons with sequences of the database were performed using the search algorithm BLAST (16). Analysis of protein sequences was carried out using the Proteomics tools provided by the ExPASy molecular biology WWW server, which contains programs such as SAPS (17) and SOPM (18). Multiple alignment of amino acid sequences was performed using the CLUSTAL W package (19). The TREEVIEW software package v. 1.5 (20) was used for construction of a phylogenetic tree, which read PHYLIP style treefiles produced by CLUSTAL W.
Reverse transcription (RT)–PCR
RT–PCR of total RNA was performed using a non-degenerate sense primer, 5′-CGGGA TCCCAACGCTATATAAGTATCAGTTTCTGTACTTTATTG-3′, designed to the L.donovani mini-exon (21) and an antisense primer, 5′-CGGAATTCTGCTACGCACA ATGGGGA -3′, corresponding to nucleotide position 301–318 of the LdTOP2 ORF. Restriction sites are underlined. The PCR program consisted of an initial reverse transcription at 42°C for 1 h, followed by amplification of the cDNA product for 30 cycles at 94°C for 1 min, 50°C for 1 min and 68°C for 2 min. The TitanTM one tube RT–PCR kit from Boehringer Mannheim was used. The resultant RT–PCR product (∼940 bp) was cloned at BamHI–EcoRI sites of pBluescript SK(+) and sequenced.
The plasmid constructs used in recombinant expression
A 1.1 kb fragment was amplified from the 4.1 kb genomic clone, using a sense primer with a flanking BamHI site, 5′-CGGGATCCTGACAGAGGGTGACTCGGCG-3′, that coded for the conserved amino acid sequence TEGDSA at position 1382–1397, and an antisense primer with a flanking XhoI site, 5′-CCGCTCGAGCGGCAGGATGGGAACGTA-3′, which corresponded to amino acid residues YVPILP at position 2498–2515. The amplified fragment was cloned into the BamHI–XhoI site of pGEX-5X2 vector (Amersham Pharmacia Biotech). The resultant construct, pGEX–GST–LdTOP2 was transformed into BL21 (DE3) strain of Escherichia coli.
The complete 3.7 kb LdTOP2 ORF was amplified from the 4.1 kb genomic clone using a sense primer, 5′-GGGAATTCCATATGACAGACGCTTCCAAG-3′, containing an NdeI site created at the initiation codon, and an antisense primer, 5′-CGGGATCCCCCGCTTCACATGCTCCCCTTACA-3′, with a BamHI site immediately downstream from the termination codon. The amplified DNA was cloned into the NdeI–BamHI sites of the pET3a vector (Novagen) to generate the construct pET3a–LdTOP2, which was subsequently transformed into BL21 (DE3) pLysS strain of E.coli.
Expression and purification procedures
Expression from the construct pGEX–GST–LdTOP2 was induced at OD = 0.6 with 0.5 mM IPTG at 37°C for 4 h. Bacteria were harvested by centrifugation and the cell pellets resuspended in lysis buffer (20 mM Tris–Cl pH 8, 150 mM NaCl, 1 mM EDTA, 100 µg/ml lysozyme, 5 mM DTT and protease inhibitor). Final lysis was achieved by addition of 1.5% sarkosyl and after sonication on ice, the lysate was clarified by centrifugation. Supernatants were adjusted to 2% (v/v) Triton X-100, vortexed and incubated with glutathione–Sepharose beads overnight at 4°C. After several washes with PBS, the beads were boiled with 1% SDS to completely elute the bound pGEX–GST–LdTOP2 fusion protein.
Expression from pET3a–LdTOP2 was performed as for pGEX–GST–LdTOP2, except that induction was carried out at 22°C for 8 h since it was noted that large amounts of the protein formed inclusion bodies at 37°C. The lysis buffer used in this case contained 0.01% (v/v) Triton X-100. After sonication and centrifugation, the supernatant was used for decatenation assays.
Decatenation assay
kDNA decatenation assays were done in total volumes of 25 µl containing 25 mM Tris–HCl pH 7.9, 10 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 50 mM NaCl, 10% glycerol, 1 µg of kDNA from L.donovani strain D1700 and several dilutions of either crude extracts containing the expressed proteins (i.e., induced cultures) or uninduced crude extracts. The assays were carried out at 30°C for 30 min and the reaction products analysed by electrophoresis in 1% agarose gels.
Antibody production and western blot analysis
The GST–LdTOP2 fusion protein (100 µg) was subcutaneously inoculated (22) in rabbit using Freund’s complete adjuvant, followed by injections at 2 week intervals with incomplete adjuvant to produce the polyclonal anti-GST–LdTOP2 antiserum. This serum was then used for western blot analysis with the GST–LdTOP2 fusion protein (66 kDa) and full-length expressed protein (∼132 kDa) in E.coli extracts and also with cellular extracts from L.donovani promastigotes and amastigotes. Western blot analysis was carried out using the ECL kit (Amersham Pharmacia Biotech) according to the manufacturer’s protocol.
Immunofluorescence microscopy
For total cell staining, L.donovani promastigotes (10 × 106 parasites) were harvested by centrifugation (4000 g for 10 min), suspended in 10 µl PBS, smeared on slides and air dried. Cells were fixed in chilled methanol and acetone (50:50) for 10 min. After rehydration in PBS for 5 min, slides were incubated with preimmune serum or with immune serum against recombinant LdTOP2 protein (1:25 dilution) in the presence of 3% bovine serum albumin for 30 min. Following the first antibody reactions, slides were washed three times in PBS and then reacted with fluorescein isothiocyanate (FITC)-conjugated goat-derived anti-rabbit IgG (Sigma; 1:500 dilution) for 30 min. After the last wash, cells were mounted in 10% glycerol in PBS containing 0.1% p-phenylenediamine to prevent fading. To locate the nucleus and the kinetoplast, fixed parasites were also stained with ethidium bromide (0.5 µg/ml). Cells were finally viewed and photographed under an Olympus fluorescence microscope using a 100× objective.
RESULTS
Isolation of the LdTOP2 gene, sequence analysis and characterisation of the 5′-untranslated region (UTR)
Initial attempts to isolate the L.donovani DNA topoisomerase II gene using a 2.7 kb SmaI fragment of pDC806, the recombinant plasmid corresponding to the T.brucei TOP2 gene (a gift from Prof. P.T.Englund, Johns Hopkins University) was not successful. Therefore, PCR was employed to pick up a gene internal probe from L.donovani using degenerate primers. For this, amino acid sequences from known eukaryotic type II topoisomerases available in the database were compared by multiple alignment and, based on this, oligonucleotides corresponding to two highly conserved motifs were used for PCR amplification with L.donovani (D1700) genomic DNA. The sense primer (Oligo1), 5′-CGGAATTCCACIGARGGIGAYYSIGCIAARGC-3′, was constructed to a degenerate oligonucleotide coding for the conserved heptapeptide sequence, LIMTDQD. The antisense primer (Oligo2), 5′-CGGGATCCCGTTYTGIAARTAIATRKCIGGICG-3′, was also synthesised to a degenerate oligonucleotide corresponding to amino acid residues APRYIFT, which included the active site tyrosine. The 800 bp PCR-amplified fragment, when blotted, gave a positive result with the T.brucei probe. BLAST analysis of the 800 bp amplicon cloned into pBluescript (SK+) showed that its sequence shared extensive homology with other eukaryotic topoisomerase II genes.
The 800 bp TOP2 gene internal probe from Leishmania was then used to screen the L.donovani genomic DNA library to isolate the full-length gene. Restriction DNA digestion of recombinant λ clones as well as the subsequent genomic clones in pBluescript (SK+) along with hybridisation with the 800 bp gene internal probe could map the entire coding sequence of the LdTOP2 gene (GenBank accession no. AF150876).
The sequence revealed a single ORF of 3711 bp predicting a protein of 1236 amino acids with a calculated molecular mass of 139 kDa. The sequence is preceded by an inframe termination codon immediately upstream from the first ATG codon. This sequence is not interrupted by introns and, like other Leishmania genes, is GC-rich, 60.5% being the average GC content and 84% of the codons containing G or C at the third nucleotide position.
Figure 1 shows regions of an alignment of the predicted amino acids from L.donovani topoisomerase II with homologous sequences from a number of other species, including the kinetoplastid protozoa. The highest density of homology is observed between L.donovani and C.fasciculata topoisomerase II, i.e., identity of 86% and similarity of 91%. The values are 68 and 78% for L.donovani and the other trypanosomes (T.brucei and T.cruzi), respectively. With its human counterpart, the L.donovani topoisomerase II sequence shares a much lower identity of 32% and similarity of 47%. The degree of conservation is generally greater in the N-terminal two-thirds of the coding sequence and falls off markedly towards the C-terminus.
Figure 1.

Multiple alignment of conserved regions of topoisomerase II sequences from L.donovani (Ld, GenBank accession no. AF150876), C.fasciculata (Cf, SWISS-PROT no. P27570), T.cruzi (Tc, SWISS-PROT no. P30190), S.cerevisiae (Sc, SWISS-PROT no. P06786), D.melanogaster (Dm, SWISS-PROT no. P15348), Human (Ha, SWISS-PROT no. P11388), P.falciparum (Pf, SWISS-PROT no. P41001) and African swine fever virus (V, SWISS-PROT no. P34203) using CLUSTAL W. The amino acids are numbered to the right of the respective sequences. Conserved regions are shown in shaded boxes with ‘c’ representing the consensus. Asterisks indicate identity; colons indicate strong similarity and full stops indicate weak similarity. Hyphens represent the gap used to optimise the alignment. The domains from which degenerate primers were designed for picking up the PCR clone have been overlined.
A number of important features identified in LdTOP2 are found to be conserved as in other type II topoisomerases, such as the Gly140 residue homologous with the ATPase domain of gyrase B (23), and Tyr775 corresponding to the active site residue. The predicted protein product of the LdTOP2 gene differs from eukaryotic topoisomerase II sequences, including other kinetoplastids, in that it possesses two tandem periodic repeats of five amino acids, TSTTA, in the N-terminal half at positions 132–136 and 598–602. The significance of these repeats is not clear. Like other type II topoisomerases, the C-terminal domain (CTD) of the LdTOP2 sequence contains putative signals for phosphorylation sites of kinases. However, the CTD of LdTOP2 is shorter, with fewer charged residues making the Leishmania TOP2 gene smaller, like other trypanosomatid topoisomerase II genes. The percentage of basic amino acid residues in the deduced protein of LdTOP2 is 13.3%, which also form an integral part of the predicted nuclear localisation signals (NLS) present at amino acid positions 997 (KRRR), 1164 (PPPSKRR) and 1168 (KRRP).
All DNA topoisomerase II genes code for ubiquitous proteins and their alignments and comparative analysis can form a strong base for molecular phylogeny. A phylogenetic tree has been constructed (Fig. 2) using the LdTOP2 and other representative topoisomerase II sequences. The tree indicates the close evolutionary relationship of L.donovani and C.fasciculata among the kinetoplastid protozoa.
Figure 2.

Phylogenetic tree using the amino acid sequences of type II topoisomerases from L.donovani and other organisms. The phylogram is displayed on TREEVIEW that reads Phylip format treefiles under the CLUSTAL W program.
Northern blotting of the total L.donovani RNA with the PCR-generated 800 bp gene internal probe for LdTOP2 revealed a single transcript of ∼5 kb (data not shown). However, northern analysis did not provide any information about the 5′-end of the transcript. It is known that mature trypanosomatid mRNAs contain a ubiquitous sequence, the 35 nt mini-exon or spliced leader, that is added post-transcriptionally by trans-splicing to the 5′-end of primary mRNA transcripts (21,24). The 5′-end of the mature LdTOP2 mRNA was thereby determined by performing RT–PCR amplification of L.donovani RNA using the medRNA (sense) primer and a LdTOP2 sequence-specific (antisense) primer. A 940 bp product was obtained which, when hybridised, probed positive with a subclone encompassing the 5′ portion of the LdTOP2 gene. Cloning and sequencing of the RT–PCR product demonstrated that the site for trans-splicing of the mini-exon was located at nucleotide –639 from the predicted translation initiation site.
Molecular characterisation of the LdTOP2 locus
In order to verify the copy number of the LdTOP2 gene, genomic DNA from L.donovani promastigotes was analysed by Southern hybridisation (Fig. 3A). For this, genomic DNA was digested with restriction endonucleases which cleave at least once within the ORF of the gene, blotted and probed with the 800 bp TOP2 gene internal probe corresponding to positions 1537–2335. Restriction digests yielded expected sized fragments that were compatible with fragments observed upon digestion of the λ genomic clones. This led us to conclude that no heterogeneity existed on the 5′ and 3′ flanking regions of the gene and the LdTOP2 locus contained only a single gene copy. Next, to determine the exact chromosomal location of the gene, chromosomes were separated by PFGE, blotted and probed with the randomly labelled 800 bp TOP2 gene internal probe. The probe hybridised to a chromosome in the 0.7 Mb region in the L.donovani genome (Fig. 3B).
Figure 3.
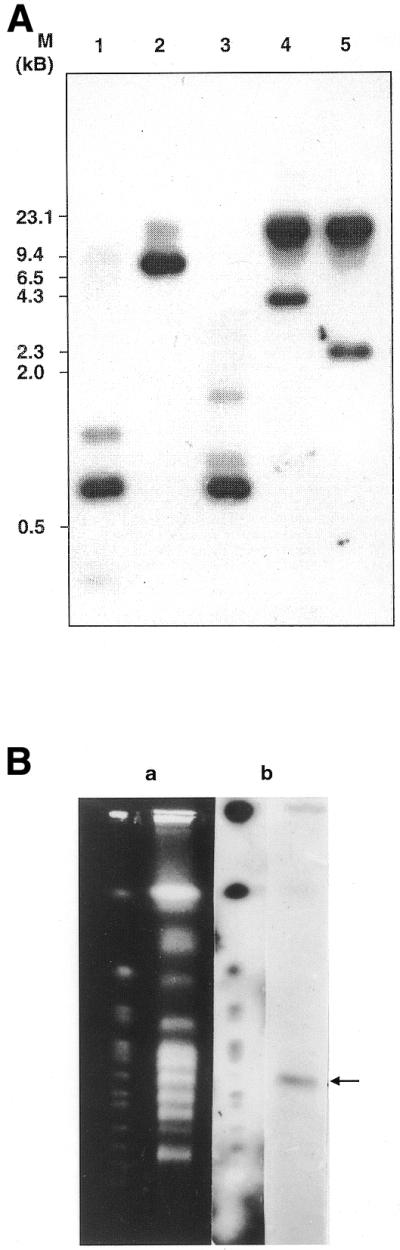
Molecular characterisation of the LdTOP2 locus. (A) Southern blot showing that the LdTOP2 is a single copy gene. Lanes 1–5, restriction digests of L.donovani D1700 genomic DNA with SalI, HindIII, PstI, EcoRV and EcoRV–XbaI, respectively, and their hybridisations with the 800 bp gene internal probe of LdTOP2 corresponding to positions 1537–2335. The positions of DNA molecular weight markers are indicated on the left of the figure. (B) Chromosomal location of the LdTOP2 gene. a, Chromosomes of S.cerevisiae and L.donovani D1700 separated and visualised with ethidium bromide before blotting; b, the same blots probed with yeast genomic DNA for S.cerevisiae and LdTOP2 gene probe for L.donovani. Arrow shows the hybridising band of L.donovani. PFGE running conditions as follows: initial switch time, 1800 s; final switch time, 1800 s; run time, 72 h; current, 2 V/cm; including angle, 106°; actual current, 30 mAmp.
Overexpression of full-length LdTOP2 enzyme and decatenating activity
To authenticate the reading frame and characterise the recombinant protein, the encoding LdTOP2 sequence was cloned inframe in pET3a expression vector with its own start ATG codon, under the control of a bacterial Shine–Dalgarno sequence and a phage T7 RNA polymerase promoter. The resultant pET3a–LdTOP2 construct was transformed into E.coli and protein overexpression induced as described in Materials and Methods. A protein with a molecular weight that matched the estimated ∼132 kDa according to amino acid composition of LdTOP2 was detected in the crude extract from induced cells carrying pET3a–LdTOP2 (Fig. 4). Such a protein was not detected in crude extract from induced cells carrying the vector pET3a alone. Decatenating activity analogous to that displayed by the ATP-dependent topoisomerase II isolated from L.donovani promastigotes could be detected in the supernatant from crude bacterial extracts overproducing the LdTOP2 protein (Fig. 5) while no such activity was observed in non-induced bacterial cells harbouring the same chimeric plasmid or in cells harbouring pET3a only (data not shown). That the observed decatenation of kDNA was not due to nuclease activity was substantiated by the inhibition of activity by etoposide, a eukaryotic topoisomerase II inhibitor (Fig. 5).
Figure 4.
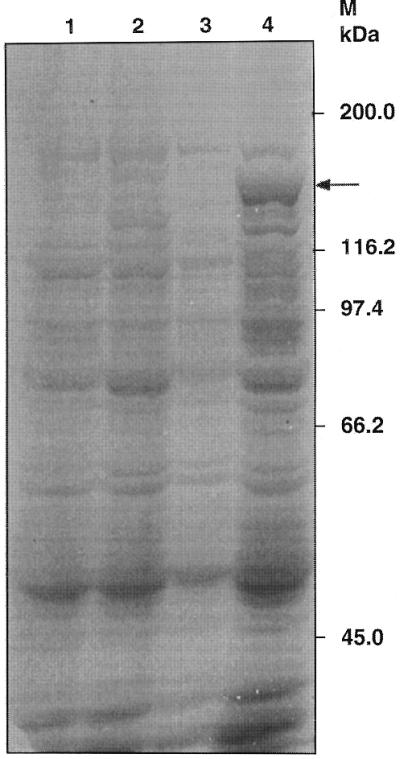
Coomassie-stained SDS–PAGE showing the overexpression of full-length LdTOP2 protein in E.coli. The pET3a bacterial extract is shown, before induction (lane 1) and after induction (lane 2) with 0.5 mM IPTG. Lanes 3 and 4 show the uninduced and induced extracts of pET3a–LdTOP2, respectively. Arrow shows the induced recombinant LdTOP2 protein. The sizes of the broad range protein MW markers (Bio-Rad) are shown on the right.
Figure 5.
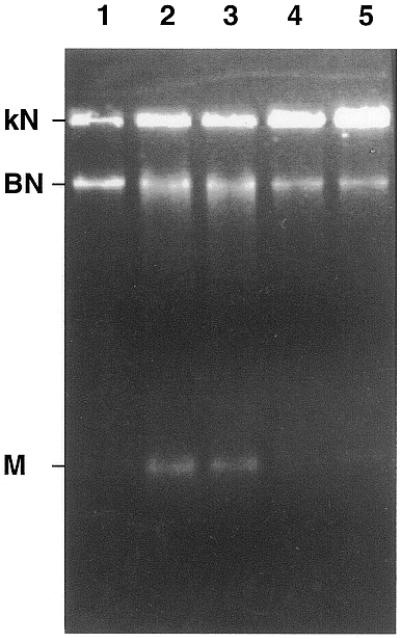
Biochemical characterisation of the bacterially expressed LdTOP2 protein: in vitro decatenation activity and its inhibition by etoposide. Lane 1, 1 µg of kDNA network; lanes 2 and 3, incubation of 1 µg of kDNA with 0.1 and 0.2 µl of induced pET3a–LdTOP2 crude extract in the decatenating mixture containing 2 mM ATP; lanes 4 and 5, same as lanes 2 and 3, respectively, but in the presence of 20 µg/ml of etoposide. kN, kDNA network; BN, broken network; M, released minicircles.
Purification of GST–LdTOP2 fusion protein for antiserum production and immunolocalisation
To gain insight into the intracellular localisation of LdTOP2 protein, rabbit antiserum was raised against a portion of recombinant LdTOP2 (the 1.1 kb sequence). This region was chosen because it corresponds to one of the most conserved regions in different eukaryotic type II topoisomerases, including those from other trypanosomatids. Following IPTG induction of transformed bacteria using the pGEX–GST–LdTOP2 construct, an additional band of 65 kDa (27 kDa from Schistostoma japonicum glutathione S-transferase and 38 kDa from LdTOP2) was observed after SDS–PAGE as compared to non-induced bacterial extracts. The 65 kDa recombinant polypeptide was further purified (Materials and Methods) and a polyclonal antibody raised against the purified protein in rabbit. The antiserum recognised the 65 kDa purified GST fusion protein, as well as the ∼132 kDa band on western blots of bacterial crude extracts containing the full-length expressed LdTOP2 protein (Fig. 6A).In the case of size-fractionated parasite proteins, the antiserum could detect a weak band of the anticipated LdTOP2 size in both promastigote and amastigote extracts. The polyclonal anti-LdTop2 antibody was then used to probe fixed parasites in order to determine the intracellular location of the topoisomerase II enzyme. When antiserum against recombinant LdTOP2 was used in double antibody immunofluorescence, the fluorescence pattern indicated that the antiserum recognised a nuclear as well as kinetoplast antigen (Fig. 6B). No fluorescence was observed when preimmune serum was used as the first antibody (data not shown). These results strongly suggest that the L.donovani topoisomerase II is associated with both the nucleus and the kinetoplast.
Figure 6.
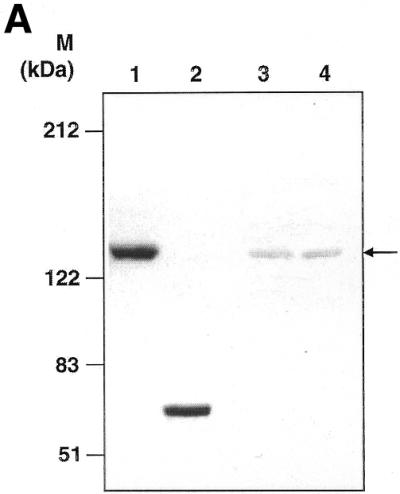

Western blotting and immunolocalisation studies using anti-GST–LdTOP2 antibody. (A) Western blot analysis of bacterial extract of induced pET3a–LdTOP2 (lane 1), purified GST–LdTOP2 fusion protein (lane 2), a leishmanial promastigote cell extract (lane 3) and a leishmanial amastigote cell extract (lane 4) reacted with the anti-LdTOP2 antibody. Arrow shows position of the L.donovani topoisomerase II. The standards for prestained broad range protein molecular weight marker (Bio-Rad) are shown on the left. (B) Immunocytochemical localisation with anti-LdTOP2 antiserum using fluorescent detection methods. Late log phase L.donovani promastigotes were fixed, and treated with the anti-LdTOP2 primary antibody and FITC-tagged secondary antibody. Parasite cells were also stained with ethidium bromide to locate the nucleus and the kinetoplast. (a) The visualisation of FITC fluorescence seen at 100× magnification in an Olympus fluorescence microscope. An enlarged view of a single promastigote after FITC staining (c) was obtained on scanning and it was compared with an ethidium bromide stained cell (b). The nucleus (N) and kinetoplast (K) are marked in b and c.
DISCUSSION
The LdTOP2 gene appeared to be present as a single copy gene on a chromosome in the 0.7 Mb region in L.donovani genome, its position corresponding to the Leishmania major Friedlin chromosome 23 (GenBank accession no. AL138973). The message size as determined using RNA prepared from L.donovani promastigotes is ∼5 kb. This is larger than the coding sequence. This could be partly accounted for by sequencing the 5′-UTR of LdTOP2 and mapping the trans-splicing junction at –639 from the initiation codon. Furthermore, in analogy with other kinetoplastida genes, no consensus eukaryotic promoter sequences such as TATA or CCAAT could be identified uptream of the initiation codon.
The nucleotide sequence of LdTOP2 and the predicted amino acid sequence deduced from it revealed few surprises. The conserved motifs present in the topoisomerases II found in other species are all present in the L.donovani sequence in similar spatial arrangement. Among the few distortions of colinearity is the presence of two tandem periodic repeats, TSTTA in the N-terminal half of the LdTOP2 protein sequence only. The homology percentages of LdTOP2 with topoisomerse II proteins from other organisms are consistent with the position of these parasites on the evolutionary ladder (25), as also indicated by the phylogenetic tree analysis whereby the kinetoplastid protozoa L.donovani and C.fasciculata appear to diverge at the same time separately as a group.
The LdTOP2 ORF reveals the presence of a putative NLS towards the C-terminal domain. This suggests nuclear localisation of L.donovani topoisomerase II, as in higher eukaryotes. A precise insight into the intracellular location of the expressed topoisomerase II protein in L.donovani was gained by indirect immunofluorescence studies. A polyclonal antibody raised against a conserved portion of LdTOP2 showed nuclear as well as kinetoplast localisations for the L.donovani topoisomerase II protein. However, no specific amino acid residues could be identified in the N-terminus of the predicted LdTOP2 sequence, those conform to the usual eukaryotic mitochondrial import signals. In fact, neither the kinetoplast topoisomerase II nor the kinetoplast structure-specific endonuclease I from the related parasite C.fasciculata have such N-terminal sequences (26), whereas DNA polymerase β and some histone-like proteins from the same organism possess a nine amino acid cleavable presequence (27), which serves as the mitochondrial import signal. Some mitochondrial precursor proteins of C.fasciculata lacking N-terminal targeting signals have been shown to be targeted to mitochondria by means of internal signals (28). As in C.fasciculata, putative internal mitochondrial targeting signals in topoisomerase II of L.donovani might also operate in conjunction with other signals to determine the kinetoplast localisation of this protein. In previous reports it has been hypothesised that the enzyme recognised by the anti-CfTOP2mt antibodies in the kinetoplast of T.cruzi is the same as that recognised by anti-TcTOP2 serum in the nuclei of T.cruzi and Crithidia, and differences in cellular localisation might be explained in terms of which epitopes are available for recognition by the two different antisera (29). Our immunolocalisation experiments indicate that the L.donovani topoisomerase II enzyme is localised in the nucleus and the kinetoplast. Western blot analysis of whole cell lysates of Leishmania, using the same antibody, shows a single band, confirming the presence of a single TOP2 enzyme. The antiserum used for probing the intracellular location of LdTOP2 was raised against a conserved region of LdTOP2 comprising 341 amino acids, which share specific sequence homology to topoisomerase II proteins of other eukaryotes and not to any other proteins. Thus, the observed immunofluorescence due to LdTOP2 antibody in the nucleus and kinetoplast is due to the existence of the same topoisomerase II protein in these organelles, rather than cross-reactivity of antibody with other epitopes.
It is worth mentioning that an ATP-independent type II topoisomerase activity from L.donovani promastigotes had been previously reported from our laboratory (13), similar to the 200 kDa ATP-independent topoisomerase II purified from T.cruzi (30) and the 60 kDa topoisomerase II activity described in C.fasciculata (31). When overexpressed, however, the LdTOP2 gene product showed ATP-dependent activity in vitro, similar to the Mg(II) and ATP-dependent activity found in cell extracts of L.donovani (12) that catalyses the decatenation of kinetoplast DNA into covalently closed mini-circles.
Since LdTOP2 is a single copy gene and the antibody raised against the recombinant gene product recognises epitopes for topoisomerase II in the nucleus as well as in the kinetoplast, the presence of a second distinct enzyme in this parasite seems less probable. Moreover, in kinetoplastid parasites, replication of kinetoplast DNA is in approximate synchrony with that of nuclear DNA (32). Therefore, it is likely that the same enzyme might be involved in both nuclear and kinetoplast DNA replication activities.
Finally, the economic and health hazards posed by L.donovani in spreading visceral leishmaniasis make the search for potential drug targets in these organisms very demanding. In view of the importance of both prokaryotic and eukaryotic DNA topoisomerase II as targets of therapeutics, overexpression and functional characterisation of the gene encoding the L.donovani type II topoisomerase should facilitate the screening and designing of drugs against this parasite.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Prof. S.Bhattacharyya, the Director of our Institute, for his interest in this work. This work is supported by a Grant (BT/PRO493/MED/09/096/96) from the Department of Biotechnology, Government of India.
DDBJ/EMBL/GenBank accession no. AF150876
References
- 1.Wang J.C. (1996) DNA topoisomerases. Annu. Rev. Biochem., 65, 635–692. [DOI] [PubMed] [Google Scholar]
- 2.Nitiss J.L. (1998) Investigating the biological functions of DNA topoisomerases in eukaryotic cells. Biophys. Biochem. Acta, 1400, 63–81. [DOI] [PubMed] [Google Scholar]
- 3.Dinardo S., Voelkel,K. and Sternglanz,R. (1984) DNA topoisomerase II mutant of S.cerevisiae: topoisomerase II is required for the segregation of daughter molecules of the termination of DNA replication. Proc. Natl Acad. Sci. USA, 81, 2616–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nitiss J.L. (1994) Roles of DNA topoisomerases in chromosomal replication and segregation. Adv. Pharmacol., 29, 103–134. [DOI] [PubMed] [Google Scholar]
- 5.Earnshaw W.C., Halligan,B., Cooke,C.A., Heck,M.M.S. and Liu,L.F. (1985) Topoisomerase II is a structural component of mitotic chromosome scaffolds. J. Cell Biol., 100, 1706–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adachi Y., Luke,M. and Laemmli,U.K. (1991) Chromosome assembly ‘in vitro’: topoisomerase II is required for condensation. Cell, 64, 137–148. [DOI] [PubMed] [Google Scholar]
- 7.Schneider E., Hsiang,Y.H. and Liu,L.F. (1991) DNA topoisomerases as anticancer drug targets. Adv. Pharmacol., 21, 149–183. [DOI] [PubMed] [Google Scholar]
- 8.Shapiro T.A. (1993) Inhibition of topoisomerases in African trypanosomes. Acta Trop., 54, 251–260. [DOI] [PubMed] [Google Scholar]
- 9.Strauss P.R. and Wang,J.C. (1990) The TOP2 gene of Trypanosoma brucei: a single copy gene that shares extensive homology with other TOP2 genes encoding eukaryotic DNA topoisomerase II. Mol. Biochem. Parasitol., 38, 141–150. [DOI] [PubMed] [Google Scholar]
- 10.Fragoso S.P. and Goldenberg,S. (1992) Cloning and characterization of the gene encoding Trypanosoma cruzi DNA topoisomerase II. Mol. Biochem. Parasitol., 55, 127–134. [DOI] [PubMed] [Google Scholar]
- 11.Pasion S.G., Hines,J.C., Aebersold,R. and Ray,D.S. (1992) Molecular cloning and expression of the gene encoding the kinetoplast-associated type II DNA topoisomerase of Crithidia fasciculata. Mol. Biochem. Parasitol., 50, 57–68. [DOI] [PubMed] [Google Scholar]
- 12.Chakraborty A.K. and Majumder,H.K. (1987) Decatenation of kinetoplast DNA by an ATP-dependent DNA topoisomerase from the kinetoplast hemoflagellate Leishmania donovani. Mol. Biochem. Parasitol., 26, 215–224. [DOI] [PubMed] [Google Scholar]
- 13.Chakraborty A.K. and Majumder,H.K. (1991) An ATP-independent catenating enzyme from the kinetoplast hemoflagellate Leishmania donovani. Biochem. Biophys. Res. Commun., 180, 279–285. [DOI] [PubMed] [Google Scholar]
- 14.Jaffe C.L., Grimaldi,G. and McMahon-Pratt,D. (1984) The cultivation and cloning of Leishmania. In Morel,C.M. (ed.), Genes and Antigens of Parasites, A Laboratory Manual. UNDP/World Bank/WHO, 2nd edition, pp. 47–91.
- 15.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 16.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 17.Brendel V., Bucher,P., Nourbaksh,I., Blaisdell,B.E. and Karlin,S. (1992) Methods and algorithms for statistical analysis of protein sequences. Proc. Natl Acad. Sci. USA, 89, 2002–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geourjon C. and Deleage,G. (1994) SOPM: a self-optimized method for protein secondary structure prediction. Protein Eng., 7, 157–164. [DOI] [PubMed] [Google Scholar]
- 19.Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Page R.D.M. (1996) TREEVIEW: an application to display phylogenetic trees on personal computers. Comput. Appl. Biosci., 12, 357–358. [DOI] [PubMed] [Google Scholar]
- 21.Wilson K., Hanson,S., Landfear,S. and Ullman,B. (1991) Nucleotide sequence of the Leishmania donovani medRNA gene. Nucleic Acids Res., 19, 5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harlow E. and Lane,D. (1988) Antibodies Manual: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 23.Reece R.J. and Maxwell,A. (1991) DNA gyrase: structure and function. Crit. Rev. Biochem. Mol. Biol., 26, 335–375. [DOI] [PubMed] [Google Scholar]
- 24.Van der Ploeg L.H.T., Liu,A.Y.C., Michels,P.A.M., De Lange,T., Borst,P., Majumder,H.K., Weber,H., Weeneman,G.H. and Van Boom,J. (1982) RNA splicing is required to make the messenger RNA for a variant surface antigen in trypanosomes. Nucleic Acids Res., 10, 3591–3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michels P.A.M. (1986) Evolutionary aspects of trypanosomes: analysis of genes. J. Mol. Biol., 24, 45–52. [DOI] [PubMed] [Google Scholar]
- 26.Engel M.L. and Ray,D.S. (1999) The kinetoplast structure-specific endonuclease I is related to the 5′ exo/endonuclease domain of bacterial DNA polymerase I and colocalizes with the kinetoplast topoisomerase II and DNA polymerase β during replication. Proc. Natl Acad. Sci. USA, 96, 8455–8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Torri A.F. and Englund,P.T. (1995) A DNA polymerase β in the mitochondrion of the trypanosomatid Crithidia fasciculata. J. Biol. Chem., 270, 3495–3497. [DOI] [PubMed] [Google Scholar]
- 28.Folsch H., Guiard,B., Neupert,W. and Stuart,R.A. (1996) Internal targeting signal of the BCS1 protein: a novel mechanism of import into mitochondria. EMBO J., 15, 479–487. [PMC free article] [PubMed] [Google Scholar]
- 29.Fragoso S.P., Mattei,D., Hines,J.C., Ray,D.S. and Goldenberg,S. (1998) Expression and cellular localization of Trypanosoma cruzi type II DNA topoisomerase. Mol. Biochem. Parasitol., 94, 197–204. [DOI] [PubMed] [Google Scholar]
- 30.Douc-Rasy S., Kayser,A., Riou,J.F. and Riou,G. (1986) ATP independent type II topoisomerase from trypanosomes. Proc. Natl Acad. Sci. USA, 83, 7152–7156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shlomai J. and Zadok,A. (1983) Reversible decatenation of kinetoplast DNA by a DNA topoisomerase from trypanosomatids. Nucleic Acids Res., 11, 4019–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pasion S.G., Hines,J.C., Ou,X., Mahmood,R. and Ray,D.S. (1996) Sequences within the 5′ untranslated region regulate the levels of a kinetoplast DNA topoisomerase mRNA during the cell cycle. Mol. Cell Biol., 16, 6724–6735. [DOI] [PMC free article] [PubMed] [Google Scholar]