

Abstract
Chronic kidney disease (CKD) is epidemic around the world and desparately needs new therapies. Peritubular capillary (PTC) rarefaction, along with interstitial fibrosis and tubular atorophy, is one of the major hallmarks of CKD and predicts renal outcome in patients with CKD. PTC endothelial cells (ECs) undergo apoptosis during CKD, leading to capillary loss, tissue hypoxia, and oxidant stress. Although the mechanisms of PTC rarefaction are not well understood, the process of PTC rarefaction depends on multiple events that happen during CKD. These events, which lead to an antiangiogenic environment, include deprivation of EC survival factors, increased production of vascular growth inhibitors, malfunction of ECs, dysfunction of endothelial progenitor cells, and loss of EC integrity via pericyte detachment from vasculature. In this review, we focus on major factors regulating angiogenesis and EC survival and describe roles of these factors in PTC rarefaction during CKD and possible therapeutic applications.
Keywords: endothelial cell, angiogenesis, angiogenic growth factors, CKD, pericytes
Introduction
Chronic kidney disease (CKD) affects one in every seven adults in the United States, and its prevalance appears to be 10–13% around the world. CKD is a leading cause of death due to premature cardiovascular diseases [1–3]. While a variety of conditions can lead to CKD, the final common pathway of renal destruction involves interstitial fibrosis, tubular atrophy, and peritubular capillary (PTC) rarefaction. Although numerous published studies have investigated the mechanisms of renal fibrosis, the mechanisms of PTC rarefaction are not well understood. Therefore, in this review we focus on PTC rarefaction. Based on several studies in human CKD and animal models of CKD, it is known that PTCs disappear in association with progressive interstitial fibrosis and tubular atrophy. Although the sequence of events connecting PTC loss to fibrosis and impaired tubular function is poorly characterized, it has been suggested that interstitial hypoxia due to arteriolar vasoconstriction and/or PTC regression is a primary event in CKD [4]. In this review, we describe (1) the contribution of PTC rarefaction to CKD progression, (2) the mechanisms of PTC rarefaction, (3) major factors regulating EC survival and angiogenesis in CKD, and (4) putative therapeutic targets for PTC rarefaction.
Contribution of peritubular cepillary rarefaction to CKD progression
Loss of renal PTCs is correlated with the severity of fibrosis in human patients during CKD [5–7] and chronic allograft rejection [8]. The extent of PTC loss predicted both interstitial damage and glomerular filtration rate (GFR) in human subjects [6]. Rodent CKD models such as experimental glomerulonephritis [9], the remnant kidney model [10], unilateral ureteral obstruction (UUO, obstructive nephropathy model) [11–14], and the aging kidney [15] also showed a negative correlation between capillary density and the severity of fibrosis. Furthermore, damage to PTCs influenced the long-term outcome after ischemia-reperfusion injury (IRI) [16]. In treated rats, GFR returned to the normal range 1–2 weeks after bilateral IRI. However, over 40 weeks the animals developed prominent fibrosis, which was accompanied by a 30–50% reduction in PTC density [16]. Similar results were obtained in mice [17]. These experimental results were similar to outcomes of clinical studies, in which acute kidney injury dramatically increased the risk of developing end-stage renal disease (ESRD) [18, 19]. Taken together, these findings strongly suggest that PTC rarefaction contributes to the progression of CKD. Currently it is speculated that PTC rarefaction is a both a cause and a result of CKD progression.
Mechanisms of PTC rarefaction
In healthy kidneys, there is a tightly controlled balance between the expression of proangiogenic and antiangiogenic molecules. However, this balance is disrupted in CKD, leading to an antiangiogenic environment that favors loss of PTCs.
Angiogenesis has been investigated in several models of CKD [10, 11, 20, 21]. In the rat UUO model, Ohashi et al. [11] observed early angiogenesis (EC proliferation) in PTCs, accompanied by intense expression of vascular endothelial growth factor (VEGF), a major proangiogenic factor, within the tubular epithelium. This was followed by EC apoptosis in PTCs, a decrease in VEGF and its major angiogenic receptor VEGF receptor 2 (VEGFR-2), and capillary regression. Vascular growth inhibitors may also modulate angiogenesis and tissue damage during chronic injury [22]. Thrombospondin-1 (TSP-1) was up-regulated in the rat remnant kidney at the time when early EC proliferation declined [10]. In the mouse UUO and IRI models, another anti-angiogenic factor, endostatin, was elevated after kidney injury [23, 24].
In response to kidney injury, ECs of PTCs may initially proliferate and subsequently disapper due to apoptosis. Endothelial apoptosis is induced by deprivation of survival growth factors such as VEGF and by increased proapoptotic stimuli such as FasL, interleukin-1, and tumor necrosis factor α (TNF-α) [25, 26]. Apoptotic ECs become pro-coagulant and pro-adhesive, leading to capillary occlusion by thrombosis and enhanced inflammation by extravasation of leukocytes. Impairment of blood flow decreases laminar shear stress on ECs, resulting in further endothelial apoptosis [27]. Furthermore, following kidney injury, vascular pericytes, which structually and functionally stabilize PTC ECs, promptly migrate away and differentiate into myofibroblasts [28]. In the absence of pericytes, PTCs are destabilized, resulting in further PTC loss [28].
It has been reported that PTC density was positively correlated with proximal tubular density in patients with CKD [5]. Renal tubular epithelial cells make up the bulk of the renal mass, and these cells disappear during the progression of CKD. Without any additional insults, genetic ablation of proximal tubles caused PTC loss and fibrosis in vivo [29]. However, how reduction of tubular mass induces PTC loss is still poorly understood.
Major factors regulating angiogenesis and EC survival in CKD
(1) Vascular endothelial growth factor (VEGF)
Despite its complexity, angiogenesis is predominantly regulated by a single growth factor, VEGF-A [30]. VEGF-A is expressed in podocytes and renal tubular epithelial cells in the developing kidneys and continues to be expressed in the adult kidneys. VEGFR-1 (Flt1) and VEGFR-2 (Flk1/KDR) are expressed predominatly in ECs. VEGFR-1 is thought to act as a decoy receptor that sequesters VEGF-A from VEGFR-2 binding. Recently, VEGF-A was identified as a risk locus related to CKD by a genome-wide association study [31], indicating that VEGF-A has a pivotal role in the development of CKD. The dose-dependent deletion or overexpression of VEGF-A in podocytes led to a variety of glomerular capillary diseases including mesangiolysis and endotheliosis, suggesting that VEGF-A is an essential growth factor for development and maintenace of glomerular capillaries [32–34]. VEGF-A is also found to be important for tubular development [35]. VEGF expression continuously increased in tubular epithelial cells in neonatal rats between 14 and 28 days after birth, but was reduced after UUO [36]. VEGFR-2 was expressed strongly in the microvasculature in neonatal kidneys in both sham and UUO groups, whereas in adult rat kidneys VEGFR-2 expression was low in sham operated kidneys but was increased after UUO [36]. These results suggest that the vascular response to kidney injury differs between developing kidneys and mature kidneys. In human kidney biopsies, VEGF was dominantly expressed in podocytes in normal adult kidneys; however, a marked increase of VEGF expression in the renal tubules was observed in CKD [5].
(2) Angiopoietins
Angiopoietins are another critical family of angiogenic factors that play a significant role in renal vascular devleopment as well as renal vascular homeostasis. Angiopoietin-1 (Ang-1), angiopoietin-2 (Ang-2), and the receptor tyrosine kinases Tie-2 (also known as TEK) and Tie-1 (a homolog to Tie-2 that we know relatively little about) are all highly expressed in developing kidneys. Their expression peaks at birth, and they continue to be expressed in the mature kidneys. In the adult kidney, Ang-1 is expressed in tubular epithelial cells, podocytes, and pericytes, whereas Ang-2 is detected in ECs, with lower levels in tubular epithelial cells. Tie-2 is expressed in glomerular and peritubular ECs in addition to hematopoietic cells [37–39]. In general, Ang-1 binds to Tie-2, enhancing EC survival, reducing vascular permeability, and stabilizing capillaries, whereas Ang-2 competitively inhibits the binding of Ang-1 to Tie-2. The biological effects of Ang/Tie signaling may depend on an interaction with VEGF. It was demonstrated in the eye that Ang-2 induced angiogenesis in the presence of VEGF, but led to vascular regression in the absence of VEGF [40]. Recent studies suggest that Ang-1 and Ang-2 can affect an inflammatory process [38]. It is possible that Ang-1 and Ang-2 affect leukocyte transmigration stimulated by TNF-α [41]. Overexpression of Ang-2 in podocytes in mice produced a similar phenotype in diabetic nephropathy, where glomerular endothelial apoptosis is prominent [42]. Ang-2 is also strongly expressed in tubules surrounding mature vasa recta, which express Tie-2 [43], suggesting that Ang/Tie-2 signaling contributes to the maintenance of vasa recta. In patients with CKD, the serum level of Ang-1 was decreased and that of Ang-2 was elevated [44], generating an anti-angiogenic environment. The renal Ang-1 level was also decreased in the UUO model [45]. In contrast, other renal injury models (folic acid nephropathy, IRI, and angiotensin II infusion) showed increased Ang-1 expression in the kidneys [44]. This difference in Ang-1 expression indicates a unique profile in each animal model of CKD. Studies using inducible Ang-1 deletion after E13.5 produced no immediate vascular phenotype [46]. However, Ang-1 deficiency after E13.5 resulted in profound renal damage including affected vasculature after kidney insult, indicating a protective role of Ang-1 in kidney injury [46].
(3) Fibroblast growth factor-2 (FGF-2, basic FGF)
FGF-2 is another potent angiogenic growth factor, which binds to four high-affinity FGF receptors (FGFR-1, 2, 3 and 4). In the normal human kidney, FGF-2 is expressed in glomerular parietal cells, podocytes, distal tubular epithelial cells, ECs, and vascular smooth muscle cells [47]. In mouse cornea, FGF-2 stimulated VEGF-A expression in ECs and stromal cells, which was required for its angiogenic activity [48]. Furthermore, ECs lacking FGF signaling were found to be unresponsive to VEGF-A, because disruption of FGF signaling down-regulated VEGFR2 expression [49]. These results indicate that FGF signaling and VEGF signaling work together for angiogenesis. Blocking FGF signaling in mice using an adenovirus encoding soluble FGFR resulted in loss of arteriogenic response and vascular integrity [49, 50]. Although FGF-2 expression was reported to correlate with the degree of interstitial fibrosis in human kidney samples [51], the role of FGF-2 in PTC rarefaction has not been investigated.
(4) Nitric oxide (NO) and endothelial nitric oxide synthase (eNOS, NOS3)
NO is synthesized from L-arginine by eNOS and functions to maintain ECs in a quiescent state, as opposed to a stressed/damaged (“activated”) state that promotes vasoconstriction as well as coagulation and inflammation (“endothelial activation”). Reduced availability of NO and concurrent endothelial dysfunction have been documented in patients with CKD [52]. Moreover, circulating levels of asymmetric dimethylarginine (ADMA), an endogenous NOS inhibitor, were elevated in CKD patients [53]. Recently, ADMA accumulation was found to reduce eNOS phosphrylation and inhibit eNOS activity in the remnant kidney model of CKD [54]. One recent study showed that administration of L-arginine increased NO synthesis, eNOS expression, and PTC density, and attenuated fibrosis in the rat UUO model, while PTC loss and fibrosis were more severe after treatment with the NOS inhibitor N-nitro-L-arginine methyl ester (L-NAME) [55]. In another study using the remnant kidney model, eNOS-deficient mice showed accelerated PTC rarefaction due to decreased EC proliferation and increased EC apoptosis, accompanied by enhanced macrophage infiltration and fibrosis [56]. These results suggest that NO protects against PTC rarefaction in CKD.
NO is also involved in the renoprotective effects of angiotensin converting enzyme (ACE) inhibitors in CKD. Angiotensin II uncouples the eNOS-catalyzed oxidation of NADPH from the reduction of L-arginine, leading to the production of superoxide (harmful for ECs) rather than NO (beneficial to ECs) [57]. Therefore, treatment of CKD patients with ACE inhibitors or angiotensin receptor blockers (ARBs) can shift the balance back toward NO production and reduce endothelial activation; this action is independent of the blood pressure lowering effect of these agents.
(5) Hypoxia-inducible factor (HIF)
One of the most obvious consequences of PTC rarefaction is a decreased oxygen supply to the tubulointerstitial compartment. Various response pathways are activated during hypoxia in the kidney. On the molecular level, the most important adaptations to hypoxia are mediated by HIF. Mammalian HIFs function as heterodimeric transcription factors composed of either HIF-1α or HIF-2α bound to HIF-1β (also known as aryl hydrocarbon nuclear translocator) [58]. In the hypoxic kidney, HIF-1α is expressed in tubular and glomerular epithelial cells, whereas HIF-2α is detected in glomerular and peritubular ECs and fibroblasts [59]. The primary mechanism of regulating HIF activity is oxygen-dependent proteasomal degradation of the α subunit. In normoxia, the α subunit is rapidly subjected to prolyl hydroxylation, binds to the von Hippel-Lindau tumor suppressor protein, and undergoes proteasomal degradation. In hypoxia, however, the α subunit escapes prolyl hydroxylation and binds to the β subunit. The functional heterodimeric HIF then translocates to the nucleus and promotes angiogenesis, erythropoiesis, and energy metabolism [58].
Increased HIF expression has been reported in kidney biopsies of patients with diabetic nephropathy, IgA nephropathy, polycystic kidney disease, and chronic allograft nephropathy [4]. However, the role of increased HIF in CKD is controversial. Several studies using genetically altered mice demonstrated that HIF-1α expression promoted kidney fibrosis in the UUO model [60] and the remnant kidney [61]. In agreement with these results, one clinical study confirmed a negative effect of HIF-1α activation on renal and patient survival in patients with acute kidney injury [62]. In contrast, the activation of HIFs appears to be protective form interstitial fibrosis and preserves PTC area in various different experimental renal diseases (the remnant kidney, IRI, cisplatin nephropathy, and diabetic nephropathy) [63–65]. Although HIF-1α null or HIF-2α null ECs constituted dysfunctional vasculature in other organs [66, 67], no study has been performed to investigate the role of HIF-1α or HIF-2α specifically in kidney ECs during CKD. For regeneration of PTCs in CKD, strategies involving HIF need to be directed to specific cell types in the kidney to avoid adverse effects.
(6) Other factors
Several other factors have been reported to affect angiogenesis following kidney injury. Thrombospondin-1 (TSP-1) is a 450 kDa homotrimeric extracellular matrix protein and a potent anti-angiogenic factor. TSP-1 expression in the normal kidney is limited to Bowman’s capsule, whereas in the damaged kidney TSP-1 is expressed in all glomerular cell types as well as in tubular epithelial cells, myofibroblasts, and infiltrating macrophages [68]. In UUO models, TSP-1 null mice displayed less inflammation and had better PTC preservation than wild-type controls [68], indicating that TSP-1 has a deleterious role in PTC rarefaction during CKD. However, another study demonstrated that TSP-1 deficient ECs exhibited decreased phosphorylation of VEGFR-2 in response to VEGF-A stimulation compared with wild-type ECs [69]. This result suggests that TSP-1 promotes the inflammatory response and inhibits angiogenesis via its pro-inflammatory activity rather than via impairment of VEGF signaling after kidney injury. Although previous studies reported that TSP-1 enhanced apoptosis of ECs [70] and inhibited EC responses to NO [71], the mechanisms by which TSP-1 inhibits angiogenesis in CKD are still not clear.
CD44 is a glycoprotein that binds to hyaluronic acid (HA). After UUO treatment, CD44 expression was increased in ECs located in PTCs, and CD44-deficient mice preserved more PTC area than wild-type mice and showed decreased endothelial apoptosis [12]. An interaction between CD44 and HA enhanced the pro-apoptotic effect of transforming growth factor-β 1 on ECs [12].
ADAMTS-1 (a disintegrin and metalloproteinase with thrombospondin motif-1), a secreted VEGF inhibitor, appears to be an anti-angiogenic factor [72] and destabilizes vessels after IRI [73].
In addition to VEGF-A and FGF-2, hepatocyte growth factor (HGF) was shown to function as an angiogenic factor through binding to its receptor, c-Met [74]. In the remnant kidney model, infusion of HGF remarkably ameriorated macrophage adhesion to ECs, whereas neutralizing endogenous HGF worsened macrophage adhesion [75, 76]. These results suggest that HGF may preserve PTCs through an anti-inflammatory action.
Considerations for therapy to limit PTC rarefaction in CKD
(1) Treatment with angiogenic growth factors
Multiple studies have demonstrated that VEGF-A may help to preserve PTCs in CKD. In the remnant kidney model, administration of recombinant VEGF-A121 improved renal function, lowered mortality, decreased PTC rarefaction, and reduced fibrosis [77]. Similarly, in a pig model of renal artery stenosis, intrarenal infusion of VEGF-A attenuated microvascular rarefaction, prevented fibrosis, and improved renal blood flow [78]. However, other results did not support the beneficial effects of VEGF-A in CKD. In kidney injury induced by angiotensin II infusion followed by a high-salt diet, administration of VEGF-A121 did not improve PTC rarefaction [79]. In addition, VEGF-A121 treatment did not improve PTC rarefaction in neonatal mouse kidneys subjected to UUO [80], and systemic overexpression of a mutant form of VEGF-A that can bind only to VEGFR-2 but not to VEGFR-1 enhanced glomerular injury as well as interstitial fibrosis in mice with uninephrectomy [81]. Furthermore, the timing of angiogenic treatment is crucial. In the rat remnant kidney model, VEGF-A121 delivered 4–8 weeks after surgery ameliorated CKD, perhaps because VEGF-A levels in the kidney are very low during this period and PTC rarefaction is severe [77]. By contrast, the first 4 weeks after surgery in the remnant kidney model were characterized by glomerular hypertrophy and increased VEGF-A levels [82]. During this initial period, neutralizing VEGF-A, but not VEGF-A injection, could improve renal function and glomerular hypertrophy [82]. In contrast, secondary CKD induced by a salt-rich diet following recovery from IRI was ameriorated by VEGF-A121 only when it was injected immediately (within 3 days) after IRI [83]. These discrepancies in the effect of VEGF-A on CKD raise important considerations for future therapeutic strategies that target angiogenesis in CKD.
A soluble, stable, and more potent form of Ang-1 (COMP-Ang-1) was successfully used in the mouse UUO and IRI models to preserve PTC area and renal blood flow and to decrease inflammation and fibrosis [45, 84], suggesting the possible use of Ang-1 as a novel treatment for patients with CKD. However, Long et al. reported contradictory findings [38]. In mice with folic acid-induced kidney injury, these authors systemically delivered a recombinant form of Ang-1 (Ang-1*) using adenoviruses, which improved PTC rarefaction but enhanced inflammation and fibrosis [38]. Further studies are necessary to reveal the relationship between angiogenesis and inflammation in CKD. Combination therapy with VEGF-A and Ang-1 could potentially overcome this problem, as Ang-1 stabilizes blood vessels and prevents VEGF-induced endothelial leakiness [85].
Finally, there is one caveat against inhibition of VEGF signaling. As reported in clinical settings such as cancer therapy, blockade of VEGF signaling causes proteinuria, hypertension and renal thrombotic microangiopathy due to disruption of the glomerular filtration barrier [86, 87].
(2) Endothelial progenitor cells (EPCs)
EPCs have been suggested to be useful for maintaining the integrity of ECs and for repairing ECs after injury [88]. EPCs are identified by a consensus combination of markers and characteristics (individually not unique to EPCs) including CD34, VEGFR-2, Tie-2, CD133, Ulex Europeus lectin binding, uptake of acetylated LDL, and the ability to form colonies (colony-forming units) [88]. Chade et al. demonstrated that intra-renal infusion of autologous EPCs attenuated PTC rarefaction and fibrosis, improving renal function by stimulating angiogenesis [89]. The adoptive transfer of EPCs could be one option to promote de novo angiogenesis and renal regeneration.
One may wonder why resident EPCs are insufficient to trigger neo-angiogenesis after kidney injury. This deficiency may arise from disease-related changes in EPC function. EPCs taken from pigs with unilateral renal artery stenosis in the early and later phases of disease progression had different characteristics [90]. EPCs isolated from peripheral blood in the early phase exhibited increased proliferation, tube formation, VEGF-A and eNOS expression, and augmented expression of C-X-C chemokine receptor type 4 (CXCR4) [90]. Homing of EPCs expressing CXCR4 is regulated by the ligand of CXCR4, α-chemokine stromal cell-derived factor 1 (SDF-1) [91]. However, these enhanced functions were not maintained in EPCs isolated in the later phase of this disease [90]. Furthermore, EPCs from patients with CKD displayed functional impairments (i.e. hampered adherence, reduced endothelial outgrowth potential, and reduced anti-thrombotic function) [92]. These impairments became more apparent when CKD progressed [92]. These results provide evidence that endogenous EPCs become dysfunctional as CKD advances, which leads to an impairment of the repair process by endogenous EPCs. One way to overcome this deficiency is the adoptive transfer of exogenous EPCs isolated from healthy donors. It should be noted that the role of EPCs in endothelial repair remains controversial. Although EPCs could form vascular structures, these may not be stable. Alternatively, EPCs may orchestrate angiogenic processes, while proliferation of existing ECs is quantitatively more significant for vessel formation or repair following kidney injury [93, 94].
(3) Endothelial-pericyte interactions
Interactions between ECs and intimately associated pericytes were recently demonstrated to be important to preserve PTCs and to alleviate fibrosis [28, 95]. Endothelial-pericyte cross-talk has been shown to be critical for angiogenesis and vascular stabilization, and involves multiple ligand/receptor interactions including platelet-derived growth factor (PDGF)-B/PDGF receptor-β (PDGFRβ) and Ang-1/Tie-2 [96]. PDGF-B/PDGFRβ signaling was essential for recruitment of pericytes to vasculature, and ECs were not integrated properly into vessels without this signaling, leading to endothelial hyperplasia, abnormal EC junctions, microaneurysms, hemorrhages, and capillary rarefaction [96]. Endothelial-pericyte inteactions were disrupted in IRI and UUO models of CKD, and pericytes detached and migrated away from microvasculature and finally differentiated into myofibroblasts [95]. When either PDGFR signaling in pericytes or VEGFR-2 signaling in ECs was blocked by circulating soluble receptor ectodomains, both PTC rarefaction and fibrosis were markedly attenuated during CKD progression [95]. The important findings of this study were that (1) bidirectional signaling between pericytes and ECs was necessary to prevent pericyte detachment from vessels, and (2) pericyte detachment was responsible for both PTC rarefaction and fibrosis. Thus, targeting the cross-talk between these two types of cells may provide a novel therapeutic opportunity to treat PTC rarefaction and fibrosis in CKD.
In the clinical setting, patients with idiopathic pulmonary fibrosis (IPF) were treated for 1 year with BIBF 1120, which can block signaling by multiple tyrosine kinase receptors including PDGFRβ and VEGFR-2 [97]. BIBF 1120 significantly prevented exacerbation of IPF, suggesting a beneficial effect of the drug on organofibrosis. The most frequent adverse effect of the drug was gastrointestinal isuues such as diarrhea and vomiting, although no information was shown about capillary density in any organ [97].
The concept of pericyte detachment suggests a connection between PTC rarefaction and kidney fibrosis. Zeisberg et al. also suggested another concept of endothelial-to-mesenchymal transition (EndMT) based on studies of fate-mapped ECs using Tie2-Cre;Rosa26 Reporter-stop-EYFP bigenic mice [98]. Further studies are required to evaluate the roles of pericytes as well as EndMT in capillary rarefaction in CKD.
Conclusion
The collective data from both clinical and experimental studies support the concept that PTC rarefaction is not only a prominent histological characteristic of CKD but also a central driving force that contributes to the progression of CKD. As multiple factors contribute to PTC rarefaction in each phase of the disease process, we need to tailor the treatment according to the stage of CKD.
Figure 1.
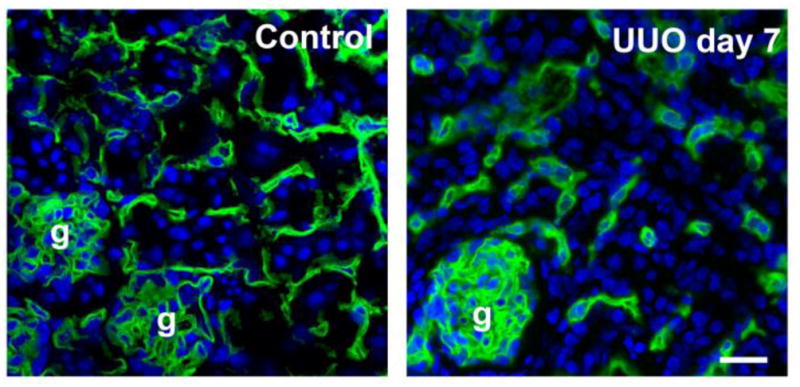
Immunofluorescence staining for CD31 (green) to visualize peritubular capillaries (PTCs) in the mouse kidney cortex. Control (left) and day 7 after UUO surgery (right). Note that the CD31+ PTC density is profoundly reduced in the UUO kidney comapred with the control kidney. g: glomerulus. Bar: 50 μm.
Figure 2.
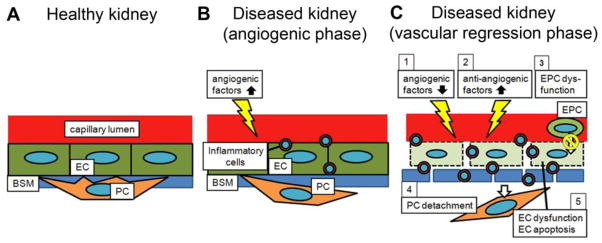
Schematic of mechanisms of peritubular capillary (PTC) rarefaction following kidney injury. (A) In the healthy kidney, pericytes (PC) are embedded into the capillary basement membrane (BSM) and attached to capillary endothelial cells (EC). This close relationship between PCs and ECs supports capillary integrity. (B) Subsequent to an initial insult, angiogenic factors (e.g. VEGF) are up-regulated and ECs are proliferative. PCs promptly start to migrate away from the capillary area. Simultaneously, inflammatory cells infiltrate into the interstitium. (C) In response to continuous insults, (1) angiogenic factors are down-regulated, (2) anti-angiogenic factors (e.g. thrombospondin-1) are up-regulated, (3) endothelial progenitor cells (EPCs) do not promote neoangiogenesis, (4) PC detachment from vasculature destabilizes ECs, and (5) EC dysfunction and EC apoptosis progress. All of these mechanisms contribute to an anti-angiogenic environment, resulting in PTC rarefaction. The capillary BSM is also degraded. Additionally, a large number of inflammatory cells migrate from the capillary lumen into the extracapillary space. Note that some of the angiogenic factors (e.g. angiopoietin-1) and anti-angiogenic factors (e.g. thrombospondin-1) induce infiltration of inflammatory cells, which impairs the angiogenic response.
Table.
Roles of major factors that regulate angiogenesis and capillary regression in CKD.
| Factor | Major mechanisms of action | Other biological effects | |
|---|---|---|---|
| Proangiogenic factors | Angiopoietin-1 |
|
|
| Fibroblast growth factor-2 |
|
|
|
| Hepatocyte growth factor |
|
||
| Hypoxia-inducible factor |
|
|
|
| Nitric oxide |
|
||
| PlGF |
|
|
|
| Platelet-derived growth factor B |
|
|
|
| VEGF-A |
|
|
|
| Antiangiogenic factors | ADAMTS-1 |
|
|
| Angiopoietin-2 |
|
|
|
| Angiostatin |
|
|
|
| Asymmetric dimethylarginine (ADMA) |
|
||
| CD44 |
|
||
| Endostatin |
|
||
| Soluble fms-like tyrosine kinase-1 (sFlt-1) |
|
||
| Thrombospondin-1 |
|
|
Abbreviations: CKD, chronic kidney disease; ADAMTS-1, a disintegrin and metalloproteinase with thrombospondin motif-1; EC, endothelial cell; PC, pericyte; PlGF, placental growth factor; VEGF: vascular endothelial growth factor; VEGFR-2, VEGF receptor-2
Acknowledgments
The authors acknowledge research grant support from the National Institutes of Health (DK080926 to I.Y.) and the American Society of Nephrology (Carl W. Gottschalk Research Scholar Grant to I.Y.). We apologize for any omission of relevant literature in this review due to limited space.
Footnotes
Disclosures
None
References
- 1.Collins AJ, Foley RN, Chavers B, Gilbertson D, Herzog C, Johansen K, Kasiske B, Kutner N, Liu J, St Peter W, Guo H, Gustafson S, Heubner B, Lamb K, Li S, Peng Y, Qiu Y, Roberts T, Skeans M, Snyder J, Solid C, Thompson B, Wang C, Weinhandl E, Zaun D, Arko C, Chen SC, Daniels F, Ebben J, Frazier E, Hanzlik C, Johnson R, Sheets D, Wang X, Forrest B, Constantini E, Everson S, Eggers P, Agodoa L. United States Renal Data System 2011 Annual Data Report: Atlas of chronic kidney disease & end-stage renal disease in the United States. Am J Kidney Dis. 2012;59:A7, e1–420. doi: 10.1053/j.ajkd.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 2.Himmelfarb J, Shankland SJ. Creating research infrastructure and functionality to address chronic kidney disease: the Kidney Research Institute. Semin Nephrol. 2009;29:457–466. doi: 10.1016/j.semnephrol.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Levey AS, Coresh J. Chronic kidney disease. Lancet. 2012;379:165–180. doi: 10.1016/S0140-6736(11)60178-5. [DOI] [PubMed] [Google Scholar]
- 4.Fine LG, Norman JT. Chronic hypoxia as a mechanism of progression of chronic kidney diseases: from hypothesis to novel therapeutics. Kidney Int. 2008;74:867–872. doi: 10.1038/ki.2008.350. [DOI] [PubMed] [Google Scholar]
- 5.Choi YJ, Chakraborty S, Nguyen V, Nguyen C, Kim BK, Shim SI, Suki WN, Truong LD. Peritubular capillary loss is associated with chronic tubulointerstitial injury in human kidney: altered expression of vascular endothelial growth factor. Hum Pathol. 2000;31:1491–1497. doi: 10.1053/hupa.2000.20373. [DOI] [PubMed] [Google Scholar]
- 6.Seron D, Alexopoulos E, Raftery MJ, Hartley B, Cameron JS. Number of interstitial capillary cross-sections assessed by monoclonal antibodies: relation to interstitial damage. Nephrol Dial Transplant. 1990;5:889–893. doi: 10.1093/ndt/5.10.889. [DOI] [PubMed] [Google Scholar]
- 7.Bohle A, Mackensen-Haen S, Wehrmann M. Significance of postglomerular capillaries in the pathogenesis of chronic renal failure. Kidney Blood Press Res. 1996;19:191–195. doi: 10.1159/000174072. [DOI] [PubMed] [Google Scholar]
- 8.Ishii Y, Sawada T, Kubota K, Fuchinoue S, Teraoka S, Shimizu A. Injury and progressive loss of peritubular capillaries in the development of chronic allograft nephropathy. Kidney Int. 2005;67:321–332. doi: 10.1111/j.1523-1755.2005.00085.x. [DOI] [PubMed] [Google Scholar]
- 9.Ohashi R, Kitamura H, Yamanaka N. Peritubular capillary injury during the progression of experimental glomerulonephritis in rats. J Am Soc Nephrol. 2000;11:47–56. doi: 10.1681/ASN.V11147. [DOI] [PubMed] [Google Scholar]
- 10.Kang DH, Joly AH, Oh SW, Hugo C, Kerjaschki D, Gordon KL, Mazzali M, Jefferson JA, Hughes J, Madsen KM, Schreiner GF, Johnson RJ. Impaired angiogenesis in the remnant kidney model: I. Potential role of vascular endothelial growth factor and thrombospondin-1. J Am Soc Nephrol. 2001;12:1434–1447. doi: 10.1681/ASN.V1271434. [DOI] [PubMed] [Google Scholar]
- 11.Ohashi R, Shimizu A, Masuda Y, Kitamura H, Ishizaki M, Sugisaki Y, Yamanaka N. Peritubular capillary regression during the progression of experimental obstructive nephropathy. J Am Soc Nephrol. 2002;13:1795–1805. doi: 10.1097/01.asn.0000018408.51388.57. [DOI] [PubMed] [Google Scholar]
- 12.Rouschop KM, Claessen N, Pals ST, Weening JJ, Florquin S. CD44 disruption prevents degeneration of the capillary network in obstructive nephropathy via reduction of TGF-beta1-induced apoptosis. J Am Soc Nephrol. 2006;17:746–753. doi: 10.1681/ASN.2005080808. [DOI] [PubMed] [Google Scholar]
- 13.Eddy AA, Lopez-Guisa JM, Okamura DM, Yamaguchi I. Investigating mechanisms of chronic kidney disease in mouse models. Pediatr Nephrol. 2011 doi: 10.1007/s00467-011-1938-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamaguchi I, Tchao BN, Burger ML, Yamada M, Hyodo T, Giampietro C, Eddy AA. Vascular endothelial cadherin modulates renal interstitial fibrosis. Nephron Exp Nephrol. 2012;120:e20–31. doi: 10.1159/000332026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang DH, Anderson S, Kim YG, Mazzalli M, Suga S, Jefferson JA, Gordon KL, Oyama TT, Hughes J, Hugo C, Kerjaschki D, Schreiner GF, Johnson RJ. Impaired angiogenesis in the aging kidney: vascular endothelial growth factor and thrombospondin-1 in renal disease. Am J Kidney Dis. 2001;37:601–611. doi: 10.1053/ajkd.2001.22087. [DOI] [PubMed] [Google Scholar]
- 16.Basile DP, Donohoe D, Roethe K, Osborn JL. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am J Physiol Renal Physiol. 2001;281:F887–899. doi: 10.1152/ajprenal.2001.281.5.F887. [DOI] [PubMed] [Google Scholar]
- 17.Horbelt M, Lee SY, Mang HE, Knipe NL, Sado Y, Kribben A, Sutton TA. Acute and chronic microvascular alterations in a mouse model of ischemic acute kidney injury. Am J Physiol Renal Physiol. 2007;293:F688–695. doi: 10.1152/ajprenal.00452.2006. [DOI] [PubMed] [Google Scholar]
- 18.Ishani A, Xue JL, Himmelfarb J, Eggers PW, Kimmel PL, Molitoris BA, Collins AJ. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol. 2009;20:223–228. doi: 10.1681/ASN.2007080837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wald R, Quinn RR, Luo J, Li P, Scales DC, Mamdani MM, Ray JG. Chronic dialysis and death among survivors of acute kidney injury requiring dialysis. JAMA. 2009;302:1179–1185. doi: 10.1001/jama.2009.1322. [DOI] [PubMed] [Google Scholar]
- 20.Konda R, Sato H, Sakai K, Sato M, Orikasa S, Kimura N. Expression of platelet-derived endothelial cell growth factor and its potential role in up-regulation of angiogenesis in scarred kidneys secondary to urinary tract diseases. Am J Pathol. 1999;155:1587–1597. doi: 10.1016/S0002-9440(10)65475-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pillebout E, Burtin M, Yuan HT, Briand P, Woolf AS, Friedlander G, Terzi F. Proliferation and remodeling of the peritubular microcirculation after nephron reduction: association with the progression of renal lesions. Am J Pathol. 2001;159:547–560. doi: 10.1016/S0002-9440(10)61726-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nyberg P, Xie L, Kalluri R. Endogenous inhibitors of angiogenesis. Cancer Res. 2005;65:3967–3979. doi: 10.1158/0008-5472.CAN-04-2427. [DOI] [PubMed] [Google Scholar]
- 23.Maciel TT, Coutinho EL, Soares D, Achar E, Schor N, Bellini MH. Endostatin, an antiangiogenic protein, is expressed in the unilateral ureteral obstruction mice model. J Nephrol. 2008;21:753–760. [PubMed] [Google Scholar]
- 24.Bellini MH, Coutinho EL, Filgueiras TC, Maciel TT, Schor N. Endostatin expression in the murine model of ischaemia/reperfusion-induced acute renal failure. Nephrology (Carlton) 2007;12:459–465. doi: 10.1111/j.1440-1797.2007.00850.x. [DOI] [PubMed] [Google Scholar]
- 25.Sanz AB, Santamaria B, Ruiz-Ortega M, Egido J, Ortiz A. Mechanisms of renal apoptosis in health and disease. J Am Soc Nephrol. 2008;19:1634–1642. doi: 10.1681/ASN.2007121336. [DOI] [PubMed] [Google Scholar]
- 26.Winn RK, Harlan JM. The role of endothelial cell apoptosis in inflammatory and immune diseases. J Thromb Haemost. 2005;3:1815–1824. doi: 10.1111/j.1538-7836.2005.01378.x. [DOI] [PubMed] [Google Scholar]
- 27.Dimmeler S, Assmus B, Hermann C, Haendeler J, Zeiher AM. Fluid shear stress stimulates phosphorylation of Akt in human endothelial cells: involvement in suppression of apoptosis. Circ Res. 1998;83:334–341. doi: 10.1161/01.res.83.3.334. [DOI] [PubMed] [Google Scholar]
- 28.Kida Y, Duffield JS. Pivotal role of pericytes in kidney fibrosis. Clin Exp Pharmacol Physiol. 2011;38:417–423. doi: 10.1111/j.1440-1681.2011.05531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grgic I, Campanholle G, Bijol V, Wang C, Sabbisetti VS, Ichimura T, Humphreys BD, Bonventre JV. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012;82:172–183. doi: 10.1038/ki.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kottgen A, Pattaro C, Boger CA, Fuchsberger C, Olden M, Glazer NL, Parsa A, Gao X, Yang Q, Smith AV, O’Connell JR, Li M, Schmidt H, Tanaka T, Isaacs A, Ketkar S, Hwang SJ, Johnson AD, Dehghan A, Teumer A, Pare G, Atkinson EJ, Zeller T, Lohman K, Cornelis MC, Probst-Hensch NM, Kronenberg F, Tonjes A, Hayward C, Aspelund T, Eiriksdottir G, Launer LJ, Harris TB, Rampersaud E, Mitchell BD, Arking DE, Boerwinkle E, Struchalin M, Cavalieri M, Singleton A, Giallauria F, Metter J, de Boer IH, Haritunians T, Lumley T, Siscovick D, Psaty BM, Zillikens MC, Oostra BA, Feitosa M, Province M, de Andrade M, Turner ST, Schillert A, Ziegler A, Wild PS, Schnabel RB, Wilde S, Munzel TF, Leak TS, Illig T, Klopp N, Meisinger C, Wichmann HE, Koenig W, Zgaga L, Zemunik T, Kolcic I, Minelli C, Hu FB, Johansson A, Igl W, Zaboli G, Wild SH, Wright AF, Campbell H, Ellinghaus D, Schreiber S, Aulchenko YS, Felix JF, Rivadeneira F, Uitterlinden AG, Hofman A, Imboden M, Nitsch D, Brandstatter A, Kollerits B, Kedenko L, Magi R, Stumvoll M, Kovacs P, Boban M, Campbell S, Endlich K, Volzke H, Kroemer HK, Nauck M, Volker U, Polasek O, Vitart V, Badola S, Parker AN, Ridker PM, Kardia SL, Blankenberg S, Liu Y, Curhan GC, Franke A, Rochat T, Paulweber B, Prokopenko I, Wang W, Gudnason V, Shuldiner AR, Coresh J, Schmidt R, Ferrucci L, Shlipak MG, van Duijn CM, Borecki I, Kramer BK, Rudan I, Gyllensten U, Wilson JF, Witteman JC, Pramstaller PP, Rettig R, Hastie N, Chasman DI, Kao WH, Heid IM, Fox CS. New loci associated with kidney function and chronic kidney disease. Nat Genet. 2010;42:376–384. doi: 10.1038/ng.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eremina V, Cui S, Gerber H, Ferrara N, Haigh J, Nagy A, Ema M, Rossant J, Jothy S, Miner JH, Quaggin SE. Vascular endothelial growth factor a signaling in the podocyte-endothelial compartment is required for mesangial cell migration and survival. J Am Soc Nephrol. 2006;17:724–735. doi: 10.1681/ASN.2005080810. [DOI] [PubMed] [Google Scholar]
- 33.Eremina V, Quaggin SE. The role of VEGF-A in glomerular development and function. Curr Opin Nephrol Hypertens. 2004;13:9–15. doi: 10.1097/00041552-200401000-00002. [DOI] [PubMed] [Google Scholar]
- 34.Eremina V, Sood M, Haigh J, Nagy A, Lajoie G, Ferrara N, Gerber HP, Kikkawa Y, Miner JH, Quaggin SE. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tufro A, Norwood VF, Carey RM, Gomez RA. Vascular endothelial growth factor induces nephrogenesis and vasculogenesis. J Am Soc Nephrol. 1999;10:2125–2134. doi: 10.1681/ASN.V10102125. [DOI] [PubMed] [Google Scholar]
- 36.Burt LE, Forbes MS, Thornhill BA, Kiley SC, Chevalier RL. Renal vascular endothelial growth factor in neonatal obstructive nephropathy. I. Endogenous VEGF. Am J Physiol Renal Physiol. 2007;292:F158–167. doi: 10.1152/ajprenal.00293.2005. [DOI] [PubMed] [Google Scholar]
- 37.Woolf AS, Yuan HT. Angiopoietin growth factors and Tie receptor tyrosine kinases in renal vascular development. Pediatr Nephrol. 2001;16:177–184. doi: 10.1007/s004670000509. [DOI] [PubMed] [Google Scholar]
- 38.Long DA, Price KL, Ioffe E, Gannon CM, Gnudi L, White KE, Yancopoulos GD, Rudge JS, Woolf AS. Angiopoietin-1 therapy enhances fibrosis and inflammation following folic acid-induced acute renal injury. Kidney Int. 2008;74:300–309. doi: 10.1038/ki.2008.179. [DOI] [PubMed] [Google Scholar]
- 39.Lancrin C, Sroczynska P, Serrano AG, Gandillet A, Ferreras C, Kouskoff V, Lacaud G. Blood cell generation from the hemangioblast. J Mol Med (Berl) 2010;88:167–172. doi: 10.1007/s00109-009-0554-0. [DOI] [PubMed] [Google Scholar]
- 40.Lobov IB, Brooks PC, Lang RA. Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc Natl Acad Sci U S A. 2002;99:11205–11210. doi: 10.1073/pnas.172161899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, Gale NW, Witzenrath M, Rosseau S, Suttorp N, Sobke A, Herrmann M, Preissner KT, Vajkoczy P, Augustin HG. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat Med. 2006;12:235–239. doi: 10.1038/nm1351. [DOI] [PubMed] [Google Scholar]
- 42.Davis B, Dei Cas A, Long DA, White KE, Hayward A, Ku CH, Woolf AS, Bilous R, Viberti G, Gnudi L. Podocyte-specific expression of angiopoietin-2 causes proteinuria and apoptosis of glomerular endothelia. J Am Soc Nephrol. 2007;18:2320–2329. doi: 10.1681/ASN.2006101093. [DOI] [PubMed] [Google Scholar]
- 43.Yuan HT, Suri C, Yancopoulos GD, Woolf AS. Expression of angiopoietin-1, angiopoietin-2, and the Tie-2 receptor tyrosine kinase during mouse kidney maturation. J Am Soc Nephrol. 1999;10:1722–1736. doi: 10.1681/ASN.V1081722. [DOI] [PubMed] [Google Scholar]
- 44.Woolf AS, Gnudi L, Long DA. Roles of angiopoietins in kidney development and disease. J Am Soc Nephrol. 2009;20:239–244. doi: 10.1681/ASN.2008020243. [DOI] [PubMed] [Google Scholar]
- 45.Kim W, Moon SO, Lee SY, Jang KY, Cho CH, Koh GY, Choi KS, Yoon KH, Sung MJ, Kim DH, Lee S, Kang KP, Park SK. COMP-angiopoietin-1 ameliorates renal fibrosis in a unilateral ureteral obstruction model. J Am Soc Nephrol. 2006;17:2474–2483. doi: 10.1681/ASN.2006020109. [DOI] [PubMed] [Google Scholar]
- 46.Jeansson M, Gawlik A, Anderson G, Li C, Kerjaschki D, Henkelman M, Quaggin SE. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J Clin Invest. 2011;121:2278–2289. doi: 10.1172/JCI46322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Floege J, Hudkins KL, Eitner F, Cui Y, Morrison RS, Schelling MA, Alpers CE. Localization of fibroblast growth factor-2 (basic FGF) and FGF receptor-1 in adult human kidney. Kidney Int. 1999;56:883–897. doi: 10.1046/j.1523-1755.1999.00637.x. [DOI] [PubMed] [Google Scholar]
- 48.Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, Robbins ES, Shapiro RL, Galloway AC, Rifkin DB, Mignatti P. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J Cell Biol. 1998;141:1659–1673. doi: 10.1083/jcb.141.7.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murakami M, Nguyen LT, Hatanaka K, Schachterle W, Chen PY, Zhuang ZW, Black BL, Simons M. FGF-dependent regulation of VEGF receptor 2 expression in mice. J Clin Invest. 2011;121:2668–2678. doi: 10.1172/JCI44762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murakami M, Nguyen LT, Zhuang ZW, Moodie KL, Carmeliet P, Stan RV, Simons M. The FGF system has a key role in regulating vascular integrity. J Clin Invest. 2008;118:3355–3366. doi: 10.1172/JCI35298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strutz F, Zeisberg M, Hemmerlein B, Sattler B, Hummel K, Becker V, Muller GA. Basic fibroblast growth factor expression is increased in human renal fibrogenesis and may mediate autocrine fibroblast proliferation. Kidney Int. 2000;57:1521–1538. doi: 10.1046/j.1523-1755.2000.00997.x. [DOI] [PubMed] [Google Scholar]
- 52.Wever R, Boer P, Hijmering M, Stroes E, Verhaar M, Kastelein J, Versluis K, Lagerwerf F, van Rijn H, Koomans H, Rabelink T. Nitric oxide production is reduced in patients with chronic renal failure. Arterioscler Thromb Vasc Biol. 1999;19:1168–1172. doi: 10.1161/01.atv.19.5.1168. [DOI] [PubMed] [Google Scholar]
- 53.Zoccali C, Bode-Boger S, Mallamaci F, Benedetto F, Tripepi G, Malatino L, Cataliotti A, Bellanuova I, Fermo I, Frolich J, Boger R. Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: a prospective study. Lancet. 2001;358:2113–2117. doi: 10.1016/s0140-6736(01)07217-8. [DOI] [PubMed] [Google Scholar]
- 54.Kajimoto H, Kai H, Aoki H, Yasuoka S, Anegawa T, Aoki Y, Ueda S, Okuda S, Imaizumi T. Inhibition of eNOS phosphorylation mediates endothelial dysfunction in renal failure: new effect of asymmetric dimethylarginine. Kidney Int. 2012;81:762–768. doi: 10.1038/ki.2011.476. [DOI] [PubMed] [Google Scholar]
- 55.Sun D, Wang Y, Liu C, Zhou X, Li X, Xiao A. Effects of nitric oxide on renal interstitial fibrosis in rats with unilateral ureteral obstruction. Life Sci. 2012;90:900–909. doi: 10.1016/j.lfs.2012.04.018. [DOI] [PubMed] [Google Scholar]
- 56.Nakayama T, Sato W, Kosugi T, Zhang L, Campbell-Thompson M, Yoshimura A, Croker BP, Johnson RJ, Nakagawa T. Endothelial injury due to eNOS deficiency accelerates the progression of chronic renal disease in the mouse. Am J Physiol Renal Physiol. 2009;296:F317–327. doi: 10.1152/ajprenal.90450.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Satoh M, Fujimoto S, Arakawa S, Yada T, Namikoshi T, Haruna Y, Horike H, Sasaki T, Kashihara N. Angiotensin II type 1 receptor blocker ameliorates uncoupled endothelial nitric oxide synthase in rats with experimental diabetic nephropathy. Nephrol Dial Transplant. 2008;23:3806–3813. doi: 10.1093/ndt/gfn357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 59.Rosenberger C, Mandriota S, Jurgensen JS, Wiesener MS, Horstrup JH, Frei U, Ratcliffe PJ, Maxwell PH, Bachmann S, Eckardt KU. Expression of hypoxia-inducible factor-1alpha and -2alpha in hypoxic and ischemic rat kidneys. J Am Soc Nephrol. 2002;13:1721–1732. doi: 10.1097/01.asn.0000017223.49823.2a. [DOI] [PubMed] [Google Scholar]
- 60.Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M, Haase VH. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kimura K, Iwano M, Higgins DF, Yamaguchi Y, Nakatani K, Harada K, Kubo A, Akai Y, Rankin EB, Neilson EG, Haase VH, Saito Y. Stable expression of HIF-1alpha in tubular epithelial cells promotes interstitial fibrosis. Am J Physiol Renal Physiol. 2008;295:F1023–1029. doi: 10.1152/ajprenal.90209.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kolyada AY, Tighiouart H, Perianayagam MC, Liangos O, Madias NE, Jaber BL. A genetic variant of hypoxia-inducible factor-1alpha is associated with adverse outcomes in acute kidney injury. Kidney Int. 2009;75:1322–1329. doi: 10.1038/ki.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tanaka T, Kojima I, Ohse T, Ingelfinger JR, Adler S, Fujita T, Nangaku M. Cobalt promotes angiogenesis via hypoxia-inducible factor and protects tubulointerstitium in the remnant kidney model. Lab Invest. 2005;85:1292–1307. doi: 10.1038/labinvest.3700328. [DOI] [PubMed] [Google Scholar]
- 64.Nangaku M, Inagi R, Miyata T, Fujita T. Hypoxia and hypoxia-inducible factor in renal disease. Nephron Exp Nephrol. 2008;110:e1–7. doi: 10.1159/000148256. [DOI] [PubMed] [Google Scholar]
- 65.Hill P, Shukla D, Tran MG, Aragones J, Cook HT, Carmeliet P, Maxwell PH. Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J Am Soc Nephrol. 2008;19:39–46. doi: 10.1681/ASN.2006090998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tang N, Wang L, Esko J, Giordano FJ, Huang Y, Gerber HP, Ferrara N, Johnson RS. Loss of HIF-1alpha in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell. 2004;6:485–495. doi: 10.1016/j.ccr.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 67.Skuli N, Majmundar AJ, Krock BL, Mesquita RC, Mathew LK, Quinn ZL, Runge A, Liu L, Kim MN, Liang J, Schenkel S, Yodh AG, Keith B, Simon MC. Endothelial HIF-2alpha regulates murine pathological angiogenesis and revascularization processes. J Clin Invest. 2012;122:1427–1443. doi: 10.1172/JCI57322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bige N, Shweke N, Benhassine S, Jouanneau C, Vandermeersch S, Dussaule JC, Chatziantoniou C, Ronco P, Boffa JJ. Thrombospondin-1 plays a profibrotic and pro-inflammatory role during ureteric obstruction. Kidney Int. 2012;81:1226–1238. doi: 10.1038/ki.2012.21. [DOI] [PubMed] [Google Scholar]
- 69.Zhang X, Kazerounian S, Duquette M, Perruzzi C, Nagy JA, Dvorak HF, Parangi S, Lawler J. Thrombospondin-1 modulates vascular endothelial growth factor activity at the receptor level. FASEB J. 2009;23:3368–3376. doi: 10.1096/fj.09-131649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med. 2000;6:41–48. doi: 10.1038/71517. [DOI] [PubMed] [Google Scholar]
- 71.Isenberg JS, Ridnour LA, Perruccio EM, Espey MG, Wink DA, Roberts DD. Thrombospondin-1 inhibits endothelial cell responses to nitric oxide in a cGMP-dependent manner. Proc Natl Acad Sci U S A. 2005;102:13141–13146. doi: 10.1073/pnas.0502977102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Basile DP, Fredrich K, Chelladurai B, Leonard EC, Parrish AR. Renal ischemia reperfusion inhibits VEGF expression and induces ADAMTS-1, a novel VEGF inhibitor. Am J Physiol Renal Physiol. 2008;294:F928–936. doi: 10.1152/ajprenal.00596.2007. [DOI] [PubMed] [Google Scholar]
- 73.Schrimpf C, Xin C, Campanholle G, Gill SE, Stallcup W, Lin SL, Davis GE, Gharib SA, Humphreys BD, Duffield JS. Pericyte TIMP3 and ADAMTS1 Modulate Vascular Stability after Kidney Injury. J Am Soc Nephrol. 2012;23:868–883. doi: 10.1681/ASN.2011080851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bussolino F, Di Renzo MF, Ziche M, Bocchietto E, Olivero M, Naldini L, Gaudino G, Tamagnone L, Coffer A, Comoglio PM. Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J Cell Biol. 1992;119:629–641. doi: 10.1083/jcb.119.3.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gong R, Rifai A, Dworkin LD. Anti-inflammatory effect of hepatocyte growth factor in chronic kidney disease: targeting the inflamed vascular endothelium. J Am Soc Nephrol. 2006;17:2464–2473. doi: 10.1681/ASN.2006020185. [DOI] [PubMed] [Google Scholar]
- 76.Gong R, Rifai A, Dworkin LD. Hepatocyte growth factor suppresses acute renal inflammation by inhibition of endothelial E-selectin. Kidney Int. 2006;69:1166–1174. doi: 10.1038/sj.ki.5000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kang DH, Hughes J, Mazzali M, Schreiner GF, Johnson RJ. Impaired angiogenesis in the remnant kidney model: II. Vascular endothelial growth factor administration reduces renal fibrosis and stabilizes renal function. J Am Soc Nephrol. 2001;12:1448–1457. doi: 10.1681/ASN.V1271448. [DOI] [PubMed] [Google Scholar]
- 78.Iliescu R, Fernandez SR, Kelsen S, Maric C, Chade AR. Role of renal microcirculation in experimental renovascular disease. Nephrol Dial Transplant. 2010;25:1079–1087. doi: 10.1093/ndt/gfp605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Long DA, Mu W, Price KL, Roncal C, Schreiner GF, Woolf AS, Johnson RJ. Vascular endothelial growth factor administration does not improve microvascular disease in the salt-dependent phase of post-angiotensin II hypertension. Am J Physiol Renal Physiol. 2006;291:F1248–1254. doi: 10.1152/ajprenal.00096.2006. [DOI] [PubMed] [Google Scholar]
- 80.Burt LE, Forbes MS, Thornhill BA, Kiley SC, Minor JJ, Chevalier RL. Renal vascular endothelial growth factor in neonatal obstructive nephropathy. II. Exogenous VEGF. Am J Physiol Renal Physiol. 2007;292:F168–174. doi: 10.1152/ajprenal.00294.2005. [DOI] [PubMed] [Google Scholar]
- 81.Sato W, Tanabe K, Kosugi T, Hudkins K, Lanaspa MA, Zhang L, Campbell-Thompson M, Li Q, Long DA, Alpers CE, Nakagawa T. Selective stimulation of VEGFR2 accelerates progressive renal disease. Am J Pathol. 2011;179:155–166. doi: 10.1016/j.ajpath.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schrijvers BF, Flyvbjerg A, Tilton RG, Rasch R, Lameire NH, De Vriese AS. Pathophysiological role of vascular endothelial growth factor in the remnant kidney. Nephron Exp Nephrol. 2005;101:e9–15. doi: 10.1159/000086034. [DOI] [PubMed] [Google Scholar]
- 83.Leonard EC, Friedrich JL, Basile DP. VEGF-121 preserves renal microvessel structure and ameliorates secondary renal disease following acute kidney injury. Am J Physiol Renal Physiol. 2008;295:F1648–1657. doi: 10.1152/ajprenal.00099.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jung YJ, Kim DH, Lee AS, Lee S, Kang KP, Lee SY, Jang KY, Sung MJ, Park SK, Kim W. Peritubular capillary preservation with COMP-angiopoietin-1 decreases ischemia-reperfusion-induced acute kidney injury. Am J Physiol Renal Physiol. 2009;297:F952–960. doi: 10.1152/ajprenal.00064.2009. [DOI] [PubMed] [Google Scholar]
- 85.Gavard J, Patel V, Gutkind JS. Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev Cell. 2008;14:25–36. doi: 10.1016/j.devcel.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 86.Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson C, Kopp JB, Kabir MG, Backx PH, Gerber HP, Ferrara N, Barisoni L, Alpers CE, Quaggin SE. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–1136. doi: 10.1056/NEJMoa0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eremina V, Quaggin SE. Biology of anti-angiogenic therapy-induced thrombotic microangiopathy. Semin Nephrol. 2010;30:582–590. doi: 10.1016/j.semnephrol.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 88.Goligorsky MS, Yasuda K, Ratliff B. Dysfunctional endothelial progenitor cells in chronic kidney disease. J Am Soc Nephrol. 2010;21:911–919. doi: 10.1681/ASN.2009111119. [DOI] [PubMed] [Google Scholar]
- 89.Chade AR, Zhu X, Lavi R, Krier JD, Pislaru S, Simari RD, Napoli C, Lerman A, Lerman LO. Endothelial progenitor cells restore renal function in chronic experimental renovascular disease. Circulation. 2009;119:547–557. doi: 10.1161/CIRCULATIONAHA.108.788653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhu XY, Urbieta Caceres VH, Favreau FD, Krier JD, Lerman A, Lerman LO. Enhanced endothelial progenitor cell angiogenic potency, present in early experimental renovascular hypertension, deteriorates with disease duration. J Hypertens. 2011;29:1972–1979. doi: 10.1097/HJH.0b013e32834ae611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Togel F, Isaac J, Hu Z, Weiss K, Westenfelder C. Renal SDF-1 signals mobilization and homing of CXCR4-positive cells to the kidney after ischemic injury. Kidney Int. 2005;67:1772–1784. doi: 10.1111/j.1523-1755.2005.00275.x. [DOI] [PubMed] [Google Scholar]
- 92.Krenning G, Dankers PY, Drouven JW, Waanders F, Franssen CF, van Luyn MJ, Harmsen MC, Popa ER. Endothelial progenitor cell dysfunction in patients with progressive chronic kidney disease. Am J Physiol Renal Physiol. 2009;296:F1314–1322. doi: 10.1152/ajprenal.90755.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Perry TE, Song M, Despres DJ, Kim SM, San H, Yu ZX, Raghavachari N, Schnermann J, Cannon RO, 3rd, Orlic D. Bone marrow-derived cells do not repair endothelium in a mouse model of chronic endothelial cell dysfunction. Cardiovasc Res. 2009;84:317–325. doi: 10.1093/cvr/cvp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li B, Cohen A, Hudson TE, Motlagh D, Amrani DL, Duffield JS. Mobilized human hematopoietic stem/progenitor cells promote kidney repair after ischemia/reperfusion injury. Circulation. 2010;121:2211–2220. doi: 10.1161/CIRCULATIONAHA.109.928796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lin SL, Chang FC, Schrimpf C, Chen YT, Wu CF, Wu VC, Chiang WC, Kuhnert F, Kuo CJ, Chen YM, Wu KD, Tsai TJ, Duffield JS. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am J Pathol. 2011;178:911–923. doi: 10.1016/j.ajpath.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 97.Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, Brown KK, Flaherty KR, Noble PW, Raghu G, Brun M, Gupta A, Juhel N, Kluglich M, du Bois RM. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365:1079–1087. doi: 10.1056/NEJMoa1103690. [DOI] [PubMed] [Google Scholar]
- 98.Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]