

Abstract
There is substantial practical interest in the mechanism by which the carbonated apatite of bone mineral can be initiated specifically in a matrix. The current literature is replete with studies aimed at mimicking the properties of vertebrate bone, teeth and other hard tissues by creating organic matrices that can be mineralized in vitro, and either functionally substitute for bone on a permanent basis, or serve as a temporary structure that can be replaced by normal remodeling processes. A key element in this is mineralization of an implant with the matrix and mineral disposed in the proper orientations and relationships. This review examines the pathway to crystallization from a supersaturated calcium phosphate solution in vitro, focusing on the basic mechanistic questions concerning mineral nucleation and growth. Since bone and dentin mineral forms within collagenous matricies we consider how the in vitro crystallization mechanisms might or might not be applicable to understanding the in vivo processes of biomineralization in bone and dentin. We propose that the pathway to crystallization from the calcium phosphate supersaturated tissue fluids involves the formation of a dense liquid phase of first-layer bound-water hydrated calcium and phosphate ions in which the crystallization is nucleated. SIBLING proteins and their in vitro analogs such as polyaspartic acids, have similar dense liquid first-layer bound water surfaces which interact with the dense liquid calcium phosphate nucleation clusters and modulate the rate of crystallization within the bone and dentin collagen fibril matrix.
INTRODUCTION
Minerals are almost ubiquitous components of living systems, found in bacteria, plants and animals of all types, in which they serve a multitude of crucial structural and biochemical functions. The vast bulk of minerals found on Earth required utilization of extreme temperatures and pressures for their formation as well as long times for their various transformations. Yet, in contrast to these physical geological processes, biominerals, minerals of biogenic origin, are created at moderate ambient temperatures and pressures, with some crystallizing in unique patterns related to the matrix in which they formed. The objective of many studies in biomimetics has been to produce in vitro, under controlled ambient conditions, that which the biological systems can produce so easily. The current literature is replete with studies aimed at mimicking the properties of bone and other hard tissues by creating matricies that can be mineralized in vitro, and either functionally substitute for bone on a permanent basis, or serve as a temporary structure that can be subsequently replaced by normal remodeling processes. A key element in this is the mineralization of an implant with the matrix and mineral phases disposed in the proper orientations and relationships. The objective of this review is to examine the processes of biomineralization in vitro with respect to the understanding of the mechanism of mineral nucleation and crystal growth within organic matrix frameworks. Although the same mechanism probably relates to all the various biominerals found in nature, here we focus on the formation of the carbonated apatite (cAp)of bone and dentin mineral. Most of the mechanistic data available has been obtained via in vitro experiments. After considering those data, we aim to connect that data to the unique aspects of biomineralization in the more complex in vivo environment as a guide to further studies.
THE BONE MATRIX
The mineralization of bone and dentin takes place within an organized matrix of type I collagen fibers, and it has been clear for many years that the crystalline phase consists of aggregates of individual platy nanocrystals of cAp, oriented and located specifically by the collagen fibril structure [1]. The type I collagen fibril is built from the staggered packing of the individual collagen molecules such that there are periodic gaps or open spaces along the fibril surface, and channels extending through the fibril [2]. The crystals may be found within the individual fibrils, with the same periodicity displayed by the collagen, and along the fibril surfaces in the extra-fibrillar spaces between packed collagen fibrils of the bone and dentin fibers [3]. Many experiments related to mineralization in vitro have indicated that collagen fibrils alone are not able to drive this organized mineralization [1]. However, two recent papers challenge this by proposing that the electrostatic landscape of collagen fibrils makes the collagen a possible heterogeneous nucleator [4] and showing that in vitro conditions can be adjusted to precipitate cAp within the collagen fibril without the use of non-collagenous macromolecular additives [5]. In the early 1960’s [6, 7, 8, 9] we found that mineralized dentin contained some unique (at that time) highly anionic phosphorylated non-collagenous proteins. The major phosphoprotein of dentin was first named “phosphophoryn” on the basis of its high content of serine phosphate residues. That discovery led to studies that showed that such proteins were probably linked to the mineralization process [10–15]. This point was confirmed by the classic radioautographic studies of Weinstock and Leblond [16, 17] which showed that in the rat incisor dentin, the collagen matrix was formed first, while the phosphoprotein (PP) was secreted and deposited directly at the mineralization front, distant from the point of deposition of the collagen. The PP was retained within the dentinal tubules, out of contact with the collagen fibrils, until released at the mineralization front. This point was confirmed and emphasized by Rabie and Veis [18] who used specific gold-labeled anti-collagen and anti-phosphophoryn antibodies to trace the routes of secretion of these two molecules from the cell. It was shown that collagen and PP were secreted in separate secretory granules. Thus, mineralization was considered to require the direct interaction of the PP and collagen to localize and induce apatite mineralization. Since those early days, many studies of the PP and closely related non-collagenous proteins of bone and dentin (now called the SIBLING proteins [19, 20]) and their participation in many aspects of mineralization have been carried out. It is quite clear that the SIBLINGS, including PP, are multifunctional proteins, having signaling properties as well as direct involvement in mineralization. In the following discussion we consider the prime question of how the protein components might direct and possibly regulate crystallization of apatite but first it is necessary to examine the crystallization mechanism in detail.
The crystallization process itself can be considered from several different perspectives: 1) the development of crystallinity in a mineral phase developed directly from a supersaturated solution of the mineral ions; 2) from the perspective of the presence of polymeric (protein) polyions in the mineralizing system and how they might interact in vitro with the free ions or with nanoclusters of Ca and PO4 ions to modulate the mineralization process; 3) from the perspective of the SIBLING proteins and how they might be delivered in vivo to regulate mineralization at specific sites, including the processes of nucleation, crystal growth, crystal morphology and size regulation; and 4) from the perspective of delivery of sequestered, vesicular nanoclusters of Ca and P directly from the cell or the mitochondria to the mineralization front. Very much larger quantities of “biominerals” are produced based on carbonates and silicates throughout the plant and animal kingdoms, and it is likely that some common mechanisms may be applicable in all cases. We will here focus on the calcium phosphates, but keep in mind that many of the basic principles discussed were developed in studies of the formation of carbonate and silicate based minerals.
I. Crystallization from a supersaturated solution of calcium phosphates
Calcium phosphate solutions are complex and the ion species present depend upon the pH and ionic strength. The phosphate species and their equilibrium constants, at 25 C in water, are [21]:
In a solution mixture containing total Ca = 1.5 × 10−3M, total phosphate = 0.9 × 10−3M at pH 7.0 and 25 C in 0.15 M KNO3 these equilibria lead to the concentrations;
Thus, around physiological pH and ionic strength 0.15, the predominant ion species are an essentially equimolar mixture of H2PO4− and HPO42− both of which interact avidly with Ca2+ ion [21]. According to the classical nucleation theory crystal formation from a supersaturated solution of the constituent ions requires local fluctuations in concentrations of the interacting ions. At some critical concentration of those ions their favorable interactions stabilize the ion cluster so that the surface free energy gain by dissociation of the ions in the cluster surface is less than the free energy gain by further addition of ions to the dense cluster or crystalline phase. That is, the process is driven thermodynamically by the difference in free energy between crystalline and solution phases, but is controlled kinetically by the magnitude of the energy barrier of critical size cluster formation, which includes the free energy of formation of a new surface. In simple 1–1 salts the crystallization process can proceed as a homogeneous reaction. The reaction free energy pathway is thus:
The generalized Gibbs equation for the free energy for crystal nucleation, ΔG(n), is given in equation 1 (Illustrated in Fig 1) in which n is the number of particles in the cluster and μ is the chemical potential per particle.
Figure 1.
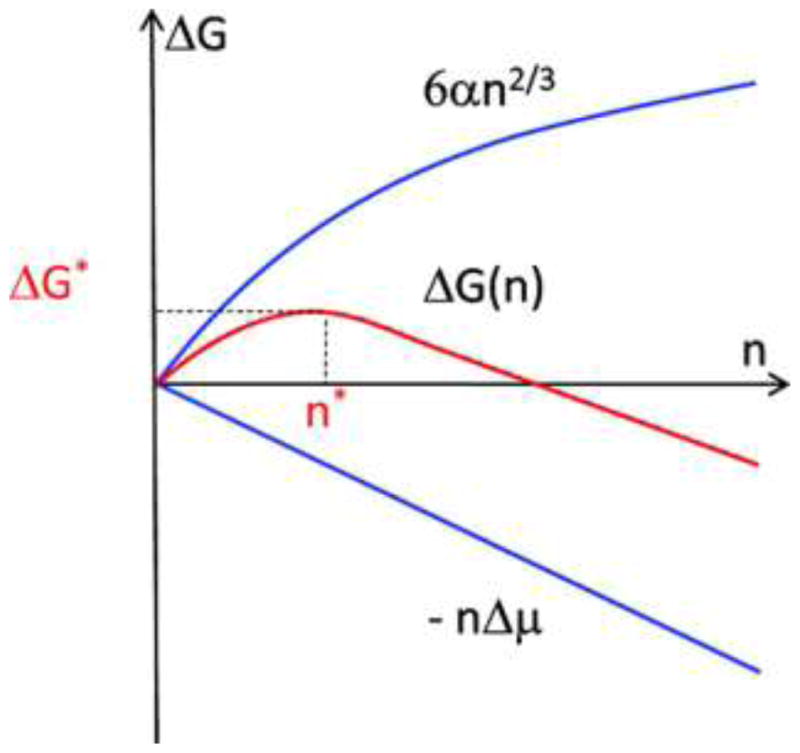
The generalized Gibbs formulation of crystal nucleation, from Equation 1. The upper blue curve is the unfavorable gain in free energy because of creation of the new surface interface, the lower blue curve is the favorable decrease in free energy because the stability of the crystal relative to the initial solution free energy. G* is the critical free energy at cluster size n*, the cluster size at which the probability of release of particles from the surface of the cluster is balanced with the probability cluster growth. When n > n* the cluster will have a greater probability of further growth. From Vekilov [26] with permission.
| (Eqn. 1) |
Thus, the aggregation of the particles from the supersaturated solution to a more stable crystalline phase becomes more favorable as more particles are added to the aggregate. This is countered in the second term which expresses the work required to create the new interface between the supersaturated solution and the crystal aggregate. This second term is problematic since it requires knowledge of the shape of the aggregate, but this is represented and generalized by scaling the surface area to volume to n2/3, taking α as an adjustable factor expressing the averaged interfacial energy per particle in the cluster. We can define the critical cluster size, n*, as the point at which the absolute value of n*Δμ equals 6αn2/3 and hence at ΔGn* = Gc-GSS the probabilities of cluster growth and cluster dissolution are balanced. To become more specific one can assume a particular shape and correct the second term more precisely. In the classical form of this generalized Gibbs equation the shape of the aggregate is assumed to be a sphere and imposes specific geometric boundary conditions. Schmelzer et al. [22] examined the thermodynamic, surface and kinetic parameters for homogeneous nucleation in detail and concluded that the classical Gibbs approach considerably overestimated the work of critical cluster formation but also demonstrated that the generalized Gibbs equation, with the adjustable parameter α, was still applicable. Some specific terms need definition before undertaking further mechanistic interpretation of the nucleation process. These are provided in Table 1 and utilized in the ensuing discussion.
Table 1.
Essential Definitions of Nucleation Intermediate States
| Aggregate: A collection or cluster of ions, molecules and/or particles. |
| Prenucleaction cluster: A cluster of ions, molecules and/or particles, stable enough to exist (resisting decay)but have neither the size and structure sufficient enough to grow into a crystal on its own. |
| Posner cluster: A prenucleation cluster that is an assembly of calcium and phosphate ions with a ratio of 1.5:1, with a size 0.9–1.0nm. |
| Dense Liquid: A prenucleation cluster that exists as a separate amorphous liquid-like precursor phase distinct from either the supersaturated solution or crystal phases. The dense liquid has a defined interface, and is more dense than the surrounding solution, but less dense than the crystal form. The dense liquid is a hydrated aggregate and can be composed of ions, molecules, and/or prenucleation clusters. The water present is bound as hydration layers. The dense liquid is larger than a crystal nucleus. Crystal nuclei can form within the dense liquid. |
| Critical Cluster: A cluster of ions, molecules and/or particles of sufficient size (n = n*) and structure to have an equal probability of growth or dissolution |
| Nucleation cluster: A stable cluster of ions, molecules and/or particles of sufficient size (n ≥ n*+1) and structure to continue to grow rather than decay. Has a greater probability of growth over decay. This represents the nuclei of a crystal phase. |
The next interpretative advance in understanding crystal nucleation was by ten Wolde and Frenkel [23] who introduced the concept that the crystallization could more readily take place within the prenucleation cluster rather than directly from the supersaturated mother liquor. Since the initial cluster is presumed to be disordered the initial prenucleation clusters can be considered as “dense liquid” (DL) phases [23, 24] and it has been suggested that crystallization may take place within that phase. Vekilov [25, 26] has provided a visualized description of the formation of the dense phase, which we have further elaborated upon as shown in Figure 2. The red dashed line (ΔG1*) in Fig. 2a depicts the formation of transient, unstable, unstructured aggregates which readily disassociate when n < n*. When fluctuations take place that lead to clusters larger than the critical value crystals may form (green dash-dot line). The diagonal arrow represents the path of cluster formation directly from solution (the solid line in Fig. 2a ) and corresponds to the free energy landscape in Fig. 1 (dashed curves in Fig. 2c) whereas the heavy green dash-dot line in Fig 2a follows the free energy landscapes in Fig. 2c. Fig. 2b depicts the formation of the DL cluster, and the subsequent growth of the crystal within the DL aggregate. Two possible two step mechanisms describing the free energy reaction coordinate are also shown in Fig. 2c. In both upper and lower reaction coordinate schemes of Fig. 2c the free energy barrier to critical cluster n1* formation is the same, ΔG1*, and further aggregation continues to form a DL phase. If the DL is more ordered and marginally more stable than the initial supersaturated solution the lower reaction coordinate applies, then the DL intermediate will be long-lived. Regardless of the stability of the DL compared to the supersaturated solution, the development of crystallinity within the DL will be governed by the value of ΔG2* < ΔG1*.
Figure 2.

Schematic illustration of the two-step mechanism of nucleation of crystals, modified from work presented by Vekilov [25,26]. In this two step mechanism a dense liquid cluster forms, and a crystal nucleus may form inside the cluster. (a) Microscopic viewpoint shows crystal formation in the plane of two order parameters (Concentration and Structure) inspired by the work of ten Wolde and Frenkel [23, 24]; (b) macroscopic viewpoint of events along the energy pathways (solid lines) depicted in (c). (i) The supersaturated solution. (ii) The formation of the dense liquid depicted in most of (a). (iii) The formation of the critical cluster for the crystal at n2* within the dense liquid, also shown in (a). (iv) The creation of the nucleation cluster and subsequent growth of the crystal. (v) The fully formed crystal. (c) The pathway for the change in free-energy ΔG, along two possible versions of the two-step nucleation mechanism. If dense liquid is unstable and ΔG0 DL > GSS(ΔG0DL standard free energy of formation of dense liquid phase), the upper curve applies; if dense liquid is stabilized with the introduction of a foreign interface, ΔG0 DL < 0, the lower curve applies. ΔG1* is the barrier for formation of a cluster of dense liquid, ΔG2* is the barriegr for a formation of a crystalline nucleus inside the dense liquid.
To move from these generalizations to the mechanism of crystal nucleation from a prenucleation, DL precursor, we need to consider the experimental verification of the idea of the prenucleation cluster in solution as part of a homogeneous nucleation process. The best direct demonstration of this came from studies of the calcium carbonate system. Wolf et al. [27] suspended a droplet of a calcium carbonate solution between a piezoelectric vibrator that generated an acoustic wave and a concentrically adjusted sonic reflector. The droplet could be held in place without any extraneous constraints other than the droplet air-water interface. The concentration of the droplet increased as the solvent evaporated and the state of the solute was followed by in situ measurement of the wide angle x-ray scattering determination of crystallinity over time. In parallel transmission electron microscopy (TEM) of droplets collected at suitable time points, along with scanning electron microscopy (SEM) and cryogenic SEM, data led to the conclusion that “in this completely contact-free system a homogeneous formation of CaCO3 proceeded via an amorphous liquid-like state, unambiguously without artifacts.” These data demonstrated the development of nanoscale liquid-like cluster formation in pure calcium carbonate solution, free of any organic polyions or proteins, showing unequivocally that metastable liquid-like cluster formation generated by density fluctuations in the inorganic ions in the bulk phase reflected the direct interactions of the inorganic ions involved. The intervention of organic polyions or other heterogeneous particles or surfaces could perhaps alter kinetics of liquid-like prenucleation complex formation, but clearly the heterogeneous factors are not required for crystal nucleation. Mahamid et al. [28] and Beniash et al. [29] proposed that amorphous calcium phosphate precursor phases were involved, respectively, in mineralizing zebra fish fin bone and tooth enamel formation. Much earlier Posner [30, 31] had found that that biological apatites had an amorphous component, and proposed that a cluster of Ca and PO4 ions with composition Ca9(PO4)6 and a diameter of 0.9 to 1.0 nm was the stable prenucleation cluster.
Recently, Dey et al. [32] have shown that stable prenucleation clusters of calcium phosphate of the size predicted as Posner clusters, can be observed in a simulated body fluid (SBF [33]) at 37°C. The clusters began to aggregate and after 24 h in the presence of a monolayer of arachidic acid, the clusters aggregated and densified on the monolayer. Initially, selected area electron diffraction (SAED) showed the clustered calcium phosphates to be amorphous as ACP, but ultimately the SAED indicated that crystallinity developed with an orientation determined by the monolayer surface. Thus, the clustering is a multistep process that begins as a homogeneous reaction. Dey et al. [32] proposed that “the presence of the nucleating surface induces structural and compositional changes that enable the denser packing of the clusters and their subsequent fusion to form ACP” and ultimately apatitic crystals. However, crystallization could also occur in the absence of heterogeneous nucleators.
II. Crystallization enhancement by heterogeneous surfaces and polymeric additives
In a companion paper from the same laboratory as the Dey work, Nudelman et al. [34] directly examined the potential role of collagen fibrils as a templating surface for the deposition of amorphous clusters or ACP directly on the collagen to nucleate apatitie crystal formation. They examined the in vitro mineralization of reconstituted collagen fibrils isolated from horse tendon. This is a collagen which does not normally, in vivo, undergo mineralization and thus should be free of mineralization related non-collagenous SIBLING proteins. In the absence of any additives other than SBF, deposition of needles of apatite was random along the surfaces of in vitro formed fibrils. When polyaspartic acid (polyAsp), used as a model for the SIBLING proteins, was added to the mineralizing solution, calcium phosphate mineral was found on the surface of the fibrils after 24h and the mineral deposits were loosely associated with the collagen gap and overlap zones. Cryo energy dispersive X-ray spectroscopy and low-dose selected-area electron diffraction (LDSAED) showed the presence of ACP. Within 48 hours, the ACP was partly converted to apatite crystals, and these developed in 72 h to elongated hydroxyapatite (HAp) crystals, but still within a bed of ACP, comparable to typical bone apatite. Remarkably, however, if the polyAsp was first absorbed on the collagen, before the collagen was exposed to the mineralizing buffer, no mineralization took place except on the surface. The in vitro mineralization with apatite deposition within the fibrils was found only when the polyAsp and SBF solution were mixed before placement on the collagen fibrils. This directly implicated the participation of the SIBLING analog polyAsp in the formation of cAp prenucleation clusters, or some other aggregate, and their interaction with the collagen.
The formation of the prenucleation cluster [35, 36] and its subsequent crystal growth in the presence of anionic polymer has been treated as a Polymer-Induced Liquid-Precursor (PILP) process, and a very complete discussion of the phenomenology of the PILP process has been given recently by Gower [36], particularly focusing on the in vitro production of biomimetic minerals. Although the nature of the PILP process is clear, the mechanism of enhancement of the nucleation by the participation of the inducing polymer, for example the polyAsp in the heterogeneous nucleation process described above, has not been described in equal depth. How this is accomplished has great importance in the actual or real biological systems in which biomineralization takes place. The questions posed above as to where the prenucleation clusters form, the specificity of placement of the mineral phase in the collagen matrix, the regulation of crystal growth within the fibrous matrix, are general to all systems, but in each case of mineral deposition we know that bone, dentin and even mineralizing tendon utilize different members of the SIBLING family proteins to promote and/or inhibit mineral deposition and produce bones and teeth with near equivalent physical properties, but with different crystal sizes and arrangements tuned to the required mechanical and dynamic properties of the particular tissue involved.
Nudelman et al. [34] have argued that the placement of the prenucleation clusters on the collagen and their role in moving the mineral into the collagen fibrils as well as leading to mineralization along the fibril surfaces, is related to the observation that the presence of the SIBLINGS and acidic non-collagenous proteins slows rather than accelerates the precipitation of the calcium phosphate prenucleation clusters. It has been observed many times that the SIBLING proteins may act to accelerate or inhibit mineralization depending on the factors of concentration and temperature. From this point of view, the SIBLING proteins may all be considered as mineralization inhibitors. The duality of activity of these proteins depending on the conditions and context illustrates their role as regulators of crystal nucleation/growth kinetics. It is certainly clear from the work mentioned earlier [16, 17, 18] odontoblasts do sequester dentin phosphophoryn (DPP) from the collagen of the unmineralized predentin, and that the DPP is delivered directly to the mineralization front so that the mixing of non-collagenous polyions and collagen is closely controlled.
III. SIBLING proteins and their interaction with collagen fibrils
The SIBLINGs are among the class of proteins now designated as Intrinsically Disordered Proteins (IDP) [37], that is, in solution they are flexible polypeptides with no long range regular structures although they can have limited sequences exhibiting, for example, local α or β conformations, and further structural order may be induced by interaction with other proteins. Electron micrographs of 0.1 μg/mL of DPP in 10 mM Ca2+ at pH 7.5 showed the DPP as roughly globular structures of about 16–18 nm in diameter [38]. When the same preparations were added to EM grids which had turkey tendon collagen fibers deposited on them, the DPP decorated the surface of the fibrils with similar sized DPP structures, distributed along the fibril with perhaps 75% of the collagen bound DPPs associated with the fibril “e”-bands [38]. Thus it was suggested that this e-band position, corresponding to the collagen fibril gap region, was a favored site for DPP binding. Further EM studies of the collagen-DPP interaction products in ammonium formate buffers without Ca2+ ions and using soluble monomeric collagen and purified DPP at different mixing ratios showed that there was a preferred DPP binding site near the amino-terminal region of the collagen monomers. This specific binding was differentially detectable only at very high collagen/DPP ratios [39, 40]. As the concentration of DPP was increased relative to the soluble collagen, additional DPP particles were seen bound non-specifically along each single collagen molecule. These collagen monomers +DPP readily aggregated into asymmetric fibrillar structures [41]. The intensity of binding at a molar ratio of 1 to 1 rather than at electrostatic equivalence suggested that a non-electrostatic binding was involved at the primary specific interaction site, emphasizing the probable specificity in DPP sequence responsible for its primary collagen localization. Though polyAsp can do the in vitro job of nucleating cAp, the evidence is strong that, although the sequence of DPP contains approximately 40% Asp residues, the phosphorylation of the Ser residues of DPP ( 50% of the amino acids) is crucial for its function in mediating mineralization [42] and, most recently, it has been shown that the introduction of a DPP cDNA into a non-mineralizing fibroblast cell leads to expression and secretion of phosphorylated DPP and the induction of HAp mineralization in the fibroblast cultures [43].
IV. Delivery of Ca and PO4 ions to the mineralization front
The in vivo situation is much more complex than the in vitro model experiments suggest, and relatively few biomineralization discussions include the problem of the Ca and PO4 delivery to the mineralization front, although such discussions have appeared since the pioneering studies of Bonnucci, [44, 45], Anderson [46, 47], Lehninger [48] and Greenwalt et al. [49] regarding the role of the mitochondria in taking in large amounts of Ca and phosphate ions, accumulating them and delivering calcium phosphate in membrane bound vesicles (Matrix Vesicles, MV) to the extracellular spaces of mineralizing cartilage and bone. The mechanisms by which vesicles may be broken open, and their mineral contents transferred and localized to the collagen are still a subject of current study and debate [50]. There is, indeed, abundant evidence that the mineral containing intracellular vesicles do exist, and their contents in different situations contain amorphous calcium phosphate, or apatitic needle-like crystals. The questions which remain unresolved deal with the mechanisms by which the mineral is transferred from the mitochondria to the mineralizing matrix, and then reordered, in essence dissolved and recrystallized, on the collagen fibril matrix on the fibril surfaces, or within the fibrils themselves.
V. Transiting from the in vitro to the in vivo situation
All of the preceeding discussion has examimed the mineral ions and other components in idealized and controlled mineralization conditions in vitro. It is worthwhile to consider these same factors in the in vivo situation and determine how that might modulate the various component interactions. Particularly first from the perspective of the cytoplasm and ECM themselves, and the crowding of the macromolecules within them. Different types of cells vary in macromolecular content, over the range of 17 to 35% by weight, and the cell water content is divided between two phases, about half as the macromolecular/protein-bound water of hydration and the other half “bulk” or free water [51]. Other components of the cell, such as the inorganic cations and anions in aqueous solution in the cell also have layers of bound water of hydration that are more ordered than in the free water. Thus, one must specifically ask about the role of the hydration layers in the aggregation of the ions into clusters, and their continued densification during crystallization, as well as the question of how the ion clusters are localized to the collagen matrix. The unusual properties of water are directly related to the fact that at ambient temperatures free (bulk) water molecules form extensive hydrogen bonded networks, albeit with energies of only a few kcal/mole (H-bond between two waters ~4.3 kcal/moL, van der Waals interaction ~1 kcal/mol, ionic bond 4–7 kcal/mol, a C-C bond ~84.0 kcal/mol). In liquid water the time scale of randomization of the networks by the rapid formation and dissociation of hydrogen bonds is on the order of approximately 2.6 ps. Fayer [52] has shown that the water orientational relaxation requires H-bond rearrangements, and that these relaxations are slowed by the presence of interfaces of all sorts, from hydrophobic surface films, to organic macromolecules (regardless of being charged or uncharged), and in the structure of the hydration layers around inorganic cations and anions. By studying the state of the water molecules in nano-confined systems, created as the water held within spherical reverse micelles, or water held between the layers of water confined between lipid monolayers, it is clear that the first shell of bound water has substantially different properties than that in the bulk water, and much longer relaxation times, on the order of 18 ps, although liquid water clearly has no long range stability.
The first hydration shell of 6 H2O molecules coordinated to a Ca2+ ion plus the next shell of 12 H2O molecules is depicted in Figure 3 for the most stable conformation of (Ca(H20)6)2+(H20)12 ) [53]. The oxygen atoms of the first shell water molecules are at 2.35 Å from the central Ca atom, whereas the second shell O atoms are at 2.79 Å from the first shell O forming a significantly less stable H-bonded network. The relaxation time for the second shell water is 2 to 3 times longer than the bulk water relaxation time, while the relaxation time for the first shell water can be upwards of 50 times longer than the bulk water [54]. In addition to the stability difference, the water layers shield the electrostatic potential of the Ca ion. The water of the first hydration shell of the phosphate ion is similarly more tightly held than the second shell water with a retention time about 2 times longer than the bulk water, Figure 4 [55], but the first shell arrangement is more complicated. Two types of water are evident, a strongly bonded P=Op- H-Ow of a water molecule in a linear arrangement, equivalent for each phosphate Op (Fig. 4a), and set of interstially located waters that appear to bridge between two Op resulting in weaker bonds to the water hydrogens with a length of 2.0–2.2 Å. In total the first shell of bound water is comprised of 12 water molecules (Fig. 4b), although molecular dynamics predicted that on average, 13 water molecules would be included [55]. The polarity of the H-bonded bound water around phosphate is opposite to that of the hydration layer around the Ca ion. It is interesting that in the simulations, all 4 phosphate O’s appear to be equivalent in bonding to water. Similar water binding and polarity organization would be found in the arrangement of first layer water in organic phosphates on phosphoserine and on carboxyl groups on Asp or Glu in polypeptides. Thus phosphorylated polypeptides and proteins such as phosphophoryn, with sequences such as (DS*S*)n or (DS*DS*)m (S* = phosphorylated serine) should, even for IDP, exhibit structured ionic patches[56] of hydrated surface with similar strongly bound first shell water. At the nano-scale where the initial nucleation clusters are forming all water molecules are not equivalent and the water should be treated as a direct reactant, as important as the Ca or phosphate ions or protein side chains.
Figure 3.

Structured water around a Ca2+ ion. The central Ca ion is indicated in yellow, the 6 oxygen atoms of the first water shell, coordinated directly to the Ca are in red. The oxygen atoms of the 12 second shell waters are in green. The polarity of the water molecules is quite well fixed and reduces the effective charge on the central Ca ion in so far as long range electrostatic interactions between ions is considered. Pavlov et al. [53], with permission.
Figure 4.
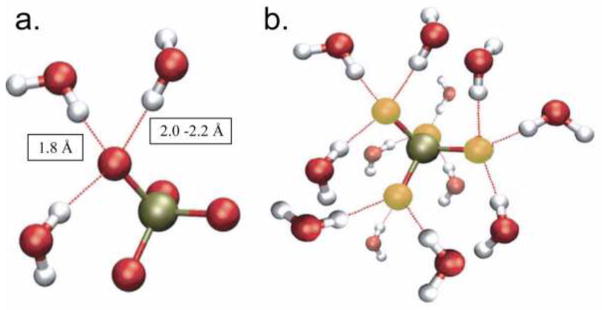
Structured water around a PO43− ion. (a) The central P atom is indicated in gold, 3 waters of the first shell are shown coordinated directly to the phosphate molecule. (b) The central P atom is indicated in gold with the oxygen atoms on the phosphate indicated in yellow. The 12 waters of the first shell are illustrated. (a) From Pribil et al. [55] with permission. (b) Is our construction of the complete first shell, as suggested [55], with the understanding that the orientations of the non-axial waters will be affected by the waters of the second shell.
VI. The Nucleation Mechanism
As depicted schematically in Figures 3 and 4 and the related discussions, the Ca and phosphate ions in the supersaturated aqueous solution are now seen as each carrying closely bonded water, differentiated from the bulk water in stability. We propose that, as the electrostatic interactions attract the oppositely charged ions (such as Ca2+ and PO43−), the interaction is modulated by the restrictions on the mobility of the bound first layer water. Thus, the initial aggregates remain in a solvated state without long range order, but now in the DL state with the free, 2nd shell water released. The enhanced stability of the DL water allows time for the final steps of densification to establish order and crystallinity from the nucleation clusters. From this perspective it is easy to see why and how the introduction of a stable but compatible polymeric polyion interface also having a restricted water layer, can be so important in modulating and /or guiding the final location and rate of formation of the final stable crystals.
There are many unanswered questions that need to be examined in the light of the mechanism proposed above. The specificity of the nucleation must reside in the nature of the interfaces presented to the physiological, supersaturated calcium phosphate solution. Obviously, heterogeneous nucleation can be enhanced by the presence of interfaces which allow the aggregation and densification of the liquid-like prenucleation clusters. The polymeric polyions, as exemplified by the SIBLING proteins of the ECM, that are both interactive with the Ca ions and localize their presence to the matrix fibrils need to be examined for their dual roles of localization of the crystals and control of the rate of deposition within the structural matrix. The effect of the presence of other ions, such as Mg2−, HCO3−, CO32−, should be studied with respect to the rate of formation of the prenucleation clusters, and their effect on the stepwise densification to the DL state. The question of the role of the collagen fibrils, and the mechanism of entry of the crystal nuclei or the DL phase is an important target for further studies. Finally, the basic step of the delivery of the calcium and phosphate ions into the extracellular space needs to be reexamined. If the matrix vesicle mechanism is important for delivery of the contents of partitioned vesicles into the ECM, the question of how the mineral is transferred from a MV to the inner regions of the bone and dentin collagenous matrices needs careful analysis.
Acknowledgments
We are pleased to acknowledge the continuing support of National Institutes of Health, National Institute of Dental and Craniofacial Research, Grant R01-DE01374 for all of the studies cited from our laboratory.
Footnotes
The authors have stated that they have no conflict of interest.
References
- 1.Veis A. Mineralization in organic matrix frameworks. In: Dove PM, DeYoreo JJ, Weiner S, editors. Biomineralization. Vol. 54. 2003. pp. 249–289. Reviews in Mineralogy & Geochemistry. [Google Scholar]
- 2.Landis WJ, Song MJ, Leith A, McEwen L, McEwen B. Mineral and organic matrix interaction in normally calcifying tendon visualized in three dimensions by high voltage electron microscopic tomography and graphic image reconstruction. Journal of Structural Biology. 1993;110:39–54. doi: 10.1006/jsbi.1993.1003. [DOI] [PubMed] [Google Scholar]
- 3.Orgel JPRO, San Antonio JD, Antipova O. Molecular and structural mapping of collagen fibril interactions. Connect Tissue Research. 2011;52:2–17. doi: 10.3109/03008207.2010.511353. [DOI] [PubMed] [Google Scholar]
- 4.Silver FH, Landis WJ. Deposition of apatite in mineralizing vertebrate extracellular matrices: A model of possible nucleation sites on type I collagen. Connect Tissue Research. 2011;52:242–254. doi: 10.3109/03008207.2010.551567. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Azaïs T, Robin M, Vallée A, Catania C, Legriel P, Pehau-Arnaudet G, Babonneau F, Giraud-Guille M-M, Nassif N. The predominant role of collagen in the nucleation, growth, structure and orientation of bone apatite. Nature Materials. 2012;11:724–733. doi: 10.1038/nmat3362. [DOI] [PubMed] [Google Scholar]
- 6.Veis A, Schlueter R. Presence of phosphate-mediated cross-linkages in hard tissue Collagens. Nature. 1963;197:1204. doi: 10.1038/1971204a0. [DOI] [PubMed] [Google Scholar]
- 7.Veis A, Schlueter RJ. The macomolecular organization of dentine matrix collagen. I. characterization of dentine collagen. Biochemistry. 1964;3:1650–1656. doi: 10.1021/bi00899a009. [DOI] [PubMed] [Google Scholar]
- 8.Schlueter RJ, Veis A. The macromolecular organization of dentine matrix collagen. II. periodate cegradation and carbohydrate cross-linking. Biochemistry. 1964;3:1657–1665. doi: 10.1021/bi00899a010. [DOI] [PubMed] [Google Scholar]
- 9.Veis A, Perry A. The phosphoprotein of the dentin matrix. Biochemistry. 1967;6:2409–2416. doi: 10.1021/bi00860a017. [DOI] [PubMed] [Google Scholar]
- 10.Veis A, Spector A, Carmichael DJ. The organization and polymerization of bone and dentin collagen. Clinical Orthopaedics and Related Research. 1969;66:188–211. [PubMed] [Google Scholar]
- 11.Dimuzio MT, Veis A. The biosynthesis of phosphophoryns and dentin collagen in the continuously erupting rat incisor. The Journal of Biological Chemistry. 1978;253:6845–6852. [PubMed] [Google Scholar]
- 12.Dimuzio MT, Veis A. Phosphophoryns - major non-collagenous proteins of the rat incisor dentin. Calcified Tissue Research. 1978;25:169–178. doi: 10.1007/BF02010765. [DOI] [PubMed] [Google Scholar]
- 13.Lee SL, Veis A, Glonek T. Dentin phosphoprotein: an extracellular calcium binding protein. Biochemistry. 1977;16:2971–2979. doi: 10.1021/bi00632a026. [DOI] [PubMed] [Google Scholar]
- 14.Veis A, Sharkey M, Dickson IR. Non-collagenous proteins of bone and dentin extracellular matrix and their role in organized mineral deposition. In: Wasserman RH, et al., editors. Calcium binding proteins and calcium function. Elsevier North-Holland, Inc; 1977. pp. 409–418. [Google Scholar]
- 15.Lee SL, Veis A. Studies on the structure and chemistry of dentin collagen-phosphophoryn covalent complexes. Calcified Tissue Research. 1980;31:123–134. doi: 10.1007/BF02407173. [DOI] [PubMed] [Google Scholar]
- 16.Weinstock M, Leblond CP. Radioautographic visualization of the deposition of a phosphoprotein at the mineralization front in the dentin of the rat incisor. The Journal of Cell Biology. 1973;56(3):838–45. doi: 10.1083/jcb.56.3.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weinstock M, Leblond CP. Synthesis, migration, and release of precursor collagen by odontoblasts as visualized by radioautography after (3H)proline administration. The Journal of Cell Biology. 1974;60(1):92–127. doi: 10.1083/jcb.60.1.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rabie AM, Veis A. An immunocytochemical study of the routes of secretion of collagen and phosphophoryn from odontoblasts. Connective Tissue Research. 1995;31:197–209. doi: 10.3109/03008209509010811. [DOI] [PubMed] [Google Scholar]
- 19.Fisher LW, Torchia DA, Fohr B, Young MF, Fedarko NS. Flexible structures of SIBLING proteins, bone sialoprotein, and osteopontin. Biochemical Biophysical Research Communications. 2001;280:460. doi: 10.1006/bbrc.2000.4146. [DOI] [PubMed] [Google Scholar]
- 20.Fisher LW, Fedarko NS. Six genes expressed in bones and teeth encode the current members of the SIBLING family of proteins. Connective Tissue Research. 2003;44:33. [PubMed] [Google Scholar]
- 21.Feenstra TP. Doctoral Dissertation. University of Utrecht; Utrecht, The Netherlands: 1980. The initial stages in the formation of calcium and strontium phosphates from supersaturated solutions: A study of induction times; pp. 32–33. [Google Scholar]
- 22.Schmelzer JW, Boltachev GSh, Baidakov VG. Classical and generalized Gibbs’ approaches and the work of critical cluster formation in nucleation theory. Journal of Chemical Physics. 2006;124:194503. doi: 10.1063/1.2196412. [DOI] [PubMed] [Google Scholar]
- 23.Ten Wolde PR, Frenkel D. Enhancement of crystal nucleation by critical density fluctuations. Science. 1997;277:1975–1978. doi: 10.1126/science.277.5334.1975. [DOI] [PubMed] [Google Scholar]
- 24.Talanquer V, Oxtoby DW. Crystal nucleation in the presence of a metastable critical point. Journal of Chemical Physics. 1998;109:223–227. [Google Scholar]
- 25.Vekilov PG. Dense liquid precursor for the nucleation of ordered solid phases from solution. Crystal Growth and Design. 2004;4:671–685. [Google Scholar]
- 26.Vekilov PG. Nucleation. Crystal Growth and Design. 2010;10:5007–5019. doi: 10.1021/cg1011633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolf S, Leiterer J, Kappl M, Emmerling F, Tremel W. Early homogenous amorphous precursor stages of calcium carbonate and subsequent crystal growth in levitated droplets. Journal of the American Chemical Society. 2008;130:12342–12347. doi: 10.1021/ja800984y. [DOI] [PubMed] [Google Scholar]
- 28.Mahamid J, Sharir A, Addadi L, Weiner S. Amorphous calcium phosphate is a major component of the forming fin bones of zebrafish: Indications for an amorphous precursor phase. Proceedings of the National Academy of Sciences. 2008;105:12748–12753. doi: 10.1073/pnas.0803354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beniash E, Metzler RA, Lam RS, Gilbert PU. Transient amorphous calcium phosphate in forming enamel. Journal of Structural Biology. 2009;166:133–143. doi: 10.1016/j.jsb.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Posner AS. Crystal chemistry of bone mineral. Physiological Reviews. 1969;49:760–792. doi: 10.1152/physrev.1969.49.4.760. [DOI] [PubMed] [Google Scholar]
- 31.Posner AS, Betts F. Synthetic amorphous calcium phosphate and its relation to bone mineral structure. Accounts of Chemical Research. 1975;8: 273–281. [Google Scholar]
- 32.Dey A, Bomans PHH, Muller FA, Will J, Frederik PM, de With G, Sommerdijk NAJM. The role of prenucleation clusters in surface-induced calcium phosphate crystallization. Nature Materials. 2010;9:1010–1014. doi: 10.1038/nmat2900. [DOI] [PubMed] [Google Scholar]
- 33.Müller L, Müller F. Preparation of SBF with different HCO3- cotent and its influence on the composition of biomimetic apatites. Acta Biomaterialia. 2006;2:181–189. doi: 10.1016/j.actbio.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 34.Nudelman F, Pieterse K, George A, Bomans PHH, Friedrich H, Brylka LJ, Hilbers PAJ, de With G, Sommerdijk NAJM. The role of collagen in bone apatite formation in the presence of hydroxyapatite nucleation inhibitors. Nature Materials. 2010;9:1004–1009. doi: 10.1038/nmat2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gebauer D, Völkel A, Cölfen H. Stable Prenucleation Calcium Carbonate Clusters. Science. 2008;322:1816–1822. doi: 10.1126/science.1164271. [DOI] [PubMed] [Google Scholar]
- 36.Gower L. Biomimetic model systems for investigating the amorphous precursor pathway and its role in biomineralization. Chemical Reviews. 2008;108:4551–4627. doi: 10.1021/cr800443h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uversky VN. What does it mean to be natively unfolded? European Journal of Biochemistry. 2002;269:2–12. doi: 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 38.Traub W, Jodaikin A, Arad T, Veis A, Sabsay B. Dentin phosphophoryn binding to collagen fibrils. Matrix. 1992;12:197–201. doi: 10.1016/s0934-8832(11)80062-4. [DOI] [PubMed] [Google Scholar]
- 39.Dahl T, Sabsay B, Veis A. Type I collagen-phosphophoryn interactions: Specificity of the monomer-monomer binding. Journal of Structural Biology. 1998;123:162–168. doi: 10.1006/jsbi.1998.4025. [DOI] [PubMed] [Google Scholar]
- 40.Veis A, Dahl T, Sabsay B. The specificity of phosphophoryn-collagen I interactions. In: France Vittel, Goldberg M, Robinson C, Boskey A., editors. Proc 6th Int Conf Chemistry & Biology of Mineralized Tissues. Orthopaedic Research Society; Rosemont, IL: 2000. pp. 169–173. [Google Scholar]
- 41.Dahl T, Veis A. Electrostatic interactions lead to the formation of asymmetric collagenphosphophoryn aggregates. Connective Tissue Research. 2003;44 (Suppl 1):206–213. [PubMed] [Google Scholar]
- 42.He G, Ramachandran A, Dahl T, George S, Schultz D, Cookson D, Veis A, George A. Phosphorylation of phosphophoryn is crucial for its function as a mediator of biomineralization. The Journal of Biological Chemistry. 2005;280:33109–33114. doi: 10.1074/jbc.M500159200. [DOI] [PubMed] [Google Scholar]
- 43.Sfeir C, Lee D, Li J, Zhang X, Boskey AL, Kumta PN. Expression of phosphophoryn is sufficient for the induction of matrix mineralization by mammalian cells. The Journal of Biological Chemistry. 2011;286:20228–20238. doi: 10.1074/jbc.M110.209528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonucci E. Fine structure of early cartilage calcification. Journal of Ultrastructure Research. 1967;20:33–50. doi: 10.1016/s0022-5320(67)80034-0. [DOI] [PubMed] [Google Scholar]
- 45.Bonucci E. Calcified tissue: from microstructures to nanoparticles to chemistry European. Journal of Histochemistry. 2005;49:1–10. doi: 10.4081/921. [DOI] [PubMed] [Google Scholar]
- 46.Anderson HC, Matsuzawa T, Sajdera SW, Ali SY. Membranous particles in calcifying cartilage matrix. Transactions of the New York Academy of Sciences. 1970;32:619–30. doi: 10.1111/j.2164-0947.1970.tb02737.x. [DOI] [PubMed] [Google Scholar]
- 47.Anderson HC. Electron microscopic studies of induced cartilage development and calcification. The Journal of Cell Biology. 1967;35:81–101. doi: 10.1083/jcb.35.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lehninger A. Mitochondria and calcium ion transport. Biochemical Journal. 1970;119:128–138. doi: 10.1042/bj1190129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Greenwalt JW, Rossi CS, Lehninger AL. Effect of active accumulation of calcium and phosphate ions on the structure of rat liver mitochondria. The Journal of Cell Biology. 1964;23:21–38. doi: 10.1083/jcb.23.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boonrungsiman S, Gentleman E, Carzaniga R, Evans ND, McComb DW, Porter E, Stevens MM. The role of intracellular calcium phosphate in osteoblast-mediated bone apatite formation Proceedings of the national Academy of Sciences (on line) 2012 doi: 10.1073/pnas.1208916109. www.pnas.org/gci/doi/10.1073/pnas.120891609. [DOI] [PMC free article] [PubMed]
- 51.Fulton AB. How crowded is the cytoplasm? Cell. 1982;30:345–347. doi: 10.1016/0092-8674(82)90231-8. [DOI] [PubMed] [Google Scholar]
- 52.Fayer MD. Dynamics of water interacting with interfaces, molecules and ions. Accounts of Chemical Research. 2012;45:3–14. doi: 10.1021/ar2000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pavlov M, Seigbahn PEM, Sandstrom M. Hydration of Beryllium, Magnesium, Calcium and Zinc ions using density functional theory. Journal of Physical Chemistry A. 1998;102:219–228. [Google Scholar]
- 54.Richens DT. The Chemistry of Aqua Ions. John Wiley & Sons; Chichester, England: 1997. [Google Scholar]
- 55.Pribil AB, Hofer TS, Randolf BR, Rode BM. Structure and dynamics of phosphate ion in aqueous solution: An Ab initio QMCF Study. Journal of Computational Chemistry. 2008;29:2330–2334. doi: 10.1002/jcc.20968. [DOI] [PubMed] [Google Scholar]
- 56.George A, Bannon L, Sabsay, Dillon JW, Malone J, Veis A, Jenkins NA, Gilbert DJ, Copeland NG. The carboxyl-terminal domain of phosphophoryn contains unique extended triplet amino acid repeat sequences forming ordered carboxyl-phosphate interaction ridges that may be essential in the biomineralization process. The Journal of Biological Chemistry. 1996;271:32869–32873. doi: 10.1074/jbc.271.51.32869. [DOI] [PubMed] [Google Scholar]