

Abstract
β2 adrenergic receptor (β2AR) regulated many key physiological processes by activation of a heterotrimeric GTP binding protein (Gs protein). This process could be modulated by different types of ligands. But the details about this modulation process were still not depicted. Here, we performed molecular dynamics (MD) simulations on the structures of β2AR-Gs protein in complex with different types of ligands. The simulation results demonstrated that the agonist BI-167107 could form hydrogen bonds with Ser2035.42, Ser2075.46 and Asn2936.55 more than the inverse agonist ICI 118,551. The different binding modes of ligands further affected the conformation of β2AR. The energy landscape profiled the energy contour map of the stable and dissociated conformation of Gαs and Gβγ when different types of ligands bound to β2AR. It also showed the minimum energy pathway about the conformational change of Gαs and Gβγ along the reaction coordinates. By using interactive essential dynamics analysis, we found that Gαs and Gβγ domain of Gs protein had the tendency to separate when the inverse agonist ICI 118,551 bound to β2AR. The α5-helix had a relatively quick movement with respect to transmembrane segments of β2AR when the inverse agonist ICI 118,551 bound to β2AR. Besides, the analysis of the centroid distance of Gαs and Gβγ showed that the Gαs was separated from Gβγ during the MD simulations. Our results not only could provide details about the different types of ligands that induced conformational change of β2AR and Gs protein, but also supplied more information for different efficacies of drug design of β2AR.
Introduction
The β2 adrenergic receptor (β2AR) belonged to class A G protein-coupled receptors (GPCRs) [1] and regulated many key physiologically processes such as smooth muscle relaxation in the airways and the vasculature [2]–[7]. During the past years, much progress had been made in the determination of the crystal structure of β2AR with different types of ligands. The crystal structure of β2AR in complex with the inverse agonist carazolol was determined in 2007. It revealed the inactive conformation of β2AR [8]. The neutral antagonist alprenolol bound to β2AR structure was reported in 2010. This work showed that the antagonist could block agonist signal but maintain basal signal [9]. The irreversible agonist-β2AR complex was reported in 2011. This agonist was irreversible because it was covalently tethered to a specific site of β2AR [10]. At the same time, a reversible agonist-β2AR in complex with the camelid antibody fragment that exhibited G protein-like behavior was obtained by X-ray crystallography [11]. Besides, Rasmussen et al. reported the crystal structure of agonist-occupied β2AR and nucleotide-free Gs heterotrimer (α, β and γ). This work gave a model system for understanding the detailed mechanism about the activation of Gs and also for understanding the ligands induced conformation change of β2 adrenergic receptor-Gs (β2AR-Gs) protein complex [12]. The analysis of β2AR-Gs complex could provide some information about the essential mechanism of structural events linking GPCR-Gs protein complex formation by using peptide amide hydrogen-deuterium exchange mass spectrometry [13]. Engineering and characterization of β2AR-based on ion-channel coupled receptors gave new insights into the conformational dynamics of β2AR [14]. All these studies also indicated that it was difficult to obtain the crystal structure of the agonist-bound to active conformation of β2AR if the G protein did not bind to β2AR.
Even though the active conformation of β2AR-Gs have been resolved, it was still difficult to obtain the detailed information about the dynamic process of inactive or active state of β2AR-Gs from real experiments. Compared with experimental study, all atoms molecular dynamics simulations [15]–[20] and coarse-grained molecular dynamics simulations [21], [22] methods could provide much more dynamic information at the atomic level about the activation or inactivation mechanism of β2AR. Other computational methods such as molecular docking and conformational analysis [23]–[27] were also successfully used to study the function and activation mechanism as well as to discovery the small molecular ligands of β2AR on basis of the crystal structures. The MD simulations of agonist-β2AR complex showed that agonist, inverse agonist and antagonist had different interaction modes with the active sites of β2AR. The main reason was that the waters in the cavity of β2AR had different contribution to the stabilization of the interaction network [20]. The atomic level description illuminated that drug must cross two energetic barriers to get into the active site of β2AR. The first barrier was mainly due to hydrophobic interaction. The second energetic barrier was due to dehydration and allosteric receptor when the drug moved into the binding pocket [28]. In addition, Dror et al. proposed that the agonist-β2AR could transform momentarily from active to the inactive conformation based on the results of MD simulations. This study also showed β2AR had an intermediate state. The conformation of β2AR would be induced to active or inactive state if agonist or inverse agonist bound to the cavity of receptor [29]. Provasi et al. performed free energy calculation on the crystal structure of β2AR with different ligands (either inverse agonists, neural antagonists, or agonists). The simulation results suggested that different type ligands had different free energy landscape. Especially, the agonist had opposite energy barrier to the inverse agonist. And there was nearly no energy barrier when β2AR had no ligands in the cavity [30]. Goetz et al. studied the interaction between C-terminal end of Gαs and β2AR by performing MD simulations [31]. Feng et al. carried out 20 ns MD simulations on agonist-bound part of β2AR without Gβγ domain to investigate the activation mechanism of β2AR [32].
Despite these recent remarkable advances in β2AR structure determination and molecular dynamics simulation, the detailed mechanism by which different types of ligands induced dynamic conformational changes of β2AR and Gs protein during the modulated process was still not reported. Most of the reported works mainly focused on the complex of β2AR and ligands. In order to understand the modulation of Gs by β2AR, it was more reliable to perform MD simulation based on the crystal structure of β2AR-Gs complex. The following important questions still need to be answered, such as: what is the difference of binding mode between β2AR and different kinds of ligands? which kind of ligand could induce Gαs to separate from Gβγ? How did the inactive conformation of β2AR interact with Gs protein?
In order to further explore how different types of ligands affected the behavior of Gαs and Gβγ in the β2AR-Gs complex. We performed a total of 800 ns MD simulations on the complex of β2AR-Gs bound to agonist (BI-167107), antagonist (alprenolol), inverse agonist (ICI 118,551) and their unliganded form with explicit solvent and lipids at constant pressure and constant temperature. The graphics processing unit (GPU) computer was used to accelerate the MD simulations. The analysis of energy landscape was performed to illustrate the minimum energy pathway of the conformational change of Gαs and Gβγ along the reaction coordinates when ICI 118,551 bound to β2AR. Furthermore, we used interactive essential dynamics (IED) [33] to identify the dissociation of Gαs and Gβγ by analyzing the MD simulated trajectory. Our simulated results showed that Gαs was separated from the Gβγ when the ICI 118,551 bound to active sites of β2AR. Besides, the α5-helix had fast motion relative to TM3, TM5, TM6, TM7 of β2AR if the ICI 118,551 bound to β2AR. Our results could also provide the information about the inactivation and activation mechanism of Gs protein induced by different types of ligands.
Results and Discussion
Structure of β2AR-Gs Complex
The structure of β2AR-Gs with explicit waters and lipids was shown as in Figure 1. The thickness for membrane location was about 30±1.0 Å, which was calculated by OPM database [34]. The main part of β2AR-Gs consisted of β2AR, Gαs and Gβγ. The loop between TM5 and TM6 was modeled on basis of the crystal structure of β2AR-Gs. TM3, TM5, TM6 and TM7 (TM3,5,6,7) were shown in the origin part of β2AR-Gs. The black part was α5-helix. The residues of the active site in the pocket of β2AR include Asp1133.32, Ser2035.42, Ser2075.46, Asn2936.5, Tyr3087.35 and Asn3127.39 (see Figure 2A). The space surrounded by these sites was the volume of β2AR. The crystal structure of β2AR-Gs in complex with the agonist (BI-167107) was used in our simulations In order to get β2AR-Gs in complex with different kinds of ligands, the inverse agonist (ICI 118,551) and antagonist (alprenolol) were docked into the pocket of β2AR-Gs. The 200 ns MD simulations were performed for β2AR-Gs in complex with different ligands on a workstation equipped with four pieces of graphics processing unit (GPU) and two processors with six cores (see Figure S1).
Figure 1. The structure of simulated complex.

The red points are water. The cyan lipids represent membrane. The membrane and water only show the positive part of y axis.
Figure 2. Snapshot of the hydrogen bonds between different ligands and β2AR.

(A) The binding sites of β2AR. (B) Alprenolol forms three hydrogen bonds with Asp113, Tyr308 and Asn312. (C) BI-167107 has five hydrogen bonds with Asp113, Ser203, Ser207, Asn293 and Asn312. (D) ICI 118,551 forms two hydrogen bonds with Asp113 and Asn312.
Ligands Bound to Different Sites of β2AR
After 200 ns MD simulations, the analysis of hydrogen bonds occupancy showed that inverse agonist (ICI 118,551), antagonist (alprenolol) and agonist (BI-167107) could form hydrogen bonds with different sites of β2AR-Gs (Figure 3A and 3B). We also obtained the hydrogen bond interaction between β2AR and different ligands (see Figure 2B, 2C and 2D) from the MD simulation trajectory at the same time. ICI 118,551 only had two stable hydrogen bonds with Asp1133.32 and Asn3127.39 (Figure 2D and Figure 3A). In comparison, BI-167107 had another three stable hydrogen bonds with Ser2035.42, Ser2075.46 and Asn2936.55 besides Asp1133.32 and Asn3127.39 (Figure 3A, 3B and Figure 2C). Alprenolol had a similar binding mode with ICI 118,551 except lower hydrogen bonds occupancy on Tyr3087.35 (Figure 3A, 3B and Figure 2B). The number of hydrogen bonds also showed BI-167107 could form more hydrogen bonds than alprenolol and ICI 118,551 along the simulation time (Figure 3C). The main reason was that BI-167107 had more oxygen and hydroxyl groups than Alprenolol and ICI 118,551 as shown in the black oval of Figure 4, so BI-167107 could be easy to form another three hydrogen bonds with Ser2035.42, Ser2075.46 and Asn2936.55 (see Figure 2C). The results showed that inverse agonist had different binding modes with agonist and antagonist.
Figure 3. The hydrogen bonds occupancy and volume of binding pocket.

(A–B) The column represents the percent of hydrogen bonds occupancy when the residues are as hydrogen bonds acceptor or donor in the pocket of β2AR. (C) The total number of hydrogen bonds versus the simulated time. (D) The ligands-bound pocket volume of β2AR versus the simulation time.
Figure 4. Structures of BI-167107, ICI 118,551 and alprenolol.

The oxygen and hydroxyl groups in the black oval form another three hydrogen bonds with the active sites of β2AR.
In order to measure the pocket change of β2AR during the simulations, the pocket detection plugin of VMD [35], [36] was used to calculate the lignad-bound pocket volume versus simulation time (Figure 1 and Figure 3D). The value of the pocket volume of unliganded complex showed that this conformation of β2AR was in the intermediate state. The pocket volume would become larger when the inverse agonist ICI 118,551 bound to the pocket of β2AR, while the pocket volume would shrink when the agonist BI-167107 or antagonist alprenolol bound to β2AR. These results indicated different ligands could adjust the pocket space size of the β2AR though different binding modes of β2AR. The changes of pocket volume size would further affect the conformation of β2AR.
Conformation CHANGE of β2AR Induced by Different Ligands
In order to study conformational change of β2AR induced by different ligands, the root mean square deviation (RMSD) of the backbone atoms of β2AR was measured versus simulation time (Figure 5A). The β2AR in complex with ICI 118,551 reached equilibrium phase after 5 ns MD simulations (see Figure S2). The RMSD of β2AR-ICI 118,551 still maintained about 2.7 Å until 26 ns MD simulations (Figure 5A). By comparison with the RMSD of β2AR-BI-167107, we could see that β2AR-ICI 118,551 was still in active conformation. After 26 ns, the conformation of β2AR was changed into another state. In order to make sure the conformational feature of β2AR, FATCAT rigid algorithm [37] was used to calculate the RMSD with respect to the crystal structure of inverse agonist ICI 118,551-bound β2AR (PDB code: 3NY8) (see Table S1). The RMSD values in the Table S1 indicated the simulated conformation was closer to the inactive conformation, while the increased value of RMSD after about 26 ns suggested that simulated structures had different conformation with the agonist-bound β2AR (see Figure 5A). The β2AR-alprenolol and unliganded form of β2AR had similar RMSD with β2AR-BI-167107. It suggested that β2AR did not change its active state if alprenolol, BI-167107 or no ligand bound to β2AR. The active and inactive state of β2AR could be identified by some reported sites (Ile1213.40/Phe2826.44, NPxxY region: Asn3227.49-Tyr3267.53 and Asp1925.31/Lys3057.32) [9], [29]. These sites could be used to distinguish the active and inactive conformation of β2AR.
Figure 5. Active and inactive state of β2AR.

(A) RMSD of the backbone atoms of β2AR versus simulation time. (B) Time evolution of RMSD of non-hydrogen atoms of Ile1213.40 and Phe2826.44. (C) Time evolution of RMSD of the backbone atoms of NPxxY region. (D) Distance of Cα carbons of Asp1925.31 and Lys3057.32 versus simulation time.
Figure 5B illustrated different RMSD of non-hydrogen atoms of Ile1213.40/Phe2826.44 when ICI 118,551, alprenolol, BI-167107 or no ligand bound to β2AR. With the increased time of MD simulations, RMSD of Ile1213.40/Phe2826.44 of β2AR in complex without ligand was up to the same level of agonist, antagonist-bound β2AR as shown in Figure 5B. These states represented the active conformation of β2AR. In comparison, the lower RMSD of Ile1213.40/Phe2826.44 of β2AR-ICI 118,551 represented the inactive conformation of β2AR.
Figure 5C showed the RMSD of the backbone atoms of NPxxY motif which could distinguish different states of β2AR. The RMSD of NPxxY region of β2AR-unligand was close to the level of β2AR-BI-167107 after about 148 ns MD simulations (see Figure 5C). The data also showed that β2AR-alprenolol had different RMSD of NPxxY region with unliganded, BI-167107 and ICI 118,551-bound β2AR. The possible reason was that the conserved NPxxY region could discern diverse conformations of β2AR when different types of ligands bound to β2AR.
Figure 5D described the distance of Cα carbons of Asp1925.31 and Lys3057.32 versus MD simulation time. The distance divided the conformation of β2AR into the inactive part and active part because Asp1925.31 and Lys3057.32 only represented part of extracellular surface of β2AR. ICI 118,551 and unligand belonged to inactive part while alprenolol and BI-167107 played an active role.
All these results corresponded to distinct functional behavior of different types of ligands. The inverse agonist ICI 118,551 could block the activating signaling. In contrast, unliganded and alprenolol-bound β2AR could maintain the basal activity signaling. BI-167107 could enhance the active signaling of β2AR [9].
Energy Landscape of Gαs and Gβγ
The above simulated results showed that different types of ligands could regulate the diverse states of β2AR. Besides, Gαs and Gβγ had shown some interesting conformations when BI-167107, alprenolol, ICI 118,551 or no ligand bound to the active sites of β2AR. Our molecular dynamics simulations trajectory of β2AR-Gs contained a wide range of conformational spaces. Therefore, abundant information was supplied for the energy landscape analysis of the conformations of Gαs and Gβγ. Two major motions represented the conformations of Gαs and Gβγ: one was the centroid distance of Gαs and Gβγ, the other was the RMSD of Gαs and Gβγ.
Figure 6 illustrated the energy landscape of Gαs and Gβγ corresponding to two reaction coordinates. This energy landscape contained one major deep well when the BI-167107, alprenolol or no ligand bound to β2AR (see Figure 6A, 6B and 6C). This energy part represented the stable structure of Gαs and Gβγ which was not separated from each other. However, the energy landscape consisted of three main deep wells when the ICI 118,551 combined with β2AR. The white points depicted the minimum energy pathway. It was mainly relevant to the stable conformation of Gαs and Gβγ (0∼43 ns) before the first deep well. Along with the change of simulated time, the Gαs and Gβγ complex passed over an energy barrier of ∼2.0 kcal/mol. At the same time, the stable conformation of Gαs and Gβγ became to dissociated state. It only need overcome the energy barrier of ∼0.5 kcal/mol for each neighboring deep well. These three deep wells represented the dissociated conformation of Gαs and Gβγ (see Figure 6D). In additions, Figure 6D showed the lowest energy barrier of ∼1.5 kcal/mol in the deep well, while Figure 6A, 6B, 6C showed the lowest energy barrier of deep well was ∼0.5 kcal/mol. It further indicated the domain of Gαs and Gβγ was not stable when ICI 118,551 bound to β2AR.
Figure 6. Energy landscape of Gαs and Gβγ.

(A–D) The energy landscape map of Gαs and Gβγ in complex without ligand or with alprenolol, BI-167107 and ICI 118,551. Reaction coordinates are defined two parts: the centroid distance between Gαs and Gβγ; the RMSD of Gαs and Gβγ. The white points represent the minimum energy pathway.
Gαs is Separated from Gβγ
After analysis of the energy landscape of Gαs and Gβγ, it is interesting to study the movement of Gαs and Gβγ. The motions of Gαs and Gβγ were analyzed by interactive essential dynamics (IED) analysis [33]. The two principal components of motions revealed the movements of TM5, TM6 and Gαs and Gβγ (Figure 7). The Gαs did not move away from Gβγ when BI-167107 and alprenolol bound to β2AR (Figure 7A and 7B). The Gαs and Gβγ domain was also not dissociated when there was no ligand on the β2AR (Figure 7D). In this case, TM5 and TM6 had almost no relative motion. In comparison, the Gαs domain was separated from Gβγ domain when ICI 118,551 bound to β2AR. At the same time, TM5 and TM6 had the open tendency with respect to Gβγ domain (Figure 7C).
Figure 7. IED plot of principal motions of Gαs and Gβγ.

(A–D) Unliganded, BI-167107 and alprenolol-bound β2AR has similar movement. Gαs and Gβγ keep the similar direction of motions. ICI 118,551 induces Gαs and Gβγ to separate from each other.
The α5-helix had been reported to play an important role on the interaction between β2AR and Gs protein [12], [13], [32]. The sketch of the structure of α5-helix and TM3,5,6,7 was shown in Figure 1. The centroid distance between α5-helix and TM3,5,6,7 was measured over the simulation time. As shown in black oval of Figure 8A, the centroid distance between α5-helix and TM3,5,6,7 was dropped sharply when ICI 118,551 bound to the pocket of β2AR. It indicated that α5-helix moved quickly relative to TM3,5,6,7. After about 43 ns MD simulations, the centroid distance became longer when BI-167107, alprenolol or no ligands was in the active pocket of β2AR, while the distance was shorter when ICI 118,551 bound to β2AR. We also analyzed the RMSD of the backbone atoms of α5-helix and TM3,5,6,7 (see Figure S3). It could be seen that both of the studied systems reached equilibrium in 200 ns. The β2AR-ICI 118,551 system had larger RMSD value of α5-helix and TM3,5,6,7 than the β2AR bound to alprenolol and BI-167107. It also suggested that the conformation of α5-helix and TM3,5,6,7 had a larger structural fluctuation when ICI 118,551 combined with β2AR. Besides, we also calculated the centroid distance of Gαs and Gβγ domain (Figure 8B). The centroid distance of Gαs and Gβγ domain kept in about 37 Å when alprenolol, BI-167107 or no ligand bound to β2AR. In contrast, Gαs and Gβγ domain was separated obviously from each other after 43 ns MD simulations when ICI 118,551 bound to the pocket of β2AR. Movie S1 gave a detailed animation about the separation or association of Gαs and Gβγ induced by different ligands. This dissociation was almost accompanied with the relative movement of α5-helix. When the relative motion of α5-helix stopped at about 43 ns, the Gαs and Gβγ were separated from each other (see Figure 8A and 8B). At the same time, we could see the RMSD of β2AR changed after about 26 ns (Figure 5A). After another 17 ns, Gαs moved away from Gβγ. It suggested the inverse agonist ICI 118,551 induced the separation of Gαs and Gβγ though changing the conformation of β2AR.
Figure 8. Motions of Gαs and Gβγ domain.

(A) The centroid distance of α5-helix and TM3, TM5, TM6, TM7 versus simulation time. (B) Time evolution of centroid distance of Gαs and Gβγ. (C) The cartoon representation of the dissociation mechanism of Gαs and Gβγ.
The above results indicated that different kinds of ligands could induce the different behaviors of Gαs and Gβγ through changing the conformation of β2AR. The Gαs and Gβγ domain were not stable when ICI 118,551 bound to β2AR. In contrast, Gαs and Gβγ domain would keep the stable distance if BI-167107, alprenolol or no ligand bound to β2AR [9].
Conclusions
In this study, we focused on the binding mode between β2AR and different ligands and the conformational states of β2AR in complex with Gαs and Gβγ domain. The hydrogen bonds occupancy showed that Alprenolol, BI-167107 and ICI 118,551 in the pocket of β2AR formed different number of hydrogen bonds with the binding site of β2AR. These different binding modes would affect the pocket volume size of β2AR. The changes of pocket space further affected the conformation of β2AR. The results of RMSD indicated that ICI 118,551 could induce β2AR to change from active conformation to inactive state. The other ligands were inclined to keep β2AR active. Specially, the energy landscape showed three main deep wells when the ICI 118,551 bound with β2AR. It suggested ICI 118,55 could induced the conformational change of Gαs and Gβγ. The analysis of IED and centroid distance further illustrated the inactive conformation of β2AR induced by ICI 118,551 could lead to the dissociation of Gαs and Gβγ. In comparison, the Gαs and Gβγ would maintain the relative stable distance if there was alprenolol, BI-167107 or no ligand in the active site of β2AR (Figure 8C). In total, our MD simulations and energy landscape results demonstrated that different ligands-bound β2AR induced the dissociation of downstream Gαs and Gβγ. These results not only depicted the detail dissociation mechanism of Gαs and Gβγ domain which was adjusted indirectly by different ligands, but also could give more clues for the design of potential ligands with different modulating functions.
Materials and Methods
Protein Structures Preparation
The agonist-bound model of β2AR was prepared beginning from the crystal structure (PDB ID: 3SN6) [12] by removing T4 lysozyme and nanobody (Nb35). Because TM5 and TM6 played an important role in the interaction between β2AR and Gs, the missing intracellular loop 3 was added by using the loop model algorithm of MODELLER [38] (see Protocol S1). The neutral antagonist (alprenolol) was extracted from the model (PDB ID: 3NYA) [39]. The inverse agonist (ICI 118,551) was obtained from the crystal structure (PDB ID: 3NY8) [39]. In order to obtain the protein-ligand complex, the inverse agonist and neutral antagonist were docked into the pocket of β2AR using AutoDock Vina program [40]. The docking complexes were then used as the starting models for membrane location. The model of β2AR-Gs was embedded into an explicit 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) by using VMD program [36]. The orientation of membrane was described in Protocol S1 and Figure 1. The length and width of lipid box was 120 Å × 120 Å. The TIP3P water model [41] was used to build the water box which dimensions were 120 Å × 120 Å × 150 Å. Seven sodium ions were added to neutralize the system which contained about 200,010 atoms per periodic cell. The CHARMM force field parameterizations of BI-167107, alprenolol and ICI 118,551 were developed by using VMD Paratool Plugin v1.2 [42] and Gaussian 98 Revision A.9 [43]: The RHF/6–31G* model was used with tight SCF convergence criteria for geometry optimization calculation. The single point calculation was computed at the theory of RHF/6–31G* level with tight SCF convergence criteria.
Molecular Dynamics Simulations
The β2AR-Gs in complex with alprenolol, BI-167107, ICI 118,551 or without ligand were built with explicit lipids and water, respectively. In order to equilibrate these four systems, firstly, each system was fixed except lipid tail for minimizing 100 ps and equilibrating 1000 ps under constant temperature (300 K) and constant pressure (1 bar). Secondly, each system was minimized for 500 ps and equilibrated for 0.5 ns with protein and ligand constrained. Then, 5 ns equilibrated simulations were performed without any constraint. At last, a total of 200 ns MD simulations were performed on the each system under a constant temperature of 300 k and a constant pressure of 1 bar.
Our MD simulations were performed with time step of 2 fs in explicit water and periodically infinite lipid through using NAMD package (version 2.9b3) [44] with CHARMM27 force field [45]. The minimization was based on a conjugate gradient method. The particle-mesh Ewald (PME) [46] method was used to calculate electrostatics with a 12 Å nonbonded cutoff. Langevin piston and Langevin barostat methods were employed for the temperature and pressure respectively [47]. The frames were saved every 20.0 ps during the MD simulations.
All MD simulations were performed on the GPU workstation. In order to get the highest efficiency of GPU, the speed test of GPU workstation was carried out with different collocations of Cores and GPU (see Figure S1).The speed test results proved that running on 12 cores of an array of two 2.66-GHz Intel Xeon 5650 processors and 4 pieces of NVDIA Tesla C 2050 graphics card could get the highest speed. The wall clock time was about 3.46 ns per day.
Hydrogen Bonds and Volume Calculation
In the statistical analysis of the hydrogen bonds occupancy, the distance and angle between the acceptor and donor atoms were set less than 3.5 Å and 35°, respectively [48], [49]. The polyhedral volumetric model of the pocket detection plugin of VMD [35], [36] was used to find the pocket volume of β2AR.
Interactive Essential Dynamics Analysis
For the interactive essential dynamics (IED) analysis [33], the complex were split into three parts: β2AR, Gαs and Gβγ. 25 eigenvectors were generated for each part on the basis of trajectory file, then 25 projections were obtained from eigenvectors. The IED was calculated by equation 1:
| (1) |
Where 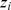 represented the ith principal component.
represented the ith principal component. 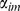 was weight coefficient.
was weight coefficient. 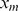 represented the position. The first two components could represent the main motions of protein. More details about IED method were described in the Text S1. Trajectory analysis was carried out using AmberTools12 and VMD [36], [50].
represented the position. The first two components could represent the main motions of protein. More details about IED method were described in the Text S1. Trajectory analysis was carried out using AmberTools12 and VMD [36], [50].
Energy Landscape Analysis
The energy landscape of the conformational change of protein complex could be estimated by an appropriate conformation sampling method. In order to get the a two dimensional (2D) energy landscape map, the centroid distance between Gαs and Gβγ, which mainly represented the motion, and the RMSD of Gαs and Gβγ, which corresponded the conformational fluctuation, were chosen as the reaction coordinates. The energy landscape could be calculated along these two reaction coordinates as equation 2 [51]–[54] shown:
| (2) |
Where 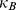 represented the Boltzmann constant, T was the simulated temperature, and
represented the Boltzmann constant, T was the simulated temperature, and 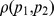 represented the normalized joint probability distribution.
represented the normalized joint probability distribution.
Supporting Information
Speed test of GPU workstation. Workstation with 12 Cores+4GPU gives the fastest speed.
(TIF)
RMSD of backbone atoms of β2AR versus 5 ns MD simulations time.
(TIF)
Time evolution of RMSD of the backbone atoms of α5-helix and TM 3,5,6,7.
(TIF)
RMSD of simulated conformational backbone atoms with respect to the crystal structure of ICI 118,551-bound β2AR.
(DOC)
Interactive Essential Dynamics.
(DOC)
Membrane building protocol.
(DOC)
Animation about the separation or association of Gαs and Gβγ induced by different ligands.
(AVI)
Acknowledgments
The authors wish to thank the Center of Communication and Network of Lanzhou University for supplying the graphics processing unit (GPU) workstation.
Funding Statement
This work was supported by the National Natural Science Foundation of China (Grant Nos. 21175063), the Fundamental Research Funds for the Central Universities (Grant Nos. lzujbky-2011-19). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Milligan G, Svoboda P, Brown CM (1994) Why are there so many adrenoceptor subtypes? Biochemical pharmacology 48: 1059–1071. [DOI] [PubMed] [Google Scholar]
- 2.Johnson M (2006) Molecular mechanisms of beta(2)-adrenergic receptor function, response, and regulation. J Allergy Clin Immunol 117: 18–24; quiz 25. [DOI] [PubMed]
- 3.McGraw DW, Liggett SB (2005) Molecular mechanisms of beta2-adrenergic receptor function and regulation. Proc Am Thorac Soc 2: 292–296; discussion 311–292. [DOI] [PMC free article] [PubMed]
- 4. Goral V, Jin Y, Sun H, Ferrie AM, Wu Q, et al. (2011) Agonist-directed desensitization of the β2-adrenergic receptor. PloS one 6: e19282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Scarselli M, Annibale P, Radenovic A (2012) Cell type-specific beta2-adrenergic receptor clusters identified using photoactivated localization microscopy are not lipid raft related, but depend on actin cytoskeleton integrity. J Biol Chem 287: 16768–16780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Greene D, Kang S, Kosenko A, Hoshi N (2012) Adrenergic regulation of HCN4 channel requires protein association with beta2-adrenergic receptor. J Biol Chem 287: 23690–23697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ma X, Zhao Y, Daaka Y, Nie Z (2012) Acute activation of beta2-adrenergic receptor regulates focal adhesions through betaArrestin2- and p115RhoGEF protein-mediated activation of RhoA. J Biol Chem 287: 18925–18936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, et al. (2007) High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318: 1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bokoch MP, Zou Y, Rasmussen SG, Liu CW, Nygaard R, et al. (2010) Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature 463: 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, et al. (2011) Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature 469: 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, et al. (2011) Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469: 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, et al. (2011) Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477: 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chung KY, Rasmussen SG, Liu T, Li S, DeVree BT, et al. (2011) Conformational changes in the G protein Gs induced by the beta2 adrenergic receptor. Nature 477: 611–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Caro LN, Moreau CJ, Revilloud J, Vivaudou M (2011) beta2-Adrenergic ion-channel coupled receptors as conformational motion detectors. PloS one 6: e18226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vanni S, Neri M, Tavernelli I, Rothlisberger U (2009) Observation of “Ionic Lock” Formation in Molecular Dynamics Simulations of Wild-Type β1 and β2 Adrenergic Receptors. Biochemistry 48: 4789–4797. [DOI] [PubMed] [Google Scholar]
- 16. Gouldson PR, Higgs C, Smith RE, Dean MK, Gkoutos GV, et al. (2000) Dimerization and domain swapping in G-protein-coupled receptors: a computational study. Neuropsychopharmacology 23: S60–77. [DOI] [PubMed] [Google Scholar]
- 17. Sadiq SK, Guixa-Gonzalez R, Dainese E, Pastor M, De Fabritiis G, et al. (2013) Molecular Modeling and Simulation of Membrane Lipid-Mediated Effects on GPCRs. Curr Med Chem 20: 22–38. [PubMed] [Google Scholar]
- 18. Bhattacharya S, Hall SE, Li H, Vaidehi N (2008) Ligand-stabilized conformational states of human beta(2) adrenergic receptor: insight into G-protein-coupled receptor activation. Biophys J 94: 2027–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Furse KE, Lybrand TP (2003) Three-Dimensional Models for β-Adrenergic Receptor Complexes with Agonists and Antagonists. Journal of Medicinal Chemistry 46: 4450–4462. [DOI] [PubMed] [Google Scholar]
- 20. Vanni S, Neri M, Tavernelli I, Rothlisberger U (2011) Predicting novel binding modes of agonists to beta adrenergic receptors using all-atom molecular dynamics simulations. PLoS Comput Biol 7: e1001053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stansfeld PJ, Sansom MSP (2011) Molecular Simulation Approaches to Membrane Proteins. Structure 19: 1562–1572. [DOI] [PubMed] [Google Scholar]
- 22. Fanelli F, De Benedetti PG (2005) Computational modeling approaches to structure-function analysis of G protein-coupled receptors. Chem Rev 105: 3297–3351. [DOI] [PubMed] [Google Scholar]
- 23. Simpson LM, Taddese B, Wall ID, Reynolds CA (2010) Bioinformatics and molecular modelling approaches to GPCR oligomerization. Current Opinion in Pharmacology 10: 30–37. [DOI] [PubMed] [Google Scholar]
- 24. Vilar S, Ferino G, Phatak SS, Berk B, Cavasotto CN, et al. (2011) Docking-based virtual screening for ligands of G protein-coupled receptors: Not only crystal structures but also in silico models. Journal of Molecular Graphics and Modelling 29: 614–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vaidehi N (2010) Dynamics and flexibility of G-protein-coupled receptor conformations and their relevance to drug design. Drug Discovery Today 15: 951–957. [DOI] [PubMed] [Google Scholar]
- 26. Ivetac A, McCammon JA (2010) Mapping the Druggable Allosteric Space of G-Protein Coupled Receptors: a Fragment-Based Molecular Dynamics Approach. Chemical Biology & Drug Design 76: 201–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gouldson PR, Snell CR, Reynolds CA (1997) A New Approach to Docking in the β2-Adrenergic Receptor That Exploits the Domain Structure of G-Protein-Coupled Receptors. Journal of Medicinal Chemistry 40: 3871–3886. [DOI] [PubMed] [Google Scholar]
- 28. Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, et al. (2011) Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc Natl Acad Sci U S A 108: 13118–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, et al. (2011) Activation mechanism of the beta2-adrenergic receptor. Proc Natl Acad Sci U S A 108: 18684–18689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Provasi D, Artacho MC, Negri A, Mobarec JC, Filizola M (2011) Ligand-induced modulation of the free-energy landscape of G protein-coupled receptors explored by adaptive biasing techniques. PLoS Comput Biol 7: e1002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Goetz A, Lanig H, Gmeiner P, Clark T (2011) Molecular dynamics simulations of the effect of the G-protein and diffusible ligands on the beta2-adrenergic receptor. J Mol Biol 414: 611–623. [DOI] [PubMed] [Google Scholar]
- 32. Feng Z, Hou T, Li Y (2012) Studies on the Interactions between β2 Adrenergic Receptor and Gs Protein by Molecular Dynamics Simulations. J Chem Inf Model 52: 1005–1014. [DOI] [PubMed] [Google Scholar]
- 33. Mongan J (2004) Interactive essential dynamics. J Comput Aided Mol Des 18: 433–436. [DOI] [PubMed] [Google Scholar]
- 34. Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI (2006) OPM: orientations of proteins in membranes database. Bioinformatics 22: 623–625. [DOI] [PubMed] [Google Scholar]
- 35. Edelsbrunner H, Koehl P (2003) The weighted-volume derivative of a space-filling diagram. Proc Natl Acad Sci U S A 100: 2203–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14: 33–38, 27–38. [DOI] [PubMed]
- 37. Ye Y, Godzik A (2003) Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics 19 Suppl 2ii246–255. [DOI] [PubMed] [Google Scholar]
- 38. Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234: 779–815. [DOI] [PubMed] [Google Scholar]
- 39. Wacker D, Fenalti G, Brown MA, Katritch V, Abagyan R, et al. (2010) Conserved binding mode of human beta2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J Am Chem Soc 132: 11443–11445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31: 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics 79: 926–935. [Google Scholar]
- 42. Saam J, Ivanov I, Walther M, Holzhutter HG, Kuhn H (2007) Molecular dioxygen enters the active site of 12/15-lipoxygenase via dynamic oxygen access channels. Proc Natl Acad Sci U S A 104: 13319–13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, et al.. (1998) Gaussian 98 (Revision A.9). Gaussian, Inc, Pittsburgh PA.
- 44. Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, et al. (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26: 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, et al. (1998) All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins†. The Journal of Physical Chemistry B 102: 3586–3616. [DOI] [PubMed] [Google Scholar]
- 46. Darden T, York D, Pedersen L (1993) Particle mesh Ewald: An N [center-dot] log(N) method for Ewald sums in large systems. The Journal of Chemical Physics 98: 10089–10092. [Google Scholar]
- 47. Feller SE, Zhang Y, Pastor RW, Brooks BR (1995) Constant pressure molecular dynamics simulation: The Langevin piston method. The Journal of Chemical Physics 103: 4613–4621. [Google Scholar]
- 48. Bai Q, Shen Y, Yao X, Wang F, Du Y, et al. (2011) Modeling a new water channel that allows SET9 to dimethylate p53. PloS one 6: e19856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Espinosa E, Molins E, Lecomte C (1998) Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chemical Physics Letters 285: 170–173. [Google Scholar]
- 50.Case DA, Darden TA, Cheatham TE III, Simmerling CL, Wang J, et al.. (2012) AMBER 12. University of California, San Francisco.
- 51. Papaleo E, Mereghetti P, Fantucci P, Grandori R, De Gioia L (2009) Free-energy landscape, principal component analysis, and structural clustering to identify representative conformations from molecular dynamics simulations: The myoglobin case. Journal of molecular graphics and modelling 27: 889–899. [DOI] [PubMed] [Google Scholar]
- 52. Zhou R, Berne BJ, Germain R (2001) The free energy landscape for β hairpin folding in explicit water. Proceedings of the National Academy of Sciences 98: 14931–14936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Du Y, Yang H, Xu Y, Cang X, Luo C, et al. (2012) Conformational Transition and Energy Landscape of ErbB4 Activated by Neuregulin1β: One Microsecond Molecular Dynamics Simulations. Journal of the American Chemical Society 134: 6720–6731. [DOI] [PubMed] [Google Scholar]
- 54. Cui Y-L, Zhang J-L, Zheng Q-C, Niu R-J, Xu Y, et al. (2013) Structural and Dynamic Basis of Human Cytochrome P450 7B1: A Survey of Substrate Selectivity and Major Active Site Access Channels. Chemistry – A European Journal 19: 549–557. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Speed test of GPU workstation. Workstation with 12 Cores+4GPU gives the fastest speed.
(TIF)
RMSD of backbone atoms of β2AR versus 5 ns MD simulations time.
(TIF)
Time evolution of RMSD of the backbone atoms of α5-helix and TM 3,5,6,7.
(TIF)
RMSD of simulated conformational backbone atoms with respect to the crystal structure of ICI 118,551-bound β2AR.
(DOC)
Interactive Essential Dynamics.
(DOC)
Membrane building protocol.
(DOC)
Animation about the separation or association of Gαs and Gβγ induced by different ligands.
(AVI)