

Abstract
The numerous processes involved in the etiology of breast cancer such as cell survival, metabolism, proliferation, differentiation, and angiogenesis are currently being elucidated. However, underlying mechanisms that drive breast cancer progression and drug resistance are still poorly understood. As we discuss here in detail, the Notch signaling pathway is an important regulatory component of normal breast development, cell fate of normal breast stem cells, and proliferation and survival of breast cancer initiating cells. Notch exerts a wide range of critical effects through a canonical pathway where it is expressed as a type I membrane precursor heterodimer followed by at least two subsequent cleavages induced by ligand engagement to ultimately release an intracellular form to function as a transcriptional activator. Notch and its ligands are overexpressed in breast cancer, and one method of effectively blocking Notch activity is preventing its cleavage at the cell surface with γ-secretase inhibitors. In the context of Notch signaling, the application of clinically relevant anti-Notch drugs in treatment regimens may contribute to novel therapeutic interventions and promote more effective clinical response in women with breast cancer.
Keywords: breast cancer, signaling pathways, γ-secretase, γ-secretase inhibitors, combination breast cancer therapy
Introduction
In recent years, there have been many advances in deciphering critical cell signaling networks and their relationship to the driving forces of cancer onset, growth, and metastasis. Moreover, in the hierarchy of signaling pathways, several pathways are considered fundamental to regulation of cell fate and having widespread survival effects, namely the Notch, Wnt/Wingless (Wnt), and Hedgehog (HH) pathways.
This review will focus on the role of the canonical Notch signaling pathway in breast cancer etiology and progression. Furthermore, we will review the current therapeutic options available for inhibiting Notch. Blockade of an upregulated Notch signaling pathway can be achieved by inhibiting the formation of the main force of Notch activity, the Notch intracellular domain (NICD). Thus, a pharmacological approach using γ-secretase inhibitors (GSIs) to prevent the final cleavage step of the precursor form of Notch, ie, transmembrane Notch (Notch™) that will decrease levels of NICD could be a novel therapeutic strategy either as a single agent or in combination with targeted or cytotoxic chemotherapy for a subset of cancer patients.
Breast cancer subtypes
Breast cancer is a heterogeneous disease divided into four major subtypes: luminal A (estrogen receptor [ER]+/progesterone receptor [PR]+), luminal B (ER+/PR+/human epidermal growth factor receptor [HER]-2+), HER-2+/neu+, and triple negative (ER-PR-/HER-2+).1,2 Breast cancer of the luminal A or B subtype is derived from the luminal epithelium of the breast ducts, and these tumors express hormone receptors, ER and PR. These subtypes comprise 70%–80% of all breast cancers. The ER+/PR+ luminal A subtype is very sensitive to current antihormonal therapy such as tamoxifen, fulvestrant, or aromatase inhibitors. Luminal B breast tumors have a higher proliferative index than those of luminal A and are inherently more resistant to current antihormonal therapy. HER-2+/neu+ designates a breast cancer subtype that contains gene amplification for the ERBB2 proto-oncogene resulting in overexpression of the HER-2 receptor tyrosine kinase protein. The HER-2+/neu+ breast cancers are very sensitive to anti-HER-2 therapy such as trastuzumab or lapatinib. The final subtype of breast cancer is triple negative, which lacks expression of ER, PR, and HER-2. Triple negative breast tumors are the most aggressive, with poor prognosis and currently no approved targeted therapy. These triple negative breast tumors are treated with cytotoxic chemotherapy such as a DNA-damaging agent (cis- or carboplatin) or tubulin-destabilizing compounds (taxanes).
Although dramatic improvements have been made to cure breast cancer, one of the major problems that continue to plague both research scientists and clinicians is drug resistance. Therefore, elucidating the critical mechanisms that contribute to drug-resistant breast cancer will hopefully prevent tumor recurrence and disease progression and ultimately provide a “cure” to women with breast cancer.
Notch signaling
Over a century of research has revealed the mechanisms that regulate canonical Notch signaling in the context of cell-to-cell signaling that controls both embryonic and adult stem cell self-renewal, stem cell quiescence, cell fate and differentiation, cell survival, apoptosis, and tumorigenesis. Investigations elucidating the Notch pathway date back to the early 20th century, when in 1913 John Smith Dexter working in the laboratory of American geneticist Thomas Hunt Morgan observed the outcome of a mutation of a gene in Drosophila ampelophila, which resulted in a notch or indentation at the ends of the fly wings. He called them “perfect notched.”3 Additional research in 1917 by Morgan identified the alleles of this fly gene which eventually became known as “Notch”4 and he published his findings in The Physical Basis of Heredity5 in 1919. The Notch gene was eventually cloned and identified for the first time in 1985–1986.6,7 Related research employing the nematode worm Caenorhabditis elegans further elucidated the Notch signaling pathway, cell-to-cell interactions, and lateral inhibition during embryogenesis. Presently, embryologists and cancer researchers are the largest groups of research scientists studying Notch signaling.
Developmental Notch
The Notch signaling pathway mediates cell fate determination in three ways: regulatory, inhibitory, and inductive action.8 In regulatory signaling, during embryogenesis, Notch regulates the development and differentiation of many organ systems (angiogenesis, hematopoiesis, homeostasis, neurogenesis, nephrogenesis, myogenesis, and somatogenesis). The importance of this function was verified in Notch-1, Notch-2, Jagged-2, and Delta-1 knockout mice, which lack each Notch receptor or ligand regulatory components.9–12 The mice exhibited severe defects which resulted in embryonic or perinatal death. Notch activation in pluripotent stem cells initiates lateral inhibition so that a certain number of cells take on a specific cell fate and those adjacent are inhibited from differentiating.13 This process is exemplified in the following experiment: nascent chick retinal neurons transiently overexpressing Deltex-1 were found to prevent adjacent neuroepithelial progenitor cells from differentiating into neurons.14 Lastly, in inductive signaling, Notch promotes or induces the development of a certain cell type, usually amongst different (nonequivalent) cells. Such interactions are important for establishing demarcated boundaries between cell types, and the signaling is aptly referred to as boundary formation. The necessity of inductive signaling is evident in developmental studies. For example, a Notch-dependent localized signal affected the formation of the dorsoventral wing organizer in Drosophila,15,16 while the expression of Radical fringe determined the position of the dorsoventral boundary of vertebrate limbs.17
Notch receptors
The Notch receptor is classified as a large single-pass type 1 transmembrane glycoprotein. It is expressed as a heterodimer at the cell membrane. There are four mammalian Notch receptors, Notch 1–4, with Notch-1 being the longest and Notch-4 the shortest.13 They are comprised of three domains, extracellular, transmembrane, and intracellular (Figure 1). Notch is synthesized as a single, relatively large (>300 kDa) polypeptide in the endoplasmic reticulum. There, it undergoes O-glycosylation. An initial addition of O-linked fucose to the epidermal growth factor (EGF)-like repeats is mediated by O-fucosyl transferase1. After the Notch preprotein is chaperoned by the guanosine triphosphate hydrolase (GTPase) Rab-protein 6 through the secretory pathway to the trans-Golgi network, it undergoes additional elongation of the O-fucose with carbohydrate chains on serine and threonine residues by the Fringe family O-fucose-specific β1,3-N acetylglucosaminyl-transferases Lunatic, Manic, or Radical.18,19 Modification of the Notch receptor by Fringe proteins controls ligand-mediated activation.20 Next is cleavage by furin-like convertase into the N-terminus and C-terminus subunits and subsequent translocation of these mature entities to the cell plasma membrane.21,22 There, the cleaved subunits are assembled into the cell membrane as a fully functional heterodimeric receptor, noncovalently linked by a calcium cation awaiting engagement with a Notch ligand.
Figure 1.

Notch receptors and ligands.
The N-terminal extracellular domain of each Notch receptor is the ligand-binding component and consists of 29–36 multiple EGF-like repeats in tandem. From each extracellular domain extend six cysteine residues, which form three intra-domain disulfide bridges. Adjacent to the extracellular domain and closer to the cell membrane is the transmembrane domain, a dual hybrid moiety. The two components of this domain are: (1) the juxtamembrane RAM23 section (the negative regulatory region) made of three Lin-12/Notch repeats, which prevent ligand-independent interactions, plus two conserved cysteine residues; and (2) the heterodimerization section, which maintains the Notch receptor in a nonactivated state. The third part of the Notch receptor is the intracellular domain (C-terminus), which extends from the inner cell membrane into the cytoplasm. It contains four separate entities: (1) the DNA-binding protein recombination signal-binding protein for immunoglobulin kappa J (RBP-JK associated molecule or RAM domain), followed by a linker with a nuclear localization sequence; (2) seven iterated cdc10/ankyrin repeats; (3) a transcription activation domain (TAD) with an additional nuclear localization sequence; and (4) polypeptide proline, glutamate, serine, and threonine-rich motifs (PEST) with degradation signals or degrons that stabilize NICD in the nucleus and target it for rapid proteolytic degradation. Lastly, TAD is found in Notch-1 and Notch-2, but not in Notch-3 and Notch-4.
Notch ligands and activation
In vertebrates, the Notch ligands are known as Delta-like 1, 3, and 413,23–26 and Jagged-1 and 213,26 (Figure 1). They are single-pass Type 1 transmembrane proteins that bind and activate the Notch receptor “in trans” (at the surface of a neighboring cell). They have extracellular and intracellular domains. The Jagged ligands are longer than the Delta-like ligands, the length determined by the 6–16 EGF-like repeats in the extracellular domain. A cysteine-rich area is located at the end of the EGF-like repeats, with Jagged ligands having an additional cysteine-rich area. The intracellular domain of each ligand has a shorter cytoplasmic tail than the extracellular domain and contains a PDZ (post synaptic density protein [PSD95], Drosophila disc large tumor suppressor [Dlg1], and zonula occludens-1 protein [zo-1])-binding motif which aids in intracellular protein–protein interactions. The ligand-activated cell-surface receptor initiates a cascade of events with two subsequent proteolytic cleavages that result in NICD entry into the nucleus to function as a transcriptional activator.27–29
Cell-cell contact mediates Notch ligand to receptor binding which initiates short-range cell-to-cell communication, a mono-directional cascade of events beginning at the cell membrane and ultimately activating the CSL (C promoter binding factor-1 [CBF-1], suppressor of hairless, Lag-1) family of transcription factors in the nucleus. The ligand engages the Notch receptor through its cognate high affinity EGF-like repeats (Figure 2). Ligand-mediated endocytosis in the ligand-expressing cell (trans-endocytosis) provides a force to pull the extracellular domain of the Notch receptor from the transmembrane domain. This mechanical pull exposes the S2 cleavage site for the α-secretases of “A disintegrin and metalloprotease” family ADAM17 (tumor necrosis factor-α-converting enzyme TACE) or ADAM10, leading to ectodomain shedding of the extracellular portion of the transmembrane portion of the Notch receptor at approximately 12 amino acids outside the transmembrane domain.30,31 This proteolytic ectodomain “release” or shedding forms a carboxyterminal fragment called Notch extracellular truncation (NEXT).32 The ligand-Notch extracellular portion undergoes trans-endocytosis into the ligand-expressing, signal-sending cell, followed by endosomal-mediated degradation or recycling. Monoubiquitination by E3 ligases Mindbomb-1 and -2 or Neuralized-1 and -2 marks the ligand for endocytosis.
Figure 2.

Significant components in the Notch signaling pathway.
Abbreviations: ADAM/TACE, a disintegrin and metalloprotease/TNF-α converting enzyme; TNF-α, tumor necrosis factor-alpha; APH1, anterior pharynx-defective 1; ER, endoplasmic reticulum; NEXT, Notch extracellular truncation; NICD, Notch intracellular domain; PEN-2, presenilin enhancer 2; EGF, epidermal growth factor; S, site.
The remaining NEXT portion now exposes the S3 and S4 cleavage sites that are mediated by the γ-secretase complex.33 Interestingly, there are many γ-secretase substrates, a great number having relevance in breast cancer.34,35 This transmembrane aspartyl proteinase, considered a large complex, is comprised of a catalytic subunit designated presenilin 1 or presenilin 2, a seven-pass transmembrane protein, and accessory subunits comprised of the transmembrane proteins nicastrin (NCT), anterior pharynx-defective 1 (APH1), and presenilin enhancer 2 (PEN-2), a two-pass transmembrane protein. Nicastrin and APH1 stabilize PEN-2, which induces endoproteolysis of presenilin.36 Following receptor activation, NICD that is still attached to the inner cell membrane is marked for proteosomal degradation by E3 ubiquitin ligases Numb and Itch. γ-secretase severs NICD from the inside of the cell membrane, allowing it to enter the cytoplasm37 and eventually translocate to the nucleus (Figure 2).38
NICD forms a transcriptional activation complex with CSL in the nucleus once the ankyrin-repeat motif of NICD docks with the Rel homology region of the DNA-binding factor CSL. Thus, CSL changes from a transcriptional repressor to a transcriptional activator. There occurs a release of transcription factor co-repressors (CoRs) like class I or II histone deacetylases, CBF-1-interacting repressor (CIR), SKI-interacting protein (SKIP), silencing mediator of retinoid and thyroid hormone receptor (SMRT), and SMRT/HDAC (histone deacetylase)-1-associated repressor protein (SHARP), and a recruitment of transcription factor co-activators (CoAs) such as mastermind-like 1–3 (MAML) protein. MAML further recruits the histone acetyltransferases, cyclic AMP (adenosine monophosphate) response element-binding (CREB) protein CBP/p300 and p300/CBP-associated factor or general control non-depressible 5 (GCN5), to acetylate histone tails for the unwinding of nucleosomes within chromatin for active transcription. This leads to an increased expression of specific genes. Some of the Notch gene targets that can be activated are: c-Myc, p21, and cyclin D1 (cell cycle progression), Bcl-2 (inhibition of apoptosis), and hairy and enhancer of split basic helix-loop-helix HES 1, 5, 6, and 7, and HEY 1 and 2, and HEY-L family of proteins (transcriptional repressors).37 NICD activity in the nucleus ends with phosphorylation triggered by cyclin c-cyclin-dependent kinase 8 (C-CDK8). Subsequently, glycogen synthase kinase 3β phosphorylates the PEST domain of the C-terminus of the NICD, which is then targeted for polyubiquitination by E3 ligase SEL10/FWB7 in the proteosome.39 Figure 3 depicts Notch-mediated nuclear transcription.
Figure 3.

Notch-mediated nuclear transcription.
Abbreviations: NICD, Notch intracellular domain; CIR, CBF-interacting repressor; CSL, C promoter binding factor-1 [CBF-1], suppressor of hairless, Lag-1; HDAC, histone deacetylase; SMRT, silencing mediator of retinoid and thyroid hormone receptor; SHARP, SMRT/HDAC1 associated repressor protein; HAT, histone acetyltransferase; MAML, mastermind-like 1–3; SKIP, ski-interacting protein; Bcl-2, B-cell lymphoma 2; HES, hairy and enhancer of split; HEY, HES related with YRPW motif protein.
Notch and cancer: general overview
One of the earliest associations between Notch signaling and cancer occurred in 1991 in human T-cell acute lymphoblastic leukemia, where the Notch-1 gene was associated with the t(7;9)(q34;q34.3) chromosomal translocation.40 Notch cell signaling defects were detected in the form of alterations in the Notch-1 negative regulatory region and a loss of the C-terminus PEST domain, both of which lead to increased Notch-1 intracellular domain (N-1ICD) activity.41 In B-cell malignancies such as chronic lymphocytic leukemia, Notch-1 mutations were linked to increased disease progression and resistance to chemotherapy.42 Inconsistencies in the role of Notch in malignant B-cells became apparent as some data indicated that Notch signaling inhibited B-cell growth,43–45 while other data reported a Notch-induced increase in B-cell proliferation.46–48 In mantle cell lymphoma, Notch-149 or Notch-250 PEST domain mutations were reported. In addition to the presence of dysfunctional Notch receptors in leukemia, the ligand Jagged-2 was found to be significantly overexpressed in multiple myeloma.51 In addition to hematologic malignancies, aberrant Notch signaling has been found in solid tumors; for example, cervical,52,53 colon,54,55 liver,56,57 lung,58 pancreatic,59–62 prostate,63,64 ovarian,65,66 and renal.67 Indeed, based on the numerous reports on the role of Notch signaling in cancer development and progression, Notch signaling has become a major target for novel therapeutic strategies.68–72 The role of Notch signaling in cancer could possibly be a double-edge sword. It was reported that Notch receptors and ligands were both oncogenic and tumor-suppressive in the same tumor.73 The possibility that Notch promotes or suppresses tumor growth has also been put forth by others.74–76 Some discrepancies in Notch signaling in cancer may be explained in part by “cell context, dose, and timing,”77 as well as Notch cross-talk with other signaling pathways, the micro-tumor environment, and the stage of cancer at the time of detection.
Notch and breast cancer
There is strong evidence that Notch signaling is dysregulated in solid tumors,28,76,78 though as reported in leukemia, it may be both a tumor oncogene and suppressor in breast and other cancers.79 In mouse studies, tissue specific expression of N-1ICD induces spontaneous mammary tumors.80–82 Furthermore, transgenic (Tg) mice expressing mammary specific N-4ICD also form spontaneous mammary tumors.83 In fact, Notch-1 and Notch-4 are categorized as bonafide breast oncogenes.84 Further studies showed that overexpression of Notch-381 and Notch-485–87 also leads to murine mammary tumor formation. Studies from human breast cancer cell lines show deregulated expression of Notch and Notch ligand messenger RNA (mRNA).88 Results from a human xenograft model for inflammatory breast carcinoma (MARY-X) implicate altered Notch-3 signaling specificalxly.89 In a study of 200 Greek women from differing breast cancer subtypes, Notch-4 mRNA levels were found to be higher in the hormone receptor and HER-2-positive breast cancers, while Notch-1 and Notch-3 mRNA levels were higher in triple-negative specimens compared with normal tissue.90 When Notch-1 and Notch-4 and Jagged-1 and Delta-like-1 expression were measured by immunohistochemistry in breast hyperplasia and carcinomas, high levels of Notch-1 were found in the hyperplasias, ductal carcinoma in situ, infiltrating ductal carcinomas (IDCs), and infiltrating lobular carcinomas (ILCs), as well as elevated expression of Notch-4 and Jagged-1 in IDCs and ILCs.91 Moreover, Notch-1 and Notch-3 NICD levels were increased in both human breast cancer specimens and cell lines, and Notch-3 activated nuclear transcription in those specimens and cells.92 Further evidence for altered Notch-1 in human breast cancer was found in the form of aberrant Notch-1 activation in various breast cancer subtypes.93 In addition, samples from breast cancer patients showed co-overexpression of the Notch-1 receptor and its ligand Jagged-1 predicting the poorest patient survival.94,95 Lastly, examination of almost 100 breast cancer specimens by immunohistochemistry and quantitative polymerase chain reaction (PCR) showed the expression of Notch-1 also correlated with poor outcomes.96
Nonetheless, Notch receptors are not a homogeneous group functionally. When the transcriptional activities of N-1ICD, N-2ICD, and N-3ICD on HES-1 and HES-5 promoters were measured using a luciferase reporter assay, some of the differences were related to the combination of receptors used and expression level of RBP-Jκ (CSL or CBF-1). Also, inhibitory Notch-2 activity was confirmed, as co-expression of N-2ICD with N-1ICD or N-3ICD reduced their activity.97 In a xenograft study using MDA-MB-231 cells, Notch-2 inhibited tumor growth.98 Similarly, a clinical study which examined Notch-2 expression in breast cancer tissue by immunohistochemistry and qualitative and quantitative PCR concluded it may function as a tumor suppressor.96
In assessing the role of Notch signaling in breast cancer stem cells, it was concluded from in vitro and in vivo experiments that Notch-4 activity was eightfold higher in breast cancer stem cells than in differentiated cells, and inhibition of Notch-4 resulted in suppression of tumor growth.99 Moreover, breast cancer stem cells exhibited increased Notch signaling as compared with bulk tumor cells, especially in levels of HES-1 mRNA, and GSIs effectively blocked mammosphere formation, which is an assay to measure survival and self-renewal of breast cancer stem cells.100
Notch and tumorigenesis
Of the more than 300 breast cancer cases examined, approximately 50% showed a loss of Numb-mediated inhibition of Notch signaling by ubiquitination and proteosomal degradation.101 Of particular interest are two germline alterations (R62H and R71W) of presenilin-2 (PS-2) that have been reported in breast cancer patients with axillary node-negative disease, resulting in PS-2 being more susceptible to degradation.102 Furthermore, nicastrin-knockout mice, which have decreased proteolytic cleavage of Notch and consequently lower NICD, developed myeloproliferative disease,103 and Notch-1 knockout mice formed spontaneous basal cell carcinoma as they grew older.104
Possible mechanisms of action for Notch-driven tumor propagation are: gain of function mutation, ligand-mediated activation of Notch, and downregulation of Notch.105 Nonetheless, Notch tumorigenicity may be organ-dependent. In self-renewing systems such as skin, intestine, and bone marrow, Notch interacts with multiple signaling pathways. Oncogenesis derails these interactions such that Notch becomes a tumor suppressor in the skin and an oncogene in the bone marrow.75 Manipulation of gene expression has been useful to study Notch receptors and ligands in tumorigenic systems. For example, MCF-10A cells, considered nonmalignant and noninvasive, when transfected with Notch-4, grew in a soft agar assay, suggesting that Notch-4 is a breast oncogene.106 Similarly, mice bred to express Notch-1ICD and Notch-3ICD in mammary epithelial cells developed mammary tumors.81 Nonetheless, Notch receptors may not be equivalent in their capacity to induce cancer. Notch-2 may suppress tumorigenicity, as MDA-MB-231 cells with constitutively expressing N-2ICD showed increased apoptosis and did not form xenograft tumors in mice.98 Notch signaling is also responsive to hormonal drivers of tumorigenicity, since estrogen was found to upregulate Notch-1 and Jagged-1 in MCF-7 cells.107 In contrast, Rizzo et al demonstrated that estrogen-mediated ER activation suppresses Notch activation, and the combination of anti-estrogen therapy with a GSI was more effective in inhibiting ER+ breast tumor growth than either therapy alone.91 Specifically, the same group identified that Notch-1ICD activates ER-responsive genes under low estrogen conditions, suggesting that Notch-1ICD could mediate activation of the ER in an estrogen-independent manner.108
Furthermore, loss of negative regulatory mechanisms contributes to neoplastic metastasis. For example, expression levels of the negative regulator of Notch signaling Numb inversely correlated with tumor aggressiveness.101 In in-vitro and in-vivo experiments examining osteolytic bone metastasis of human breast cancer cells, osteoblasts together with secretion of transforming growth factor β1 enhanced Notch-3 expression in the breast cancer cells and mediated their metastasis; this effect was inhibited by GSI L-685458.109
Notch and oncogenic crosstalk
The oncogenic reach of the Notch signaling pathway is partly due to its communication or crosstalk with other signaling pathways. Hurlbut et al110 proposed more than 50 connections for the Notch crosstalk network; for example, receptor tyrosine kinases (RTKs), HH, Janus kinase/signal transducers and activators of transcription (Jak/STAT), transforming growth factor-β/decapentaplegic (TGF-β), and Wnt pathways. In addition to HH, Wnt, and TGF-β, amongst others pertinent to Notch crosstalk are platelet-derived growth factor (PDGF/PDGFR), vascular endothelial growth factor (VEGF), phosphatidylinositol 3-kinase (PI3K/Akt), Ras, mammalian target of rapamycin (mTOR), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), hypoxia-inducible factor (HIF), and cytokines interleukin-6 (IL-6), IL-1, and leptin plus ER signaling as well as microRNAs considered operationally important for Notch crosstalk in breast cancer.111 The majority of functions which include cell proliferation, differentiation, and development, tumor angiogenesis, morphogenesis, and somitogenesis are all important during oncogenesis. One of the most critical pathways necessary for survival of cancer cells is the NF-κB pathway. It has been shown that NF-κB regulates Notch and is regulated by Notch. For example, N-1ICD or N-3ICD has been shown to activate NF-κB signaling components such as IKK (inhibitor of kappa B kinase).112 Furthermore, NF-κB has also been shown to regulate Notch indirectly by inducing Jagged-1, HES-5, and/or Deltex-1.113
The existence of Notch and RTK crosstalk in breast and other solid tumors has been established by our research group and many others since. We and others have shown that Notch-1 signaling is decreased in ErbB-2 overexpressing BT-474, SkBr3, and MCF-7/HER2 breast cancer cells and that anti-HER-2 therapy using trastuzumab or a small molecule tyrosine kinase inhibitor similar to lapatinib reactivated Notch-1. More importantly, a GSI or specifically Notch-1 knockdown increased the sensitivity of ErbB-2+ breast cancer cells to anti-HER-2-mediated growth inhibition, indicating that Notch-1 signaling might contribute to trastuzumab resistance in vitro.114 Moreover, tumor recurrence was prevented in mice injected with trastuzumab-sensitive BT-474 cells following treatment with trastuzumab and MRK-003 GSI or significantly reduced with trastuzumab and LY-411575 GSI; additionally, BT-474 breast tumors that were resistant to trastuzumab were re-sensitized by addition of MRK-003 GSI.115 An overview of the role and significance of Notch signaling in trastuzumab resistant breast cancer is reviewed by Mehta and Osipo.116
The regulation and activation of Notch signaling in triple negative breast cancer was recently eludicated by Clementz et al.117 Specifically, the investigators demonstrated that PEA3, an Ets family transcription factor, activates transcription of Notch-1 and Notch-4.117 It was identified that enrichment of PEA3 on the Notch-1 promoter was independent of AP-1 while PEA3 recruitment to the Notch-4 promoter was dependent on c-JUN and Fra-1, but negatively regulated by c-Fos. The findings from this study also showed that knockdown of PEA3 was potent in inhibiting triple negative breast cancer growth in vitro and in vivo.
GSIs: mode of action and side effects
The more than 100 GSIs synthesized to date can be divided into three classes: peptide isosteres, azepines, and sulfonamides.118,119 They are oral agents, the azepines and sulfonamides being the most popular. A list of select GSIs is presented in Table 1. GSIs currently undergoing US clinical trials are listed in Table 2.
Table 1.
Chemical structure of γ-secretase inhibitors
| Class | Name | Structure | Type |
|---|---|---|---|
| Peptide isostere | Tripeptide Z-Leu-Leu-Nle-CHO GSI-I |
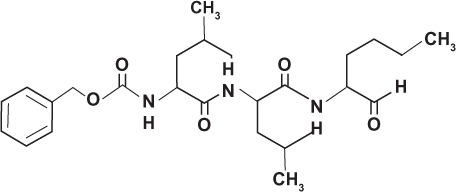
|
Transition state analog |
| Azepine | DAPT Compound 3 GSI-IX |
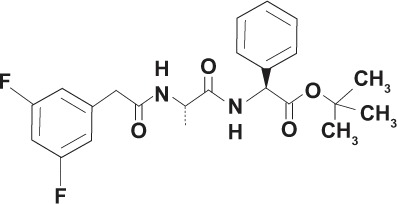
|
Non-transition state analog |
| LY-685458 GSI-X |
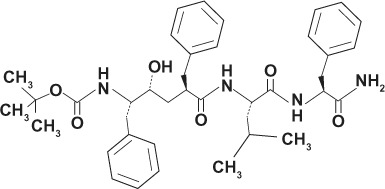
|
Transition state analog | |
| YO-01027 Dibenzazepine GSI-XX Deshydroxy LY-411575 |
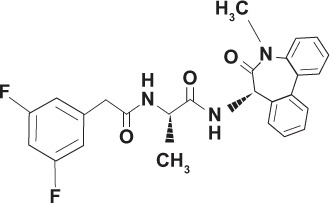
|
Transition state analog | |
| Compound E GSI-XXI |
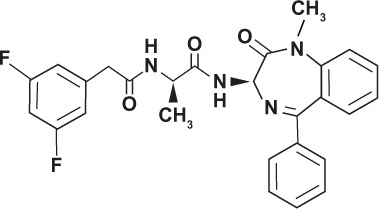
|
Non-transition state analog | |
| RO-4929097 (Roche, Nutley, NJ, USA) |
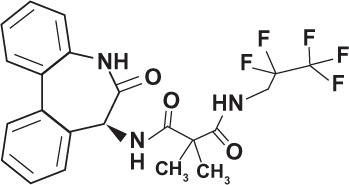
|
Transition state analog | |
| Sulfonamide | MRK-003 (Merck and Co, Boston, MA, USA) |
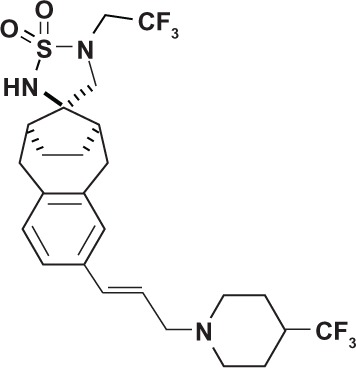
|
Non-transition state analog |
| MK-0752 (Merck and Co) |
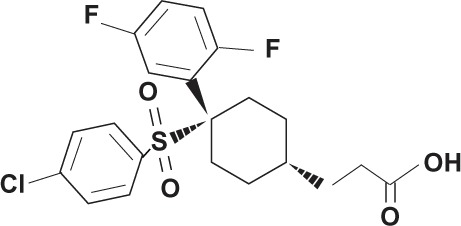
|
Non-transition state analog | |
| Selective | PF-03084014 (Pfizer Inc., Groton, CT, USA) |
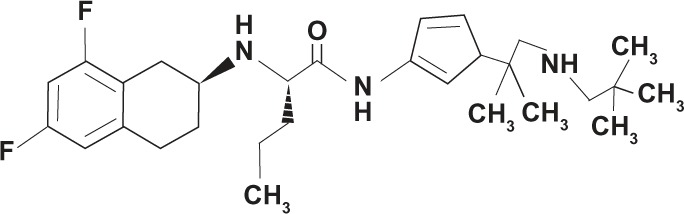
|
Non-transition state analog |
Abbreviations: GSI, gamma-secretase inhibitor; DAPT, N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester.
Table 2.
Clinical trials employing γ-secretase inhibitors in the treatment of breast cancer
| Name | Target | Type of study | Trial ID* |
|---|---|---|---|
| Individual therapy | |||
| MK-0752 (Merck and Co, whitehouse Station, NJ, USA) | Metastatic or locally advanced breast cancer | Phase I | NCT00106145 |
| PF03084014 (Pfizer, Groton, CT, USA) | Advanced solid tumors | Phase I | NCT00878189 |
| RO-4929097 (Roche, Nutley, NJ, USA) | Advanced or metastatic breast cancer or recurrent triple negative breast cancer | Phase II | NCT01151449 |
| Combination therapy | |||
| MK-0752 + Docetaxel | Locally advanced or metastatic breast cancer | Phase I/II | NCT00645333 |
| MK-0752 + Tamoxifen or Letrozole | Early stage breast cancer | Pilot study | NCT00756717 |
| Ridaforolimus (MK-8669) with either MK-0752 or MK-2206 (Akt inhibitor) | Advanced solid tumor | Phase I | NCT01295632 |
| RO-4929097 + Capecitabine | Refractory solid tumors | Phase I | NCT01158274 |
| RO-4929097 + Cediranib maleate | Advanced solid tumors | Phase I clinical trial | NCT01131234 |
| RO-4929097 + Letrozole | Post-menopausal ER+/PR+ Stage I or II breast cancer | Phase Ib clinical trial | NCT01208441 |
| RO-4929097 + Vismodegib | Metastatic breast cancer | Phase I clinical trial | NCT01071564 |
| RO-4929097 + Paclitaxel + Carboplatin | Stage II or III triple negative breast cancer | Phase I clinical trial | NCT01238133 |
Note:
clinicaltrials.gov.
Abbreviations: ER, estrogen receptor; PR, progesterone receptor.
The GSIs are classified into two types, depending on structure and binding sites: (1) aspartyl proteinase transition-state analogs as peptide isosteres that mimic the transition state of a substrate cleavage by γ-secretase and bind competitively to the catalytic active site of presenilins; and (2) small molecule non-transition-state inhibitors where the binding site is different from the active site, possibly at the interface of the γ-secretase complex dimer.120 Well known side effects of GSIs occur within the gastrointestinal tract. For example, acute treatment of TgCRND8 mice with LY-411575 for 15 days caused an increase in the number of mucin-containing goblet cells in the small and large intestines and changes in the tissue architecture of the gastrointestinal tract which resulted in severe diarrhea.121 The GSIs also cause various off-target effects in breast cancer cells and Notch signaling. An early transition-state analog GSI, IL X (cbz-IL-CHO), produced a decrease in mRNA and protein levels of HES-1, induced G0–G1 cell cycle arrest, and inhibited human tongue carcinoma Tca8113 cell growth.122 Dipeptide GSI XII (z-Ile-Leu-CHO) induced apoptosis in breast cancer cell lines by inducing Noxa, a pro-apoptotic protein.123 A later generation GSI, LY-294002 suppressed angiogenesis by blocking the epidermal growth factor (EGF)-induced upregulation of Jagged-1 in squamous cell carcinoma, thereby inhibiting EFG-Notch crosstalk.124 Tripeptide GSI I (z-Leu-Leu-Nle-CHO) suppressed cell proliferation and induced apoptosis in Notch-3 overexpressing ovarian cell lines.65
An early generation non-transition state analog is DAPT, N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester, a dipeptide inhibitor of the benzodiazepine type, also known as GSI IX and Compound 3.125 It is the most widely used in the laboratory setting.126 DAPT potentiated the apoptotic effects of the DNA-damaging drug melphalan in MCF-7 breast cancer cells.127 In cell lines with chromosomal translocations, DAPT inhibited the proliferation of truncated Notch-1 expressing an ADAM cleavage site but not of truncated Notch-2 which was without the cleavage site.128 From DAPT, numerous other GSIs have been developed that are even more effective, ie, LY-411,575 (Compound 5) 100-fold stronger than DAPT, LY-450,139 (Semagacestat or Compound 6),129 and RO-4929097 (Roche, Nutley, NJ, USA).
The small-molecule GSI classified as a tetralin imidazole PF-03084014 (Pfizer Inc., Groton, CT, USA) is in a Phase I trial to treat advanced breast cancer and other solid tumors.130 It is considered a selective or Notch-sparing GSI or GS (gamma secretase) modulator. When evaluated for Notch activity, PF-03084014 significantly decreased tumor cell migration and mammosphere formation in vitro, reduced tumor cell self-renewal ability in vivo, and decreased mRNA expression of Notch target genes HES-1, HES-4, Notch-1, and HEY-2 in HCC1599 xenograft tumors.131
Another small-molecule GSI, RO-4929097, was used in a multicenter Phase I clinical dose escalating study and continued on to Phase II and combination therapy studies. Derived from LY-411575 and containing a dibenzazepinone core, it is being tested for the treatment of breast cancer and other solid tumors.132,133 Patients with low basal levels of plasma IL-6 and IL-8 responded well, indicating that cytokines may be predictive biomarkers for response to therapy.134 In in vitro studies using a colon cancer cell line A549, RO-4929097 produced a significant decrease in mRNA levels of Notch target genes HES-1, HES-4, and HEY-1.135
Another GSI, the sulfonamide-containing non-transition-state GSI analog MK-0752 (Merck and Co, Inc, Whitehouse Station, NJ, USA) is in a Phase I study to treat metastatic or locally advanced breast cancer.
Novel combination strategies
Since single-drug therapy is ineffective for long-term use, combination therapy oftentimes becomes necessary. Such a treatment regimen is applicable to “endocrine therapy, targeted therapies, chemotherapy, or possibly even radiation therapy.”72 There are several clinical studies using RO-4929097 in combination therapy. One Phase I study is using RO-4929097 with capecitabine for patients with refractory solid tumors and another with paclitaxel and carboplatin for patients with Stage I or III triple negative breast cancer. A Phase I clinical trial is presently underway testing the efficacy of RO-4929097 and a potent HH antagonist GDC-0449 vismodegib in patients with advanced breast cancer.136 These patients may have been selected on the basis of upregulated crosstalk between Notch and the self-renewal pathway (targeted therapy). Another Phase I study with RO-4929097 is adding cediranib for post-menopausal patients with advanced solid tumors (targeted therapy). In a Phase Ib clinical trial, RO-4929097 is combined with letrazole for patients with ER+/PR+ Stage II or III breast cancer (endocrine therapy). Another GSI in combination therapy is MK-0752. In a Pilot study, MK-0752 is being combined with tamoxifen or letrazole for patients with early stage breast cancer (endocrine therapy). In a Phase I study, MK-0752 (or MK-2206 Akt inhibitor) is being combined with ridaforolimus (MK-8669) in patients with advanced solid tumors (targeted therapy). A Phase I/II study is combining MK-0752 with docetaxel in patients presenting with locally advanced or metastatic breast cancer (chemotherapy).
Gastrointestinal toxicity is a major side-effect with GSI use.137 Nonetheless, careful monitoring of treatment protocols, whether by modulating expression of Notch receptors with receptor antibody pretreatment before GSI treatment138 or development of a practical combination therapy139 should minimize problematic side-effects. Notch activation must be assessed prior to GSI treatment (mutations and/or overexpression), since GSIs are more effective against tumors with upregulated Notch signaling. In addition, close attention must be paid to the therapeutic window so that the minimally active dose needed to inhibit Notch is employed, thereby reducing adverse side effects.
Conclusion
Much progress has been made in understanding Notch signaling in breast cancer. Molecular profiling of patients, fast becoming standard of care, identifies the type and location of signaling dysfunction. Moreover, pharmacological innovations are helping produce more selective GSIs with fewer side effects. A “one problem–one solution” type of cure to breast cancer seems unlikely. Inhibition of Notch signaling with pharmacodynamically active drugs such as the GSIs is preventing metastasis and recurrence and increasing disease-free survival. The next level of care for determining the molecular signature of a breast tumor will develop therapeutic combinatorial protocols that effectively target crosstalk pathways, tumor microenvironment, tumor-initiating cells (or cancer stem cells), developmental factors, non-canonical signaling components, and possibly other additional modulating factors still unknown. Breast cancer management will require a multidisciplinary team to prepare and optimize the anticancer drug regimen, conduct the therapy, and even interpret results and treatment progress. Overall, targeting the Notch signaling pathway in breast cancer therapy and attempting its downregulation with GSIs looks promising.
Acknowledgments
The authors thank Rima M Rusinas, MDes, for the design of the figures and tables.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 2.Hu Z, Fan C, Oh DS, et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics. 2006;7(1):96. doi: 10.1186/1471-2164-7-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dexter JS. The analysis of a case of continuous variation in drosophila by a study of its linkage relations. Am Nat. 1914;48(576):712–758. [Google Scholar]
- 4.Morgan TH. The theory of the gene. Am Nat. 1917:513–544. [Google Scholar]
- 5.Morgan TH. The Physical Basis of Heredity. Philadelphia: JB Lippincott; 1919. [Google Scholar]
- 6.Kidd S, Kelley MR, Young MW. Sequence of the Notch locus of Drosophila melanogaster: relationship of the encoded protein to mammalian clotting and growth factors. Mol Cell Biol. 1986;6(9):3094–3108. doi: 10.1128/mcb.6.9.3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wharton KA, Johansen KM, Xu T, Artavanis-Tsakonas S. Nucleotide sequence from the neurogenic locus Notch implies a gene product that shares homology with proteins containing EGF-like repeats. Cell. 1985;43(3 Pt 2):567. doi: 10.1016/0092-8674(85)90229-6. [DOI] [PubMed] [Google Scholar]
- 8.Lai EC. Notch signaling: control of cell communication and cell fate. Development. 2004;131(5):965–973. doi: 10.1242/dev.01074. [DOI] [PubMed] [Google Scholar]
- 9.Swiatek PJ, Lindsell CE, Del Amo FF, Weinmaster G, Gridley T. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994;8(6):707–719. doi: 10.1101/gad.8.6.707. [DOI] [PubMed] [Google Scholar]
- 10.Conlon RA, Reaume AG, Rossant J. Notch1 is required for the coordinate segmentation of somites. Development. 1995;121(5):1533–1545. doi: 10.1242/dev.121.5.1533. [DOI] [PubMed] [Google Scholar]
- 11.Jiang R, Lan Y, Chapman HD, et al. Defects in limb, craniofacial, and thymic development in Jagged2 mutant mice. Genes Dev. 1998;12(7):1046–1057. doi: 10.1101/gad.12.7.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hrabe de Angelis M, McIntyre IJ, Gossler A. Maintenance of somite borders in mice requires the Delta homologue DII1. Nature. 1997;386:717–721. doi: 10.1038/386717a0. [DOI] [PubMed] [Google Scholar]
- 13.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284(5415):770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 14.Henrique D, Hirsinger E, Adam J, et al. Maintenance of neuroepithelial progenitor cells by Delta–Notch signalling in the embryonic chick retina. Curr Biol. 1997;7(9):661–670. doi: 10.1016/s0960-9822(06)00293-4. [DOI] [PubMed] [Google Scholar]
- 15.Neumann CJ, Cohen SM. A hierarchy of cross-regulation involving Notch, wingless, vestigial and cut organizes the dorsal/ventral axis of the Drosophila wing. Development. 1996;122(11):3477–3485. doi: 10.1242/dev.122.11.3477. [DOI] [PubMed] [Google Scholar]
- 16.de Celis JF, Garcia-Bellido A, Bray SJ. Activation and function of Notch at the dorsal-ventral boundary of the wing imaginal disc. Development. 1996;122(1):359–369. doi: 10.1242/dev.122.1.359. [DOI] [PubMed] [Google Scholar]
- 17.Gaunt SJ. Developmental biology-chick limbs, fly wings and homology at the fringe. Nature. 1997;386(6623):324–325. doi: 10.1038/386324a0. [DOI] [PubMed] [Google Scholar]
- 18.Moloney DJ, Panin VM, Johnston SH, et al. Fringe is a glycosyltransferase that modifies Notch. Nature. 2000;406(6794):369–375. doi: 10.1038/35019000. [DOI] [PubMed] [Google Scholar]
- 19.Haltiwanger RS, Stanley P. Modulation of receptor signaling by glycosylation: fringe is an O-fucose-β1, 3-N-acetylglucosaminyltransferase. Biochim Biophys Acta. 2002;1573(3):328–335. doi: 10.1016/s0304-4165(02)00400-2. [DOI] [PubMed] [Google Scholar]
- 20.Haines N, Irvine KD. Glycosylation regulates Notch signalling. Nat Rev Mol Cell Biol. 2003;4(10):786–797. doi: 10.1038/nrm1228. [DOI] [PubMed] [Google Scholar]
- 21.Blaumueller CM, Qi H, Zagouras P, Artavanis-Tsakonas S. Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell. 1997;90(2):281–291. doi: 10.1016/s0092-8674(00)80336-0. [DOI] [PubMed] [Google Scholar]
- 22.Logeat F, Bessia C, Brou C, et al. The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc Natl Acad Sci USA. 1998;95(14):8108–8112. doi: 10.1073/pnas.95.14.8108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bettenhausen B, de Angelis MH, Simon D, Guénet JL, Gossler A. Transient and restricted expression during mouse embryogenesis of Dll1, a murine gene closely related to Drosophila Delta. Development. 1995;121(8):2407–2418. doi: 10.1242/dev.121.8.2407. [DOI] [PubMed] [Google Scholar]
- 24.Dunwoodie SL, Henrique D, Harrison SM, Beddington R. Mouse Dll3: a novel divergent Delta gene which may complement the function of other Delta homologues during early pattern formation in the mouse embryo. Development. 1997;124(16):3065–3076. doi: 10.1242/dev.124.16.3065. [DOI] [PubMed] [Google Scholar]
- 25.Shutter JR, Scully S, Fan W, et al. Dll4, a novel Notch ligand expressed in arterial endothelium. Genes Dev. 2000;14(11):1313–1318. [PMC free article] [PubMed] [Google Scholar]
- 26.Lindsell CE, Shawber CJ, Boulter J, Weinmaster G. Jagged: a mammalian ligand that activates Notch1. Cell. 1995;80(6):909–917. doi: 10.1016/0092-8674(95)90294-5. [DOI] [PubMed] [Google Scholar]
- 27.Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7(9):678–689. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 28.Miele L. Notch signaling. Clin Cancer Res. 2006;12(4):1074–1079. doi: 10.1158/1078-0432.CCR-05-2570. [DOI] [PubMed] [Google Scholar]
- 29.Kopan R, Ilagan MXG. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mumm JS, Kopan R. Notch signaling: from the outside in. Dev Biol. 2000;228(2):151. doi: 10.1006/dbio.2000.9960. [DOI] [PubMed] [Google Scholar]
- 31.Brou C, Logeat F, Gupta N, et al. A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell. 2000;5(2):207–216. doi: 10.1016/s1097-2765(00)80417-7. [DOI] [PubMed] [Google Scholar]
- 32.Reiss K, Saftig P. The “a disintegrin and metalloprotease”(ADAM) family of sheddases: physiological and cellular functions. Semin Cell Dev Biol. 2009;20(2):126–137. doi: 10.1016/j.semcdb.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 33.Mumm JS, Schroeter EH, Saxena MT, et al. A ligand-induced extracellular cleavage regulates-secretase-like proteolytic activation of Notch1. Mol Cell. 2000;5(2):197–206. doi: 10.1016/s1097-2765(00)80416-5. [DOI] [PubMed] [Google Scholar]
- 34.Lleo M. Activity of gamma-secretase on substrates other than APP. Curr Top Med Chem. 2008;8(1):9–16. doi: 10.2174/156802608783334060. [DOI] [PubMed] [Google Scholar]
- 35.De Strooper B. Aph-1, Pen-2, and nicastrin with presenilin generate an active gamma-secretase complex. Neuron. 2003;38(1):9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 36.Lai EC. Notch cleavage: nicastrin helps presenilin make the final cut. Curr Biol. 2002;12(6):R200–R202. doi: 10.1016/s0960-9822(02)00749-2. [DOI] [PubMed] [Google Scholar]
- 37.Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393(6683):382–386. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- 38.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12(5):599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 39.Wu G, Lyapina S, Das I, et al. SEL-10 is an inhibitor of Notch signaling that targets Notch for ubiquitin-mediated protein degradation. Mol Cell Biol. 2001;21(21):7403–7415. doi: 10.1128/MCB.21.21.7403-7415.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ellisen LW, Bird J, West DC, et al. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. 1991;66(4):649–661. doi: 10.1016/0092-8674(91)90111-b. [DOI] [PubMed] [Google Scholar]
- 41.Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 42.Fabbri G, Rasi S, Rossi D, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208(7):1389–1401. doi: 10.1084/jem.20110921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morimura T, Goitsuka R, Zhang Y, Saito I, Reth M, Kitamura D. Cell cycle arrest and apoptosis induced by Notch1 in B cells. J Biol Chem. 2000;275(47):36523–36531. doi: 10.1074/jbc.M006415200. [DOI] [PubMed] [Google Scholar]
- 44.Romer S, Saunders U, Jäck H, Jehn B. Notch1 enhances B-cell receptor-induced apoptosis in mature activated B cells without affecting cell cycle progression and surface IgM expression. Cell Death Differ. 2003;10(7):833–844. doi: 10.1038/sj.cdd.4401253. [DOI] [PubMed] [Google Scholar]
- 45.Nefedova Y, Cheng P, Alsina M, Dalton WS, Gabrilovich DI. Involvement of Notch-1 signaling in bone marrow stroma-mediated de novo drug resistance of myeloma and other malignant lymphoid cell lines. Blood. 2004;103(9):3503–3510. doi: 10.1182/blood-2003-07-2340. [DOI] [PubMed] [Google Scholar]
- 46.Hubmann R, Schwarzmeier JD, Shehata M, et al. Notch2 is involved in the overexpression of CD23 in B-cell chronic lymphocytic leukemia. Blood. 2002;99(10):3742–3747. doi: 10.1182/blood.v99.10.3742. [DOI] [PubMed] [Google Scholar]
- 47.Jundt F, Anagnostopoulos I, Förster R, Mathas S, Stein H, Dörken B. Activated Notch1 signaling promotes tumor cell proliferation and survival in Hodgkin and anaplastic large cell lymphoma. Blood. 2002;99(9):3398–3403. doi: 10.1182/blood.v99.9.3398. [DOI] [PubMed] [Google Scholar]
- 48.Jundt F, Pröbsting KS, Anagnostopoulos I, et al. Jagged1-induced Notch signaling drives proliferation of multiple myeloma cells. Blood. 2004;103(9):3511–3515. doi: 10.1182/blood-2003-07-2254. [DOI] [PubMed] [Google Scholar]
- 49.Kridel R, Meissner B, Rogic S, et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood. 2012;119(9):1963–1971. doi: 10.1182/blood-2011-11-391474. [DOI] [PubMed] [Google Scholar]
- 50.Lee S, Kumano K, Nakazaki K, et al. Gain-of-function mutations and copy number increases of Notch2 in diffuse large B-cell lymphoma. Cancer Sci. 2009;100(5):920–926. doi: 10.1111/j.1349-7006.2009.01130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Houde C, Li Y, Song L, et al. Overexpression of the NOTCH ligand JAG2 in malignant plasma cells from multiple myeloma patients and cell lines. Blood. 2004;104(12):3697–3704. doi: 10.1182/blood-2003-12-4114. [DOI] [PubMed] [Google Scholar]
- 52.Zagouras P, Stifani S, Blaumueller CM, Carcangiu ML, Artavanis-Tsakonas S. Alterations in Notch signaling in neoplastic lesions of the human cervix. Proc Natl Acad Sci USA. 1995;92(14):6414–6418. doi: 10.1073/pnas.92.14.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weijzen S, Zlobin A, Braid M, Miele L, Kast WM. HPV16 E6 and E7 oncoproteins regulate Notch-1 expression and cooperate to induce transformation. J Cell Physiol. 2003;194(3):356–362. doi: 10.1002/jcp.10217. [DOI] [PubMed] [Google Scholar]
- 54.May R, Riehl TE, Hunt C, Sureban SM, Anant S, Houchen CW. Identification of a novel putative gastrointestinal stem cell and adenoma stem cell marker, doublecortin and CaM kinase-like-1, following radiation injury and in adenomatous polyposis coli/multiple intestinal neoplasia mice. Stem Cells. 2007;26(3):630–637. doi: 10.1634/stemcells.2007-0621. [DOI] [PubMed] [Google Scholar]
- 55.Akiyoshi T, Nakamura M, Yanai K, et al. γ-Secretase inhibitors enhance taxane-induced mitotic arrest and apoptosis in colon cancer cells. Gastroenterology. 2008;134(1):131–144. doi: 10.1053/j.gastro.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 56.Gao B, Jeong WI, Tian Z. Liver: an organ with predominant innate immunity. Hepatology. 2007;47(2):729–736. doi: 10.1002/hep.22034. [DOI] [PubMed] [Google Scholar]
- 57.Qi R, An H, Yu Y, et al. Notch1 signaling inhibits growth of human hepatocellular carcinoma through induction of cell cycle arrest and apoptosis. Cancer Res. 2003;63(23):8323–8329. [PubMed] [Google Scholar]
- 58.Konishi J, Kawaguchi KS, Vo H, et al. γ-Secretase inhibitor prevents Notch3 activation and reduces proliferation in human lung cancers. Cancer Res. 2007;67(17):8051–8057. doi: 10.1158/0008-5472.CAN-07-1022. [DOI] [PubMed] [Google Scholar]
- 59.Terris B, Blaveri E, Crnogorac-Jurcevic T, et al. Characterization of gene expression profiles in intraductal papillary-mucinous tumors of the pancreas. Am J Pathol. 2002;160(5):1745–1754. doi: 10.1016/S0002-9440(10)61121-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miyamoto Y, Maitra A, Ghosh B, et al. Notch mediates TGFα-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. 2003;3(6):565–576. doi: 10.1016/s1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- 61.Wang Z, Zhang Y, Li Y, Banerjee S, Liao J, Sarkar FH. Down-regulation of Notch-1 contributes to cell growth inhibition and apoptosis in pancreatic cancer cells. Mol Cancer Ther. 2006;5(3):483–493. doi: 10.1158/1535-7163.MCT-05-0299. [DOI] [PubMed] [Google Scholar]
- 62.Kimura K, Satoh K, Kanno A, et al. Activation of Notch signaling in tumorigenesis of experimental pancreatic cancer induced by dimethylbenzanthracene in mice. Cancer Sci. 2007;98(2):155–162. doi: 10.1111/j.1349-7006.2006.00369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Santagata S, Demichelis F, Riva A, et al. JAGGED1 expression is associated with prostate cancer metastasis and recurrence. Cancer Res. 2004;64(19):6854–6857. doi: 10.1158/0008-5472.CAN-04-2500. [DOI] [PubMed] [Google Scholar]
- 64.Ramdas L, Giri U, Ashorn CL, et al. miRNA expression profiles in head and neck squamous cell carcinoma and adjacent normal tissue. Head Neck. 2009;31(5):642–654. doi: 10.1002/hed.21017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park JT, Li M, Nakayama K, et al. Notch3 gene amplification in ovarian cancer. Cancer Res. 2006;66(12):6312–6318. doi: 10.1158/0008-5472.CAN-05-3610. [DOI] [PubMed] [Google Scholar]
- 66.Rose SL, Kunnimalaiyaan M, Drenzek J, Seiler N. Notch 1 signaling is active in ovarian cancer. Gynecol Oncol. 2010;117(1):130–133. doi: 10.1016/j.ygyno.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 67.Sjolund J, Johansson M, Manna S, et al. Suppression of renal cell carcinoma growth by inhibition of Notch signaling in vitro and in vivo. J Clin Invest. 2008;118(1):217–228. doi: 10.1172/JCI32086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miele L, Miao H, Nickoloff B. NOTCH signaling as a novel cancer therapeutic target. Curr Cancer Drug Targets. 2006;6(4):313–323. doi: 10.2174/156800906777441771. [DOI] [PubMed] [Google Scholar]
- 69.Joutel A, Tournier-Lasserve E. Notch signalling pathway and human diseases. Semin Cell Dev Biol. 1998;9(6):619–625. doi: 10.1006/scdb.1998.0261. [DOI] [PubMed] [Google Scholar]
- 70.Nickoloff BJ, Osborne BA, Miele L. Notch signaling as a therapeutic target in cancer: a new approach to the development of cell fate modifying agents. Oncogene. 2003;22(42):6598–6608. doi: 10.1038/sj.onc.1206758. [DOI] [PubMed] [Google Scholar]
- 71.Miele L, Osborne B. Arbiter of differentiation and death: Notch signaling meets apoptosis. J Cell Physiol. 1999;181(3):393–409. doi: 10.1002/(SICI)1097-4652(199912)181:3<393::AID-JCP3>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 72.Al-Hussaini H, Subramanyam D, Reedijk M, Sridhar SS. Notch signaling pathway as a therapeutic target in breast cancer. Mol Cancer Ther. 2011;10(1):9–15. doi: 10.1158/1535-7163.MCT-10-0677. [DOI] [PubMed] [Google Scholar]
- 73.Leong KG, Karsan A. Recent insights into the role of Notch signaling in tumorigenesis. Blood. 2006;107(6):2223–2233. doi: 10.1182/blood-2005-08-3329. [DOI] [PubMed] [Google Scholar]
- 74.Radtke F, Raj K. The role of Notch in tumorigenesis: oncogene or tumour suppressor? Nat Rev Cancer. 2003;3(10):756–767. doi: 10.1038/nrc1186. [DOI] [PubMed] [Google Scholar]
- 75.Wilson A, Radtke F. Multiple functions of Notch signaling in self-renewing organs and cancer. FEBS Lett. 2006;580(12):2860–2868. doi: 10.1016/j.febslet.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 76.Roy M, Pear WS, Aster JC. The multifaceted role of Notch in cancer. Curr Opin Genet Dev. 2007;17(1):52–59. doi: 10.1016/j.gde.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 77.Maillard I, Pear WS. Notch and cancer: best to avoid the ups and downs. Cancer Cell. 2003;3(3):203–205. doi: 10.1016/s1535-6108(03)00052-7. [DOI] [PubMed] [Google Scholar]
- 78.Miele L, Golde T, Osborne B. Notch signaling in cancer. Curr Mol Med. 2006;6(8):905–918. doi: 10.2174/156652406779010830. [DOI] [PubMed] [Google Scholar]
- 79.Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11(5):338–351. doi: 10.1038/nrc3035. [DOI] [PubMed] [Google Scholar]
- 80.Kiaris H, Politi K, Grimm LM, et al. Modulation of Notch signaling elicits signature tumors and inhibits hras1-induced oncogenesis in the mouse mammary epithelium. Am J Pathol. 2004;165(2):695–705. doi: 10.1016/S0002-9440(10)63333-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hu C, Diévart A, Lupien M, Calvo E, Tremblay G, Jolicoeur P. Overexpression of activated murine Notch1 and Notch3 in transgenic mice blocks mammary gland development and induces mammary tumors. Am J Pathol. 2006;168(3):973–990. doi: 10.2353/ajpath.2006.050416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Klinakis A, Szabolcs M, Politi K, Kiaris H, Artavanis-Tsakonas S, Efstratiadis A. Myc is a Notch1 transcriptional target and a requisite for Notch1-induced mammary tumorigenesis in mice. Proc Natl Acad Sci USA. 2006;103(24):9262–9267. doi: 10.1073/pnas.0603371103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Politi K, Feirt N, Kitajewski J. Notch in mammary gland development and breast cancer. Semin Cancer Biol. 2004;14(5):341–347. doi: 10.1016/j.semcancer.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 84.Diévart A, Beaulieu N, Jolicoeur P. Involvement of Notch1 in the development of mouse mammary tumors. Oncogene. 1999;18(44):5973–5981. doi: 10.1038/sj.onc.1202991. [DOI] [PubMed] [Google Scholar]
- 85.Smith GH, Gallahan D, Diella F, Jhappan C, Merlino G, Callahan R. Constitutive expression of a truncated INT3 gene in mouse mammary epithelium impairs differentiation and functional development. Cell Growth Differ. 1995;6(5):563–577. [PubMed] [Google Scholar]
- 86.Gallahan D, Callahan R. The mouse mammary tumor associated gene INT3 is a unique member of the NOTCH gene family (NOTCH4) Oncogene. 1997;14(16):1883–1890. doi: 10.1038/sj.onc.1201035. [DOI] [PubMed] [Google Scholar]
- 87.Gallahan D, Jhappan C, Robinson G, et al. Expression of a truncated Int3 gene in developing secretory mammary epithelium specifically retards lobular differentiation resulting in tumorigenesis. Cancer Res. 1996;56(8):1775–1785. [PubMed] [Google Scholar]
- 88.Robinson DR, Kalyana-Sundaram S, Wu YM, et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat Med. 2011;17(12):1646–1651. doi: 10.1038/nm.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xiao Y, Ye Y, Zou X, et al. The lymphovascular embolus of inflammatory breast cancer exhibits a Notch 3 addiction. Oncogene. 2010;30(3):287–300. doi: 10.1038/onc.2010.405. [DOI] [PubMed] [Google Scholar]
- 90.Konstantinos T. Expression of Notch receptors in primary breast cancer and correlation with pathological features. Clin Exp Pharmacol. 2012;2:1000109. [Google Scholar]
- 91.Rizzo P, Miao H, D’Souza G, et al. Cross-talk between Notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res. 2008;68(13):5226–5235. doi: 10.1158/0008-5472.CAN-07-5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yamaguchi N, Oyama T, Ito E, et al. NOTCH3 signaling pathway plays crucial roles in the proliferation of ErbB2-negative human breast cancer cells. Cancer Res. 2008;68(6):1881–1888. doi: 10.1158/0008-5472.CAN-07-1597. [DOI] [PubMed] [Google Scholar]
- 93.Stylianou S, Clarke RB, Brennan K. Aberrant activation of Notch signaling in human breast cancer. Cancer Res. 2006;66(3):1517–1525. doi: 10.1158/0008-5472.CAN-05-3054. [DOI] [PubMed] [Google Scholar]
- 94.Reedijk M, Odorcic S, Chang L, et al. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005;65(18):8530–8537. doi: 10.1158/0008-5472.CAN-05-1069. [DOI] [PubMed] [Google Scholar]
- 95.Dickson BC, Mulligan AM, Zhang H, et al. High-level JAG1 mRNA and protein predict poor outcome in breast cancer. Mod Pathol. 2007;20(6):685–693. doi: 10.1038/modpathol.3800785. [DOI] [PubMed] [Google Scholar]
- 96.Parr C, Watkins G, Jiang W. The possible correlation of Notch-1 and Notch-2 with clinical outcome and tumour clinicopathological parameters in human breast cancer. Int J Mol Med. 2004;14(5):779–786. doi: 10.3892/ijmm.14.5.779. [DOI] [PubMed] [Google Scholar]
- 97.Shimizu K, Chiba S, Saito T, Kumano K, Hamada Y, Hirai H. Functional diversity among Notch1, Notch2, and Notch3 receptors. Biochem Biophys Res Commun. 2002;291(4):775–779. doi: 10.1006/bbrc.2002.6528. [DOI] [PubMed] [Google Scholar]
- 98.O’Neill CF, Urs S, Cinelli C, et al. Notch2 signaling induces apoptosis and inhibits human MDA-MB-231 xenograft growth. Am J Pathol. 2007;171(3):1023–1036. doi: 10.2353/ajpath.2007.061029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Harrison H, Farnie G, Howell SJ, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010;70(2):709–718. doi: 10.1158/0008-5472.CAN-09-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Grudzien P, Lo S, Albain KS, et al. Inhibition of Notch signaling reduces the stem-like population of breast cancer cells and prevents mammosphere formation. Anticancer Res. 2010;30(10):3853–3867. [PubMed] [Google Scholar]
- 101.Pece S, Serresi M, Santolini E, et al. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol. 2004;167(2):215–221. doi: 10.1083/jcb.200406140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.To M, Gokgoz N, Doyle T, et al. Functional characterization of novel presenilin-2 variants identified in human breast cancers. Oncogene. 2006;25(25):3557–3564. doi: 10.1038/sj.onc.1209397. [DOI] [PubMed] [Google Scholar]
- 103.Klinakis A, Lobry C, Abdel-Wahab O, et al. A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature. 2011;473(7346):230–233. doi: 10.1038/nature09999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nicolas M, Wolfer A, Raj K, et al. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. 2003;33(3):416–421. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 105.Allenspach EJ, Maillard I, Aster JC, Pear WS. Notch signaling in cancer. Cancer Biol Ther. 2002;1(5):460–460. doi: 10.4161/cbt.1.5.159. [DOI] [PubMed] [Google Scholar]
- 106.Imatani A, Callahan R. Identification of a novel NOTCH-4/INT-3 RNA species encoding an activated gene product in certain human tumor cell lines. Oncogene. 2000;19(2):223–231. doi: 10.1038/sj.onc.1203295. [DOI] [PubMed] [Google Scholar]
- 107.Soares R, Balogh G, Guo S, Gärtner F, Russo J, Schmitt F. Evidence for the Notch signaling pathway on the role of estrogen in angiogenesis. Mol Endocrinol. 2004;18(9):2333–2343. doi: 10.1210/me.2003-0362. [DOI] [PubMed] [Google Scholar]
- 108.Hao L. Notch-1 Activates Estrogen Receptor Alpha-Dependent Transcription via IKK Alpha. Loyola University Chicago; 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang Z, Wang H, Ikeda S, et al. Notch3 in human breast cancer cell lines regulates osteoblast-cancer cell interactions and osteolytic bone metastasis. Am J Pathol. 2010;177(3):1459–1469. doi: 10.2353/ajpath.2010.090476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hurlbut GD, Kankel MW, Lake RJ, Artavanis-Tsakonas S. Crossing paths with Notch in the hyper-network. Curr Opin Cell Biol. 2007;19(2):166–175. doi: 10.1016/j.ceb.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 111.Guo S, Liu M, Gonzalez-Perez RR. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim Biophys Acta. 2011;1815(2):197–213. doi: 10.1016/j.bbcan.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Song LL, Peng Y, Yun J, et al. Notch-1 associates with IKK alpha and regulates IKK activity in cervical cancer cells. Oncogene. 2008;27(44):5833–5844. doi: 10.1038/onc.2008.190. [DOI] [PubMed] [Google Scholar]
- 113.Osipo C, Golde TE, Osborne BA, Miele LA. Off the beaten pathway: the complex cross talk between Notch and NF-κB. Lab Invest. 2007;88(1):11–17. doi: 10.1038/labinvest.3700700. [DOI] [PubMed] [Google Scholar]
- 114.Osipo C, Patel P, Rizzo P, et al. ErbB-2 inhibition activates Notch-1 and sensitizes breast cancer cells to a γ-secretase inhibitor. Oncogene. 2008;27(37):5019–5032. doi: 10.1038/onc.2008.149. [DOI] [PubMed] [Google Scholar]
- 115.Pandya K, Meeke K, Clementz A, et al. Targeting both Notch and ErbB-2 signalling pathways is required for prevention of ErbB-2-positive breast tumour recurrence. Br J Cancer. 2011;105(6):796–806. doi: 10.1038/bjc.2011.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mehta K, Osipo C. Trastuzumab resistance: role for Notch signaling. Sci World J. 2009;9:1438–1448. doi: 10.1100/tsw.2009.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Clementz AG, Rogowski A, Pandya K, Miele L, Osipo C. NOTCH-1 and NOTCH-4 are novel gene targets of PEA3 in breast cancer: novel therapeutic implications. Breast Cancer Res. 2011;13(3):R63. doi: 10.1186/bcr2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Olson RE, Albright CF. Recent progress in the medicinal chemistry of-secretase inhibitors. Curr Top Med Chem. 2008;8(1):17–33. doi: 10.2174/156802608783334088. [DOI] [PubMed] [Google Scholar]
- 119.Kreft AF, Martone R, Porte A. Recent advances in the identification of γ-secretase inhibitors to clinically test the Aβ oligomer hypothesis of Alzheimer’s disease. J Med Chem. 2009;52(20):6169–6188. doi: 10.1021/jm900188z. [DOI] [PubMed] [Google Scholar]
- 120.Clarke EE, Churcher I, Ellis S, et al. Intra-or intercomplex binding to the γ-secretase enzyme. J Biol Chem. 2006;281(42):31279–31289. doi: 10.1074/jbc.M605051200. [DOI] [PubMed] [Google Scholar]
- 121.Wong GT, Manfra D, Poulet FM, et al. Chronic treatment with the γ-secretase inhibitor LY-411,575 inhibits β-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem. 2004;279(13):12876–12882. doi: 10.1074/jbc.M311652200. [DOI] [PubMed] [Google Scholar]
- 122.Yao J, Duan L, Fan M, Wu X. γ-secretase inhibitors exerts antitumor activity via down-regulation of Notch and Nuclear factor kappa B in human tongue carcinoma cells. Oral Dis. 2007;13(6):555–563. doi: 10.1111/j.1601-0825.2006.01334.x. [DOI] [PubMed] [Google Scholar]
- 123.Séveno C, Loussouarn D, Bréchet S, Campone M, Juin P, Barillé-Nion S. γ-Secretase inhibition promotes cell death, Noxa upregulation, and sensitization to BH3 mimetic ABT-737 in human breast cancer cells. Breast Cancer Res. 2012;14(3):R96. doi: 10.1186/bcr3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zeng Q, Li S, Chepeha DB, et al. Crosstalk between tumor and endothelial cells promotes tumor angiogenesis by MAPK activation of Notch signaling. Cancer Cell. 2005;8(1):13–23. doi: 10.1016/j.ccr.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 125.Dovey H, John V, Anderson J, et al. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J Neurochem. 2009;76(1):173–181. doi: 10.1046/j.1471-4159.2001.00012.x. [DOI] [PubMed] [Google Scholar]
- 126.Purow B. Notch inhibition as a promising new approach to cancer therapy. Adv Exp Med Biol. 2012;727:305–319. doi: 10.1007/978-1-4614-0899-4_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Meurette O, Stylianou S, Rock R, Collu GM, Gilmore AP, Brennan K. Notch activation induces Akt signaling via an autocrine loop to prevent apoptosis in breast epithelial cells. Cancer Res. 2009;69(12):5015–5022. doi: 10.1158/0008-5472.CAN-08-3478. [DOI] [PubMed] [Google Scholar]
- 128.Robinson DR, Kalyana-Sundaram S, Wu Y-M, et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat Med. 2011;17(12):1646–1651. doi: 10.1038/nm.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Siemers E, Skinner M, Dean RA, et al. Safety, tolerability, and changes in amyloid beta concentrations after administration of a gamma-secretase inhibitor in volunteers. Clin Neuropharmacol. 2005;28(3):126–132. doi: 10.1097/01.wnf.0000167360.27670.29. [DOI] [PubMed] [Google Scholar]
- 130.Wei P, Walls M, Qiu M, et al. Evaluation of selective γ-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol Cancer Ther. 2010;9(6):1618–1628. doi: 10.1158/1535-7163.MCT-10-0034. [DOI] [PubMed] [Google Scholar]
- 131.Zhang CC, Pavlicek A, Zhang Q, et al. Biomarker and pharmacologic evaluation of the γ-secretase inhibitor PF-03084014 in breast cancer models. Clin Cancer Res. 2012;18(18):5008–5019. doi: 10.1158/1078-0432.CCR-12-1379. [DOI] [PubMed] [Google Scholar]
- 132.Tolcher A, Mikulski S, Messersmith W, et al. A phase I study of R04929097, a novel gamma secretase inhibitor, in patients with advanced solid tumors. J Clin Oncol (Meeting Abstracts) 2010;28(Suppl 15):2502. [Google Scholar]
- 133.Tolcher AW, Messersmith WA, Mikulski SM, et al. Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J Clin Oncol. 2012;30(19):2348–2353. doi: 10.1200/JCO.2011.36.8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.He W, Luistro L, Carvajal D, et al. High tumor levels of IL6 and IL8 abrogate preclinical efficacy of the γ-secretase inhibitor, RO4929097. Mol Oncol. 2011;5(3):292–301. doi: 10.1016/j.molonc.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Luistro L, He W, Smith M, et al. Preclinical profile of a potent γ-secretase inhibitor targeting Notch signaling with in vivo efficacy and pharmacodynamic properties. Cancer Res. 2009;69(19):7672–7680. doi: 10.1158/0008-5472.CAN-09-1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wu J, LoRusso PM, Matherly LH, Li J. Implications of plasma protein binding for pharmacokinetics and pharmacodynamics of the γ-secretase inhibitor RO4929097. Clin Cancer Res. 2012;18(7):2066–2079. doi: 10.1158/1078-0432.CCR-11-2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Barten DM, Meredith JE, Zaczek R, Houston JG, Albright CF. Gamma-secretase inhibitors for Alzheimers disease: balancing efficacy and toxicity. Drugs R D. 2006;7(2):87–97. doi: 10.2165/00126839-200607020-00003. [DOI] [PubMed] [Google Scholar]
- 138.Wu Y, Cain-Hom C, Choy L, et al. Therapeutic antibody targeting of individual Notch receptors. Nature. 2010;464(7291):1052–1057. doi: 10.1038/nature08878. [DOI] [PubMed] [Google Scholar]
- 139.Rizzo P, Osipo C, Foreman K, Golde T, Osborne B, Miele L. Rational targeting of Notch signaling in cancer. Oncogene. 2008;27(38):5124–5131. doi: 10.1038/onc.2008.226. [DOI] [PubMed] [Google Scholar]