

Abstract
Background
Intracellular deposition of tau protein is a hallmark lesion of Alzheimer’s disease. Although it is known this event is secondary to excessive tau phosphorylation, the mechanisms involved remain unknown. We previously reported that the enzyme 5-Lipoxygenase (5LO) acts as a modulator of Aβ peptides formation in vivo, and here we investigate its influence on tau protein.
Methods
Tg2576 mice overexpressing neuronal 5LO were generated and its contribution to endogenous tau levels and metabolism investigated.
Results
Although no differences were noted in the levels of total tau for both groups, compared with controls, Tg2576 mice overexpressing 5LO had a significant increase in the phosphorylation state of tau at S396 and S396/S404, as recognized by the antibodies PHF-13 and PHF-1, respectively. By contrast, no phosphorylation changes were observed in other tau epitopes. This increase was associated with a significant elevation in cyclin dependent kinase-5 but not other kinases that have been involved in tau phosphorylation. Additionally, mice overexpressing 5LO had biochemical evidence of altered synaptic integrity because they manifested a reduction in PSD-95, synaptophysin and MAP2.
Conclusions
This study demonstrates a new role for 5LO in regulating endogenous tau metabolism in the central nervous system and supports the hypothesis that its pharmacologic inhibition could be beneficial for Alzheimer’s disease-related tau neuropathology.
Keywords: 5-Lipoxygenase, Alzheimer’s disease, amyloid beta, cyclin dependent kinase-5, tau protein, transgenic mouse model
One of the hallmark features of Alzheimer’s disease (AD) brain pathology is the presence of neurofibrillary tangles, which are intracellular aggregates of conformationally abnormal forms of the tau protein. Interestingly, this type of pathology is also the major signature of a large group of other neurodegenerative diseases collectively referred to as tauopathies, which includes progressive supranuclear palsy, Pick’s disease, and corticobasal degeneration (1,2). Today we know that in all of these conditions, tau, because of an excessive phosphorylation status, acquires a pathologic conformation, detaches from the microtubules, and accumulates as neurofibrillary tangles (3,4). However, the mechanisms responsible for this phenomenon are still unknown. Recent evidence suggests that alterations in inflammatory processes occur in the brains of individuals with tauopathies and in mouse models of them. Thus, microglia activation coexists with the progression of tau pathology, pro-inflammatory stressors promote tau hyperphosphorylation, and immunosuppressant drugs lessen tau pathology and enhance survival in a model of tauopathy (5-8).
Although these studies suggest a correlative link between inflammation and tau pathology, the exact mechanism as well as the source of inflammation remains unclear. 5-Lipoxygenase (5LO) is a pro-inflammatory enzyme widely expressed in the central nervous system where its level increases in an age-dependent manner (9). Previous work has shown that, compared with control brains, 5LO is upregulated in AD brains, and its genetic absence or pharmacologic blockade results in an amelioration of the AD-like amyloidotic phenotype of a transgenic mouse model of the disease (i.e., the Tg2576 mice) (10-12). However, no data are available on the influence that this pathway might have on the metabolic fate of endogenous tau in these mice. We recently reported that adeno-associated virus (AAV)–mediated brain delivery of 5LO modulates amyloid beta levels and deposition, as well as behavior of the Tg2576 mice (13). In the current study, we sought to investigate its effects on tau level and metabolism in the same mouse model.
Methods and Materials
Construction of AAV2/1 Vector and Injection to Neonatal Mice
The construction, packaging, purification, and titering of the recombinant AAV2/1 vector expressing 5LO were performed as previously reported (13).
Tg2576 mice harboring a human mutant amyloid precursor protein (APP; KM670/671NL) used in this study were previously reported (13). The injection procedures were performed as described previously (14,15). Briefly, 2 μL of AAV2/1-5LO (1.3 × 1013 genome particles/mL) were bilaterally injected into the cerebral ventricle of newborn mice using a 5-μL Hamilton syringe. Eighteen pups were used for the study; 10 were injected with AAV2/1-5LO, and 8 were injected with empty vector. Because of the known aggressive phenotype and the need to single cage males with this genotype, only females were used for this study. Animals were then followed until they were 13 to 14 months old. All animal procedures were approved by the Institutional Animal Care and Usage Committee, in accordance with the U.S. National Institutes of Health guidelines.
Immunoblot Analyses
The primary antibodies used in this study are summarized in Table 1. Proteins were extracted in enzyme immunoassay buffer containing 250 mmol/L Tris base, 750 mmol/L NaCl, 5% NP-40, 25 mmol/L ethylenediaminetetraacetate tetraacetate, 2.5% sodium deoxycholate, .5% sodium dodecyl sulfate, and an ethylenediaminetetraacetate tetraacetate–free protease and phosphatase inhibitors cocktail tablet (Roche Applied Science, Indianapolis, Indiana); sonicated; and centrifuged at 13,000 rpm for 45 min at 4°C. Supernatants were used for immunoblot analysis as previously described (11-13). Total protein concentration was determined by using BCA Protein Assay Kit (Pierce, Rockford, Illinois). Samples were electrophoretically separated using 10% Bis-Tris gels or 3-8% Tris-acetate gel (Bio-Rad, Richmond, California) according to the molecular weight of the target molecule and then transferred onto nitrocellulose membranes (Bio-Rad). They were blocked with Odyssey blocking buffer (LI-COR Bioscience, Lincoln, Nebraska) for 1 hour and then incubated with primary antibodies overnight at 4°C. After three washing cycles with Tris-buffered saline with .1% (v/v) Tween-20 detergent, membranes were incubated with IRDye 800CW or IRDye 680CW-labeled secondary antibodies (LI-COR Bioscience, Lincoln, Nebraska) at 22°C for 1 hour. Signals were developed with Odyssey Infrared Imaging Systems (LI-COR Bioscience). Actin was always used as an internal loading control.
Table 1.
Antibodies Used in the Study
| Antibody | Immunogen | Host | Application | Source |
|---|---|---|---|---|
| Tau-1 | Purified denaturated bovine MAP | Mouse | WB, IHC | Millipore (Billerica, Massachusetts) |
| AT-8 | Peptide containing phospho-S202/T205 | Mouse | WB | Pierce (Rockford, Illinois) |
| AT-180 | Peptide containing phospho-T231/S235 | Mouse | WB | Pierce |
| AT-270 | Peptide containing phospho-T181 | Mouse | WB | Pierce |
| PHF-13 | Peptide containing phospho-Ser396 | Mouse | WB, IHC | Cell Signaling, Danvers, Massachusetts |
| PHF-1 | Peptide containing phospho-Ser396/S404 | Mouse | WB, IHC | Dr. P. Davies, Albert Einstein College of Medicine, Bronx, New York |
| GFAP | aa spinal chord homogenate of bovine origin | Mouse | WB | Santa Cruz Biotechnology, Santa Cruz, California |
| CD45 | Mouse thymus or spleen | Rat | WB | BD Pharmingen, Franklin Lakes, New Jersey |
| GSK3α/β | aa 1-420 full length GSK3β of Xenopus origin | Mouse | WB | Millipore |
| p-GSK3α/β | aa around Ser21 of human GSK3a | Rabbit | WB | Cell Signaling |
| JNK2 | aa of human JNK2 | Rabbit | WB | Cell Signaling |
| SAPK/JNK | aa of recombinant human JNK2 fusion protein | Rabbit | WB | Cell Signaling |
| Phospho-SAPK/JNK | aa Thr183/Tyr185 of human SAPK/JNK | Mouse | WB | Cell Signaling |
| Cdk5 | aa C-terminus of Cdk5 of human origin | Rabbit | WB | Santa Cruz |
| P35/P25 | aa C-terminus of p35/25 of human origin | Rabbit | WB | Santa Cruz |
| PP-2A | A 16 residue synthetic peptide corresponding to aa 295-309 of the 36kDa catalytic subunit of human protein phosphatase 2A (PP-2A) | Mouse | WB | Millipore |
| Synaptophysin | Rat retina synaptosome | Mouse | WB, IHC | Sigma-Aldrich (St. Louis, Missouri) |
| PSD-95 | Peptide corresponding to residues of human PSD-95 | Rabbit | WB, IHC | Cell Signaling |
| MAP2 | Microtubule associated protein rat brain | Rabbit | WB, IHC | Millipore |
| Actin | aa C-terminus of actin of human origin | Goat | WB | Santa Cruz |
IHC, immunohistochemistry; WB, Western blot.
Sarkosyl Insolubility Assay
The assay for insoluble tau was performed as previously described (16). Briefly, ultracentrifugation and sarkosyl extraction (30 min in 1% sarkosyl) was used to obtain soluble and insoluble fractions of tau. Insoluble fractions were washed one time with 1% sarkosyl then immunoblotted with Tau-1 antibody (Table 1).
Immunohistochemistry
The primary antibodies used in this study are summarized in Table 1. Immunostaining was performed as reported previously by our group (12,13,17). Serial 6-μm-thick coronal sections were mounted on 3-aminopropyl triethoxysilane-coated slides. Every eighth section from the habenular to the posterior commissure (8–10 sections per animal) was examined using unbiased stereo-logic principles. The sections were deparaffinized, hydrated, pretreated with 3% H2O2 in methanol, and then treated with citrate (10 mmol/L) or IHC-Tek Epitope Retrieval Solution (IHC World, Woodstock, Maryland) for antigen retrieval. Sections were blocked in 2% fetal bovine serum before incubation with primary antibody overnight at 4°C. Sections were then incubated with biotinylated anti-mouse immunoglobulin G (Vector Laboratories, Burlingame, California) and then developed by using the avidinbiotin complex method (Vector Laboratories) with 3,3′-diaminobenzidine as a chromogen. Light microscopic images were used to calculate the integrated optical density of the immunopositive reactions by using the software Image-Pro Plus for Windows version 5.0 (Media Cybernetics, Rockville, Maryland). The threshold optical density that discriminated staining from background was determined and kept constant for all quantifications.
Cell Cultures
N2A (neuro-2 A neuroblastoma) cells stably expressing human APP carrying the K670 N, M671 L Swedish mutation (kindly provided by Dr. Nikolaos K. Robakis, Mount Sinai School of Medicine, New York, New York) were grown in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin (Cellgro, Herdon, Virginia), and 400 μg/mL G418 (Invitrogen, Carlsbad, California), at 37°C in the presence of 5% CO2. For each experiment, equal numbers of cells were plated in six-well plates; 24 hours later, media were removed, and fresh media containing either 5-hydroxyeicosatetraenoic acid (5-HETE; 10 μmol/L) or vehicle were added. After 24 hours of incubation, cell pellets were harvested in lytic buffer for immunoblot analyses performed as described in the previous paragraph. In another set of experiments, before the challenge with 5-HETE, cells were incubated overnight with small interfer (si)RNA for cyclin dependent kinase-5 (cdk5) (.1 μmol/L; Cell Signaling, Danvers, Massachusetts), or the selective cdk5 inhibitor roscovitine (20 μmol/L) (Calbiochem, Billerica, Massachusetts). Cells were then harvested in lytic buffer for immunoblot analyses. Cell viability was always assessed by measuring lactate dehydrogenase release in the supernatants using a cytotoxicity detection kit (Roche Applied Science) according to the manufacturer’s instructions.
Immunofluorescence Microscopy
N2A-APPswe cells were plated on glass coverslips and the following day fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min at 22°C. After rinsing several times with PBS, cells were incubated in a blocking solution (5% normal serum/.4% TX-100) for 1 hour at 22°C and then with the appropriate primary antibody overnight at 4°C. After washing with PBS, cells were subsequently incubated for 1 hour with a secondary Alexa546-conjugated antibody (1:800 dilution, goat anti-rabbit or donkey anti-mouse; Invitrogen). Coverslips were mounted using VECTASHIELD mounting medium (Vector Laboratories) and analyzed with an Olympus BX60 fluorescent microscope (Olympus, Center Valley, Pennsylvania). Fluorescence emission was collected at 425 to 475 nm for 4′, 6-diamidino-2-phenylindole and 555 to 655 nm for Alexa546. Control coverslips were processed as described above except that no primary antibody was added to the solution (images not shown).
Cdk5 activity assay
For determination of cdk5 kinase activity, cells were rinsed with PBS once and lysed in buffer A (50 mmol/L Tris-HCl [pH 8.0], 150 mmol/L sodium chloride, 1% NP-40, .5% sodium deoxycholate, .1% sodium dodecyl sulfate, .02% sodium azide and freshly added protease inhibitors [100 μg/mL phenylmethysulfonyl fluoride and 1 μg/mL aprotinin]). Following incubation on ice for .5 hours, the samples were centrifuged at 12,000 g at 4°C for 20 min, and the supernatant was collected. The supernatant (300 μL equivalent to 150 μg protein) was incubated with 3 μg of anti-cdk5 antibody (Santa Cruz Biotechnology, Santa Cruz, California) at 4°C for 2 hours. Protein A agarose beads (50 μL) were then added and incubated for another hour. The immunoprecipitates were washed with lysis buffer three times and once with HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid)-buffered saline (10 mmol/L HEPES, pH 7.4, 150 mmol/L NaCl). The kinase activity of the immunoprecipitated cdk5 was determined by using histone H1 (Santa Cruz Biotechnology). Beads were incubated with 5 μg of histone H1 (Santa Cruz Biotechnology) in HEPES-buffered saline (20 μL) containing 15 mmol/L MgCl2, 50 μM adenosine triphosphate, 1 mmol/L dithiothreitol, and 1 μCi of [32P] adenosine triphosphate. After 30 min of incubation at 30°C, the reaction products were determined by a liquid scintillation counter.
Data Analysis
One-way analysis of variance followed by the Bonferroni multiple comparison tests were performed using GraphPad Prism 5.0 (La Jolla, California). All data are presented as mean ± SEM. Significance was set at p < .05.
Results
In Vivo Study
Tau Levels and Phosphorylation in Tg2576 Treated with AAV-Encoding 5LO
The transgene expression in animals receiving the AAV2/1-5LO was confirmed by their significantly higher 5LO levels compared with controls, as we previously reported (13). To assess the effect of 5LO gene transfer on tau level and its metabolism, we measured the steady-state levels of endogenous mouse tau and some of its phosphorylated isoforms in the Tg2576 mice. At sacrifice we observed that there was no difference in the levels of total endogenous tau between the two groups of animals (Figure 1A,B). By contrast, we found that, compared with the group receiving the empty vector, mice treated with AAV encoding for 5LO had a significant increase in the phosphorylated forms at epitopes S396 and S396/S404 as recognized by the specific antibodies PHF-13 and PHF-1 respectively (ratios PHF-13/tau: 2.14, and PHF-1/tau: 1.86; Figure 1A and 1B). However, no changes were detected for other phosphorylation sites as recognized by the antibody AT8 (S202/T205), AT180 (T231/S235), and AT270 (T181; Figure 1A,B). To further confirm the results obtained with the immunoblot analyses, we performed immunohistochemical studies in brain sections from the two groups of mice. As shown in Figure 1C–F, although we did not observe any significant changes in the immunoreactivity for endogenous total tau, we detected a significant increase in the somatodendritic accumulation of phosphorylated epitopes as recognized by PHF-13 and PHF-1 immunopositive areas both in the hippocampus and cortex regions. Finally, we observed that although the total amount of sarkosyl soluble tau was unaltered between the two groups, overexpression of 5LO resulted in a significant increase in the sarkosyl insoluble fraction of tau (Figure 1G,H).
Figure 1.
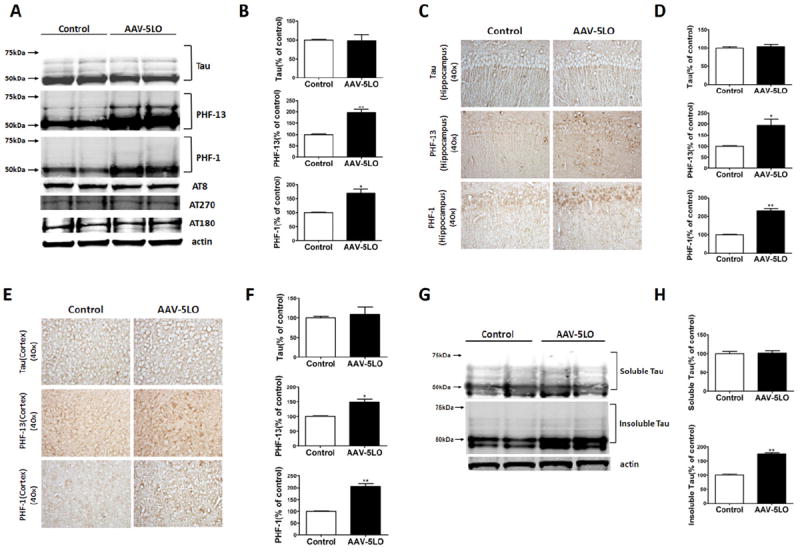
5-Lipoxygenase (5LO) gene transfer regulates endogenous tau phosphorylation in brains of Tg2576 mice. (A) Representative Western blot analyses of total tau (Tau-1), phosphorylated tau at residues S396 (PHF-13), S202/T205 (AT8), T231/S235 (AT180), at T181 (AT270), and S396/S404 (PHF-1) in brain cortex homogenates from Tg2576 mice receiving empty vector (Control) or adeno-associated virus (AAV)1/2-5LO (AAV-5LO). (B) Densitometric analyses of the immunoreactivities to the antibodies shown in the previous panel (*p = .04, **p = .02). (C, E) Representative immunohistochemical staining for Tau-1-, PHF-13-, and PHF-1-positive areas in hippocampus and cortex areas from brain sections of mice receiving empty vector control (Control) or AAV1/2-5LO. (D, F) Quantification of the positive immunoreactivities to the antibodies shown in the previous two panels (*p < .05, **p ≤ .002). (G, H) Representative Western blots and densitometric analyses for sarkosyl soluble and insoluble tau fractions in brain cortex homogenates from Tg2576 receiving empty vector (Control) or AAV1/2-5LO (**p < .02). Values represent mean ± SEM.
Tau Metabolism in Tg2576 Treated with AAV Encoding 5LO
Because we found that 5LO gene transfer results in an alteration in tau phosphorylation levels, next we sought to investigate the molecular mechanisms involved in this biological effect. To this end, we examined some of the kinases that are considered major regulators of tau posttranslational phosphorylation modification. As shown in Figure 2, we observed no differences between the two groups in the levels of total or phosphorylated glycogen synthase-3α (GSK3α) and GSK3β, JNK2 and total and phosphorylated SAPK/JNK levels. By contrast, compared with control animals, the ones receiving the AAV vector encoding for 5LO had a significant increase in the levels of cdk5 and its two coactivators p35 and p25, suggesting an activation of this pathway (Figure 2A,B). By contrast, we did not detect any significant changes between the two groups of mice in the steady-state levels of protein phosphatase-2A (PP-2A) another player implicated in the phosphorylative modification of tau (Figure 2A).
Figure 2.
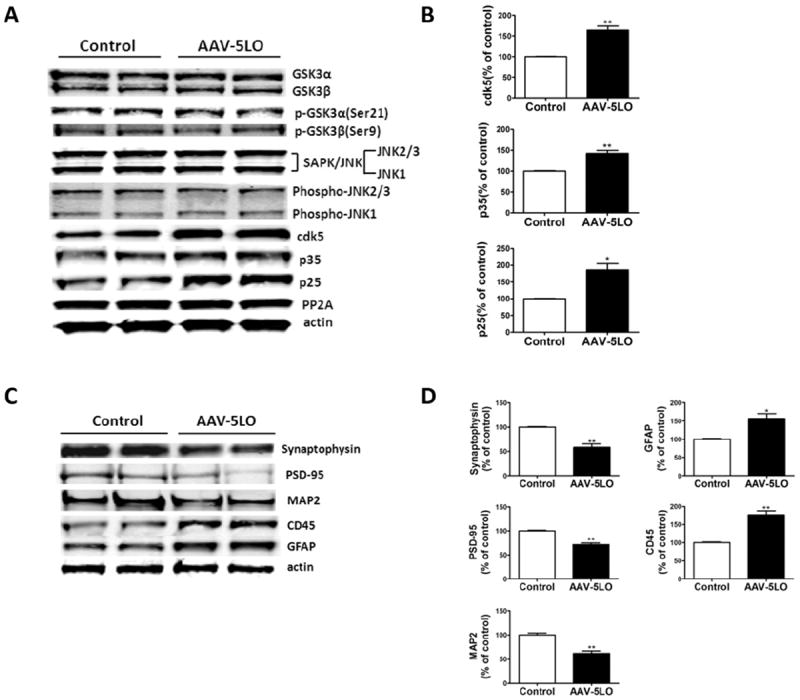
5-Lipoxygenase (5LO) gene transfer regulates endogenous tau metabolism in brains of Tg2576 mice. (A) Representative Western blots of GSK3α, GSK3β, p-GSK3α, p-GSK3β, SAPK/JNK, p-JNK2/3, p-JNK1, cdk5, p35, p25, and PP-2A in brain cortex homogenates of Tg2576 mice treated with empty vector (Tg) or adeno-associated virus (AAV)1/2-5LO (Tg-5LO). (B) Densitometric analyses of the immunoreactivities for cdk5, p35, and p-25 shown in the previous panel (*p = .04, **p = .002). (C) Representative Western blots of synaptophysin, PSD-95, MAP2, GFAP, and CD45 in brain cortex homogenates of Tg2576 mice receiving empty vector (Tg) or AAV1/2-5LO (Tg-5LO). (D) Densitometric analyses of the immunoreactivities to the antibodies shown in the previous panel (*p < .05; **p = .02). Values represent mean ± SEM. CD-45, microglial markers CD45; GFAP, glial fibrillary acidic protein; MAP2, microtubule-associated protein 2; PSD-95, postsynaptic density protein 95.
5LO Gene Transfer Modulates Synaptic Integrity
Because an increase in tau phosphorylation has been implicated in the alteration and disruption of synaptic integrity in AD, we wanted to assess this aspect in Tg2576 mice overexpressing 5LO. Compared with the control group, mice receiving the 5LO gene had a significant reduction in the steady-state levels of two main synaptic proteins: postsynaptic density protein 95 (PSD-95) and synaptophysin (Figure 2C,D). A similar result was obtained when the dendritic protein MAP2 was assayed (Figure 2C,D). These results were further confirmed in brain sections of the same mice when they were assessed by immunohistochemical analyses (Figure 3A,D). Finally, we observed that brain homogenates from mice receiving the AAV-5LO had a significant increase in glial fibrillary acidic protein and CD45 immunoreactivities, which are markers of an activation of astrocytes and microglia cells, respectively (Figure 2C,D).
Figure 3.
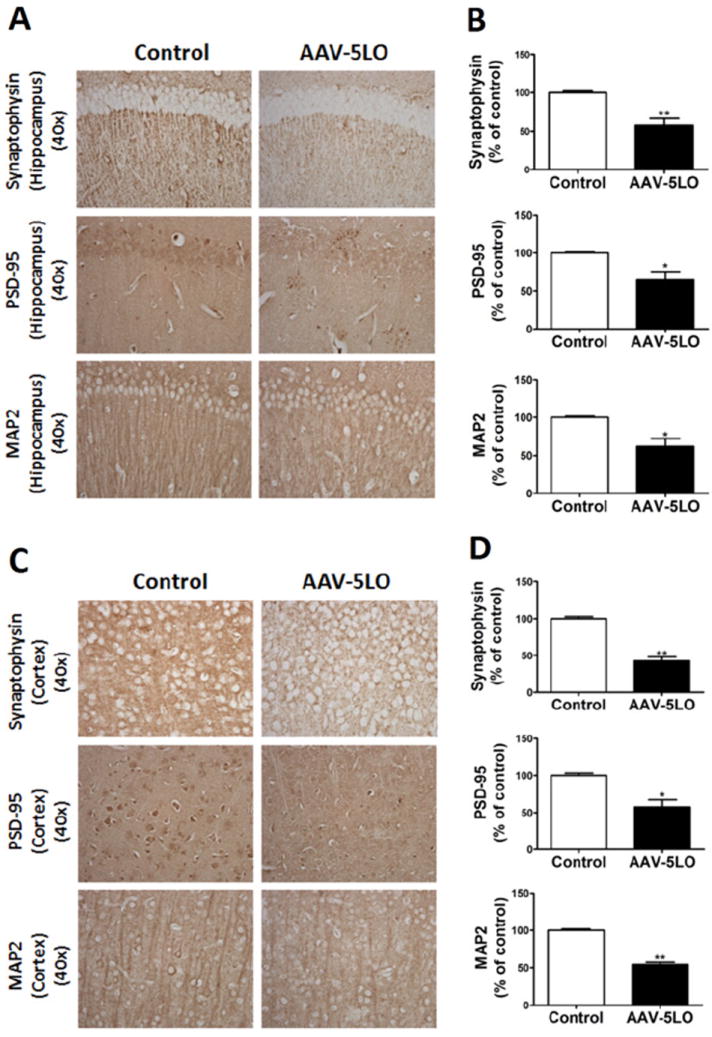
5-Lipoxygenase (5LO) gene transfer results in an alteration of brain synaptic integrity of Tg2576 mice. (A, C) Representative immunohistochemical staining for synaptophysin-, PSD-95-, and MAP2-positive areas in hippocampus and cortex of brains from mice receiving empty vector (Tg) or adeno-associated virus (AAV)1/2-5LO (Tg-5LO). (B, D) Quantitation of the positive immunoreactivities to the antibodies shown in the previous panels (*p ≤ .03, **p ≤ .002). Values represent mean ± SEM. PSD-95, postsynaptic density protein 95; MAP2, microtubule-associated protein 2.
In Vitro Study
Effect of 5-Lipoxygenase Activation on Tau Metabolism
To support our in vivo findings, we conducted a series of in vitro experiments using neuronal cells that express the same APP Swedish mutation and endogenous mouse tau as the Tg2576 mice (i.e., N2A-APPSwe neuronal cells) and incubated them with the main metabolic product of 5LO activation (i.e., 5-HETE).
Compared with controls, we observed that neuronal cells incubated with 5-HETE (10 mmol/L) had an increase in tau at S396 as recognized by PHF-13, and at S396/S404 as recognized by PHF-1 (ratios of PHF-13/tau: 1.82; PHF-1/tau: 2.45; Figure 4A,B). The same changes in tau were further documented by immunofluorescence studies in which we observed that 5-HETE induced in a significant increase in immunofluorescence reactivity for phospho tau recognized by PHF-1 (Figure 4C). Additionally, immunoblot analyses showed that although the steady-state levels of cdk5 kinase were not changed, the levels of its main coactivator, p25, were significantly increased in cells incubated with 5-HETE, suggesting an increase in the activity of this kinase (Figure 4 D,E). This increase was also documented by an in vitro assay showing a significant elevation in the activity of this kinase in lysates from neuronal cells treated with 5-HETE (Figure 4F). To support the mechanistic involvement of the cdk5 in the 5LO-dependent effect on tau phosphorylation, we used a siRNA for this kinase. First, we identified the concentrations of this siRNA that significantly reduced the steady-state protein level of cdk5 (not shown), then, by using this amount of siRNA, we tested the 5-HETE-dependent effect on tau phosphorylation. As shown in Figure 5A and B, we found that indeed by suppressing cdk5, we could prevent the increase in the immunoreactivity for PHF-1 and PHF-13. Similar results were obtained when we incubated cells with a specific cdk5 inhibitor, roscovitine (Figure 5C,D). In both experimental settings, no biochemical sign of cell toxicity as measured by lactate dehydrogenase release in the supernatants was detected (not shown).
Figure 4.
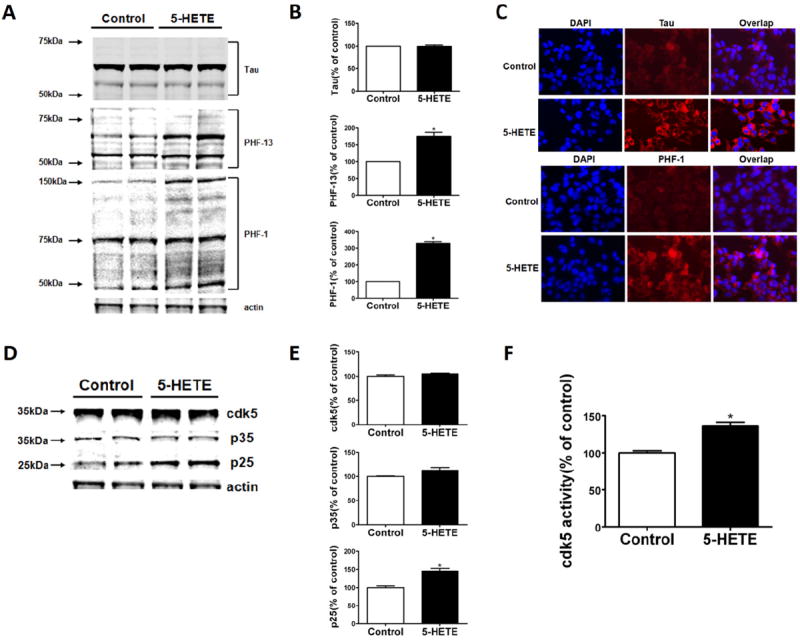
5-Lipoxygenase (5LO) activation modulates endogenous tau phosphorylation in neuronal cells. (A) Representative Western blot analysis for total tau and phosphorylated tau at S396 (PHF-13), S396/404 (PHF-1), S202/T205 (AT8), T231/S235 (AT180), and T181 (AT270) in lysates from neuronal cells incubated with 5-hydroxyeicosatetraenoic acid (5-HETE; 10 μmol/L) or vehicle. (B) Densitometric analyses of the immunoreactivities shown in the previous panel (*p = .01, **p = .02). (C) Cells were incubated overnight with 5-HETE (10 μmol/L) or vehicle and then fixed with 4% paraformaldehyde in phosphate buffered saline for 15 min at 22°C. Representative immunofluorescent pictures for cellular expression of tau (Tau1) and phosphorylated tau at S396/404 (PHF-1) in neuronal cells incubated with 5-HETE or vehicle. (D). Representative Western blot analysis for cdk5, p25, and p35 in lysates from cells incubated with 5-HETE (10 μmol/L) or vehicle. (E) Densitometric analyses of the immunoreactivities shown in the previous panel (*p = .02). (F) Cdk5 kinase activity in lysates from neuronal cells incubated with 5-HETE (10 μmol/L) or vehicle (*p = .02). Values represent mean ± SEM. DAPI, 4′,6-diamidino-2-phenylindole.
Figure 5.
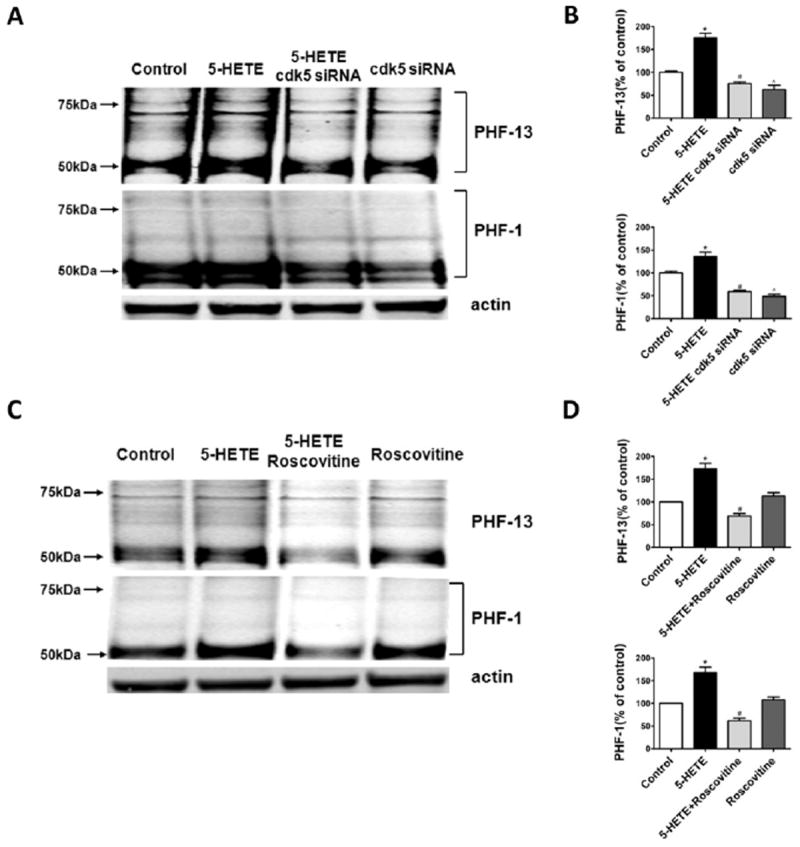
5-Lipoxygenase (5LO) activation regulates tau phosphorylation in neuronal cells via cdk5 kinase activation. (A) Representative Western blot analysis for tau phosphorylated tau at S396 (PHF-13), S396/404 (PHF-1), in lysates from neuronal cells preincubated with siRNA cdk5 (.1 μmol/L), and then challenged with 5-hydroxyei-cosatetraenoic (5-HETE; 10 μmol/L) or vehicle. (B) Densitometric analyses of the immunoreactivities shown in the previous panel (*p < .02). (C) Representative Western blots for tau phosphorylated tau at S396 (PHF-13), S396/404 (PHF-1), in lysates from neuronal cells incubated with roscovitine (20 μmol/L) and then challenged with 5-HETE (10 μmol/L) or vehicle. (D) Densitometric analyses of the immunoreactivities shown in the previous panel (*p < .02). Values represent mean ± SEM.
Discussion
This study provides experimental evidence that 5LO modulates in vivo the phosphorylation state of the microtubule associate protein tau via the activation of the cdk5 kinase pathway. Tau is a microtubule-associated protein normally found in the axons of neurons where it promotes microtubule assembly and stabilization. In postmortem brains of AD patients, tau is hyperphosphorylated and conformationally altered, resulting in the formation of intracellular aggregates called neurofibrillary tangles (18). Interestingly, this type of pathology is also the major signature lesion of a large group of neurodegenerative diseases collectively referred to as tauopathies, which among others include progressive supranuclear palsy, Pick’s disease, and corticobasal degeneration. More than 5 million Americans currently have AD, and several millions are affected by non-AD tauopathies. Tragically, the progression of these diseases is lengthy, and there is currently no effective treatment. Numerous reports from the past 2 decades have suggested that development of tau pathology strongly correlates with clinical symptoms in most of these neurodegenerative conditions (19). Although only a minority has been clearly associated with mutations in the tau gene, today we know that the majority of these cases results from the interaction between genetic risk factors with modifiable and unmodifiable environmental elements. Among them inflammatory processes have been casually linked to the development of tau neuropathology; however, the source of inflammation remains elusive.
In recent years our laboratory has been interested in the neurobiology of the 5LO, an enzyme with potent pro-inflammatory activity widely expressed within the central nervous system (20). Thus, 5LO immunoreactivity is increased in hippocampi of AD patients (10), and polymorphism of the 5LO promoter influences the age of onset of the disease (21). Work from our group showed that 5LO modulates brain amyloidosis and behavior in APP transgenic mice Tg2576 (11-13). Importantly, despite the fact that the effect of 5LO on Aβ is γ-secretase-dependent, we have also shown that it does not influence Notch processing (12). This makes the pharmacologic inhibition of this pathway a viable antiamyloid therapy. Additionally, in a more recent article, we showed for the first time that this enzymatic pathway also modulates Aβ and tau neuropathology in a mouse model with plaques and tangles, the 3xTg-AD mice (22). However, because the development of tau pathology in these mice is strictly secondary to the presence of a human mutant form of tau protein in their genome, no data are available to date on the effect that 5LO may have on the metabolic fate of endogenous tau.
To address this important issue, we used a virally mediated somatic gene transfer approach, which largely results in neuronal increase of 5LO expression, in the Tg2576 mice, which express the human Swedish mutant for the APP but retain the endogenous mouse tau. In contrast to mice expressing mutant tau genes (i.e., 3xTg), this model offers the advantage that similar to human AD, in which no mutations for tau gene have been identified, any change in tau phosphorylation state is not secondary to an abnormal tau per se.
In the current study, we showed for the first time that 5LO influences the metabolic fate of endogenous tau in vivo as well as in vitro. Thus, by using both biochemistry and immunohistochemistry approaches, we found that mice overexpressing 5LO had a significant increase in their tau phosphorylation state. This increase was selective because we found an increase in phosphorylated tau only at S396 (detected by antibody PHF-13) and S396/404 (detected by antibody PHF-1). By contrast, we did not find any significant differences when other phosphoepitopes such as AT8, AT180, and AT270 were assayed. Interestingly, in these mice the latter represent early markers of tau phosphorylation, whereas the PHF-1 and PHF-13 reactivity represents mid and late stages (23). We also observed that the fraction of insoluble tau was significantly increased in brain of mice overexpressing 5LO. Interestingly, besides these changes in tau metabolism, we also observed that overexpression of 5LO resulted in an alteration in synaptic integrity, finding that PSD-95, synaptophysin, and MAP2 were all significantly reduced. These data represent a strong biochemical mechanistic support for our previous observation that Tg2576 mice overexpressing 5LO manifest a significant worsening of their memory impairments (13).
To elucidate the molecular mechanism for the 5LO-induced selective in vivo tau hyperphosphorylation, we assayed several putative tau kinases. We measured the total and activated forms of GSK3, JNK2, and SAPK/JNK because they have been implicated in regulating tau phosphorylation (24-27). In our study, we found that 5LO overexpression did not alter the activation status of any of these kinases. By contrast, we observed that it affected specifically the cdk5 kinase pathway, the activation of which is regulated by its binding to two activators proteins p35 and p25, a cleaved product of p35 (28,29), suggesting that this kinase is responsible for the observed changes in tau phosphorylation in vivo.
To further corroborate the role of cdk5 kinase in the 5LO-dependent effect on tau metabolism, we embarked in a series of in vitro experiments. Neuronal cells incubated with the main metabolic product of 5LO activation (i.e., 5-HETE) had a significant increase in tau phosphorylated forms at S396 (recognized by the antibody PHF-13) and at S396/404 (recognized by the antibody PHF-1), as demonstrated by immunoblot analyses and immunofluorescence microscopy. Confirming the in vivo data, we observed that in the same cells the cdk5 kinase pathway was activated as shown by the selective increase of its coactivator p25 and the in vitro activity assay. The biological importance of the cdk5 in regulating 5LO-dependent effect on tau phosphorylation was also demonstrated by a genetic and a pharmacologic approach. Blockage of cdk5 transcription by siRNA or incubation with roscovitine, a selective and specific inhibitor of cdk5 activity, both resulted in a suppression of the 5LO-dependent effect on tau phosphorylation.
The novel concept that 5LO modulates tau phosphorylation via the cdk5 pathway and that this link could be pharmacologically targeted in AD-related tauopathies is also relevant in view of previous reports showing that activation of the same kinase worsens AD pathology and decreases membrane insertion of GluR1 α-amino-3-hydroxy-5-methy-4-isoxazole propionate receptors (30), whereas 5LO pharmacologic inhibition increases GluR1 phosphorylation (31).
In conclusion, our studies establish an additional and novel functional role for 5LO in the metabolism of endogenous tau protein besides the established one in the other two key pathologic changes found in AD (cognitive decline and amyloid deposition). The unique pleiotropism of this protein in AD pathogenesis and development of its phenotype is a strong biological support for the hypothesis that its pharmacologic inhibition is a real therapeutic opportunity for AD.
Acknowledgments
This study was funded in part by grants to DP (Grant Nos. AG33568, NS71096) and TG (Grant Nos. AG18454, AG20206) from the National Institutes of Health.
Footnotes
The authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Hernandez F, Avila J. Tauopathies. Cell Mol Life Sci. 2007;64:2219–2233. doi: 10.1007/s00018-007-7220-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geschwind DH. Tau phosphorylation, tangles, and neurode-generation: the chicken or the egg? Neuron. 2003;40:457–460. doi: 10.1016/s0896-6273(03)00681-0. [DOI] [PubMed] [Google Scholar]
- 3.Iqbal K, Liu F, Gong CX, Grundke-Iqbal I. Tau in Alzheimer disease and related tauopathies. Curr Alz Res. 2010;7:656–664. doi: 10.2174/156720510793611592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mederios R, Baglietto-Vargas D, Laferla F. The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci Ther. 2011;17:514–524. doi: 10.1111/j.1755-5949.2010.00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishizawa K, Dickson DW. Microglial activation parallels system degeneration in progressive supranuclear palsy and corticobasal degeneration. J Neuropathol Exp Neurol. 2001;60:647–657. doi: 10.1093/jnen/60.6.647. [DOI] [PubMed] [Google Scholar]
- 6.Kitazawa M, Oddo S, Yamasaki TR, Green KN, Laferla F. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5 mediated pathways in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25:8843–8853. doi: 10.1523/JNEUROSCI.2868-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhaskar K, Konerth M, Kokiko-Cochran O, Cardona A, Ransohoff R, Lamb B. Regulation of tau pathology by microglial fractalkine receptor. Neuron. 2010;68:19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshima Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangle in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 9.Chinnici C, Yao Y, Praticò D. The 5-Lipoxygenase enzymatic pathways in the mouse brain: Young versus old. Neurobiol Aging. 2007;28:1457–1462. doi: 10.1016/j.neurobiolaging.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 10.Ikonomovic MD, Abrahamson EE, Uz T, Manev H, Dekosky ST. Increased 5-lipoxygenase immunoreactivity in hippocampus of patients with Alzheimer’s disease. J Histochem Cytochem. 2008;56:1065–1073. doi: 10.1369/jhc.2008.951855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Firuzi O, Zhou J, Chinnici CM, Wisnieski T, Praticò D. 5-Lipoxygenase gene disruption reduces amyloid-{beta} pathology in a mouse model of Alzheimer’s disease. FASEB J. 2008;22:1169–1178. doi: 10.1096/fj.07-9131.com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chu J, Pratico D. Pharmacologic blockade of 5–Lipoxygenase improves the amyloidotic phenotype of an AD transgenic mouse model: Involvement of γ-secretase. Am J Pathol. 2011;178:1762–1769. doi: 10.1016/j.ajpath.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Chu J, Giannopoulos PJ, Ceballos-Diaz C, Golde T, Pratico D. Adeno-associated virus-mediated brain delivery of 5-Lipoxygenase modulates the AD-like phenotype of APP mice. Mol Neurodegen. 2012;7:1. doi: 10.1186/1750-1326-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levites Y, Jansen K, Smithson LA, Dakin R, Holloway VM, Das P, Golde TE. Intracranial adeno-associated virus mediated delivery of antipan amylodi Aβ, amyloid β40, and amyloid β42 single-chain variable fragments attenuates plaque pathology in amyloid precursor protein mice. J Neurosci. 2006;26:11923–11928. doi: 10.1523/JNEUROSCI.2795-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim J, Miller VM, Levites Y, Jansen West K, Zwiziski GW, Moore BD, et al. BRI2(ITM2b) inhibits Abeta deposition in vivo. J Neurosci. 2008;28:6030–6036. doi: 10.1523/JNEUROSCI.0891-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde Y, et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- 17.Joshi Y, Chu J, Pratico D. Stress hormone leads to memory deficits and altered tau phosphorylation in a mouse model of Alzheimer’s disease. J Alz Dis. 2012;31:167–176. doi: 10.3233/JAD-2012-120328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwakiri M, Mizukami K, Ikonomovic M, Ishikawa M, Abrahamson E, Dekosky S, Asada T. An immunohistochemical study of GABA receptor gamma subunits in Alzheimer’s disease hippocampus: Relationship to neurofibrillary tangle progression. Neuropathology. 2009;29:263–269. doi: 10.1111/j.1440-1789.2008.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gong CX, Iqbal K. Hyperphosphorylation of microtubule-associated protein tau: A promising therapeutic target for Alzheimer disease. Curr Med Chem. 2008;15:2321–2328. doi: 10.2174/092986708785909111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sampson AP. Five lipoxygenase activating protein inhibitors for the treatment of inflammatory disease. Curr Opin Investig Drugs. 2009;10:1163–1172. [PubMed] [Google Scholar]
- 21.Qu T, Manev R, Manev H. 5-Lipoxygenase (5-LOX) promoter polymorphism in patients with early-onset and late-onset Alzheimer’s disease. J Neuropsychiatry Clin Neurosci. 2001;13:304–305. doi: 10.1176/jnp.13.2.304. [DOI] [PubMed] [Google Scholar]
- 22.Chu J, Giannopoulos PJ, Ceballos-Diaz C, Golde T, Pratico D. 5-Lipoxygenase gene transfer worsens memory, amyloid and tau brain pathologies in a mouse model of Alzheimer disease. Ann Neurol. 2012;72:442–454. doi: 10.1002/ana.23642. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem Int. 2011;58:458–471. doi: 10.1016/j.neuint.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 24.Atzori C, Ghetti B, Piva R, Srinivasan AN, Zolo P, Delisle MB, et al. Activation of JNK/p38 pathway occurs in diseases characterized by tau protein pathology and is related to tau phosphorylation but not to apoptosis. J Neuropathol Exp Neurol. 2001;60:1190–1197. doi: 10.1093/jnen/60.12.1190. [DOI] [PubMed] [Google Scholar]
- 25.Savage MJ, Lin YG, Ciallela JR, Flood DG, Scott RW. Activation of c-Jun N-terminal kinase and p38 in an Alzheimer’s disease model is associated with amyloid deposition. J Neurosci. 2002;22:3376–3385. doi: 10.1523/JNEUROSCI.22-09-03376.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun W, Qureshi HY, Cafferty PW, Sobue K, Agarwal-Mawal A, Neufield KD, Paudel HK. Glycogen synthase kinase-beta is complexed with tau protein in brain microtubules. J Biol Chem. 2002;277:11933–11940. doi: 10.1074/jbc.M107182200. [DOI] [PubMed] [Google Scholar]
- 27.Liu SJ, Zhang AH, Li HL, Wang Q, Deng HM, Netzer WJ, et al. Overactivation of glycogen synthase kinase-3 by inhibition of phosphoinositol-3 kinase and protein kinase C leads to hyperphosphorylation of tau and impairment of spatial memory. J Neurochem. 2003;87:1333–1344. doi: 10.1046/j.1471-4159.2003.02070.x. [DOI] [PubMed] [Google Scholar]
- 28.Humbert S, Dhavan R, Tsai L. p39 activates cdk5 in neurons, and is associated with the actin cytoskeleton. J Cell Sci. 2000;113:975–983. doi: 10.1242/jcs.113.6.975. [DOI] [PubMed] [Google Scholar]
- 29.Lee MS, Tsai LH. Cdk5: One of the links between senile plaques and neurofibrillary tangles? J Alz Dis. 2003;5:127–137. doi: 10.3233/jad-2003-5207. [DOI] [PubMed] [Google Scholar]
- 30.Hsiao YH, Kuo JR, Chen SH, Gean PW. Amelioration of social isolation-triggered onset of early Alzheimer’s disease-related cognitive deficit by N-acetylcysteine in a transgenic mouse model. Neurobiol Dis. 2012;45:1111–1120. doi: 10.1016/j.nbd.2011.12.031. [DOI] [PubMed] [Google Scholar]
- 31.Imbesi M, Zavoreo I, Uz T, Sharma RP, Dimitrijevic N, Manev H, Manev R. 5-Lipoxygenase inhibitor MK-886 increases GluR1 phosphorylation in neuronal cultures in vitro and in the mouse cortex in vivo. Brain Res. 2007;1147:148–153. doi: 10.1016/j.brainres.2007.02.012. [DOI] [PubMed] [Google Scholar]