

Abstract
Apolipoprotein E (ApoE) is a major cholesterol carrier that supports lipid transport and injury repair in the brain. APOE polymorphic alleles are the main genetic determinants of Alzheimer disease (AD) risk: individuals carrying the ε4 allele are at increased risk of AD compared with those carrying the more common ε3 allele, whereas the ε2 allele decreases risk. Presence of the APOE ε4 allele is also associated with increased risk for cerebral amyloid angiopathy and age-related cognitive decline during normal ageing. ApoE–lipoproteins bind to several cell-surface receptors to deliver lipids and also to hydrophobic amyloid-β (Aβ) peptide, which is thought to initiate toxic events that lead to synaptic dysfunction and neurodegeneration in AD. ApoE isoforms differentially regulate Aβ aggregation and clearance in the brain, and have distinct functions in regulating brain lipid transport, glucose metabolism, neuronal signalling, neuroinflammation, and mitochondrial function. In this Review, we describe current knowledge on ApoE in the CNS, with a particular emphasis on the clinical and pathological features associated with carriers of different ApoE isoforms. We also discuss Aβ-dependent and Aβ-independent mechanisms that link ApoE4 status with AD risk, and consider how to design effective strategies for AD therapy by targeting ApoE.
Introduction
Alzheimer disease (AD) is a progressive neurodegenerative disease associated with cognitive decline and is the most common form of dementia in the elderly. Approximately 13% of people over the age of 65 and 45% over the age of 85 are estimated to have AD.1 Mounting evidence from genetic, pathological, and functional studies has shown that an imbalance between the production and clearance of amyloid-β (Aβ) peptides in the brain results in accumulation and aggregation of Aβ. The toxic Aβ aggregates in the form of soluble Aβ oligomers, intraneuronal Aβ, and amyloid plaques injure synapses and ultimately cause neurodegeneration and dementia.2, 3 The toxicity of Aβ seems to depend on the presence of microtubule-associated protein tau,4 the hyperphosphorylated forms of which aggregate and deposit in AD brains as neurofibrillary tangles. Aβ is composed of 40 or 42 amino acids and is generated through proteolytic cleavage of amyloid precursor protein (APP).5
Early-onset familial AD, which typically develops before the age of 65 years and accounts for only a small portion (<1%) of AD cases,2, 3 is primarily caused by overproduction of Aβ owing to mutations in either the APP gene or genes encoding presenilin 1 (PSEN1) or presenilin 2 (PSEN2), essential components of the γ-secretase complexes responsible for cleavage and release of Aβ. The majority of AD cases occur late in life (>65 years) and are commonly referred to as late-onset AD (LOAD). Although multiple genetic and environmental risk factors are involved in LOAD pathogenesis, overall impairment in Aβ clearance is probably a major contributor to disease development.6 Genetically, the ε4 allele of the apolipoprotein E (APOE) gene is the strongest risk factor for LOAD.7–9 The human APOE gene exists as three polymorphic alleles—ε2, ε3 and ε4—which have a worldwide frequency of 8.4%, 77.9% and 13.7%, respectively.10 However, the frequency of the ε4 allele is dramatically increased to ~40% in patients with AD.10
ApoE regulates lipid homeostasis by mediating lipid transport from one tissue or cell type to another.11 In peripheral tissues, ApoE is primarily produced by the liver and macrophages, and mediates cholesterol metabolism in an isoform-dependent manner. ApoE4 is associated with hyperlipidaemia and hypercholesterolemia, which lead to atherosclerosis, coronary heart disease and stroke.11, 12 In the CNS, ApoE is mainly produced by astrocytes, and transports cholesterol to neurons via ApoE receptors, which are members of the low-density lipoprotein receptor (LDLR) family.8
ApoE is composed of 299 amino acids and has a molecular mass of ~34 kDa.11 Differences between the three ApoE isoforms are limited to amino acids 112 and 158, where either cysteine or arginine is present (Figure 1a): ApoE2 (Cys112, Cys158), ApoE3 (Cys112, Arg158), and ApoE4 (Arg112, Arg158).11 The single amino acid differences at these two positions affect the structure of ApoE isoforms and influence their ability to bind lipids, receptors and Aβ.13–15 Human and animal studies clearly indicate that ApoE isoforms differentially affect Aβ aggregation and clearance. Several Aβ-independent functions are also associated with ApoE isoforms. In this Review, we provide an overview of clinical evidence for the association between APOE genotypes and the risk of cognitive decline in AD, mild cognitive impairment (MCI) and other CNS diseases with a cognitive component, and discuss our current understanding of the mechanisms underlying ApoE actions and ApoE-targeted therapies.
Figure 1. APOE ε4 is a major genetic risk factor for Alzheimer disease.
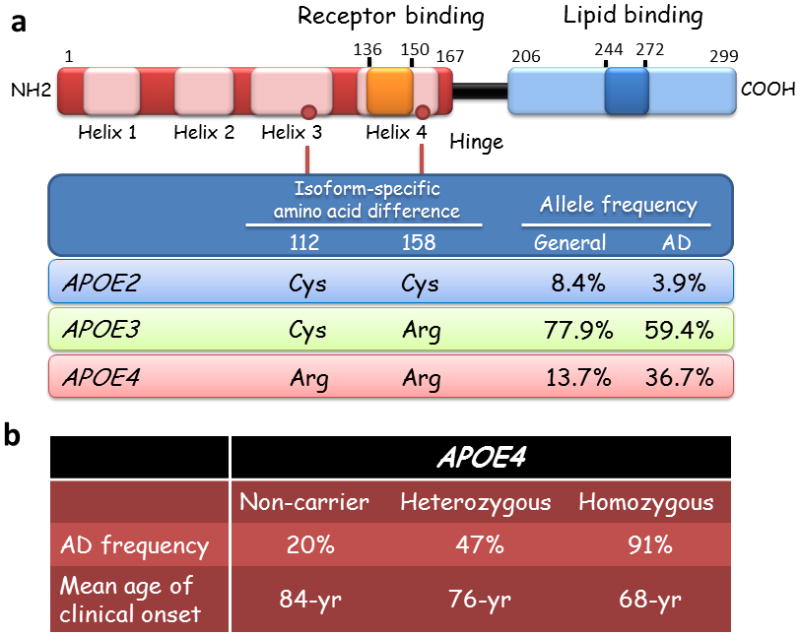
(a) The ApoE2, E3, and E4 isoforms, which are encoded by the ε2, ε3 and ε4 alleles of the APOE gene, respectively, differ from one another at amino acid residues 112 and/or 158 (red circles). ApoE has two structural domains: the N-terminal domain, which contains the receptor-binding region (residues 136–150), and the C-terminal domain, which contains the lipid-binding region (residues 244–272); the two domains are joined by a hinge region. A meta-analysis demonstrated a significant association between the ε4 allele of APOE and AD.10 (b) APOE ε4 increases the risk of AD and lowers the age of disease onset in a gene-dose-dependent manner.7, 20 Abbreviations: AD, Alzheimer disease; ApoE, Apolipoprotein E.
APOE genotypes, AD and cognition
APOE ε4 as a strong risk factor for AD
Genome-wide association studies have confirmed that the ε4 allele of APOE is the strongest genetic risk factor for AD.16, 17 The presence of this allele is associated with increased risk for both early-onset AD and LOAD.18, 19 A meta-analysis of clinical and autopsy-based studies demonstrated that, compared with individuals with an ε3/ε3 genotype, risk of AD was increased in individuals with one copy of the ε4 allele (ε2/ε4, OR 2.6; ε3/ε4, OR 3.2) or two copies (ε4/ε4, OR 14.9) among Caucasian subjects.10 The ε2 allele of APOE has protective effects against AD: the risk of AD in individuals carrying APOE ε2/ε2 (OR 0.6) or ε2/ε3 (OR 0.6) are lower than those of ε3/ε3.10 In population-based studies, the APOE4–AD association was weaker among African Americans (ε4/ε4, OR 5.7) and Hispanics (ε4/ε4, OR 2.2) and was stronger in Japanese people (ε4/ε4, OR 33.1) compared with Caucasian cases (ε4/ε4, OR 12.5).10 APOE ε4 is associated with increased prevalence of AD and lower age of onset.7, 10, 20 The frequency of AD and mean age at clinical onset are 91% and 68 years of age in ε4 homozygotes, 47% and 76 years of age in ε4 heterozygotes, and 20% and 84 years in ε4 noncarriers,7, 20 indicating that APOE ε4 confers dramatically increased risk of development of AD with an earlier age of onset in a gene dose-dependent manner (Figure 1b).
Genetic variants in the TOMM40 (translocase of outer mitochondrial membrane 40 homologue) gene, which lies adjacent to the APOE gene on chromosome 19, have been implicated as a modulator of AD age-of-onset in APOE ε3 carriers.21 A more recent study, however, has cast doubt on the strength of this association.22 Whether the effects of APOE and TOMM40 on AD risk, both genetically and functionally, are synergistic requires further investigation.
APOE and Aβ deposition
ApoE has an important role in Aβ metabolism (Figure 2). Studies show that APOE genotypes strongly affect deposition of Aβ to form senile plaques and cause cerebral amyloid angiopathy (CAA), two major hallmarks of amyloid pathology in AD brains.23 Immunohistological evidence demonstrates that ApoE is co-deposited in senile plaques in the brains of AD patients.24 The Aβ deposition in the form of senile plaques is more abundant in APOE ε4 carriers compared with noncarriers.25–27 The difference was most evident among individuals aged 50–59 years: 40.7% of APOE ε4 carriers had senile plaques compared with 8.2% of noncarriers.25 In individuals with positive Pittsburgh compound B (PiB)-PET images, which indicate fibrillar aggregates of Aβ,28 APOE ε4 was more common than in those with negative scans (65% versus 22%) in patients with AD.29
Figure 2. Apolipoprotein E and amyloid-β metabolism in the brain.
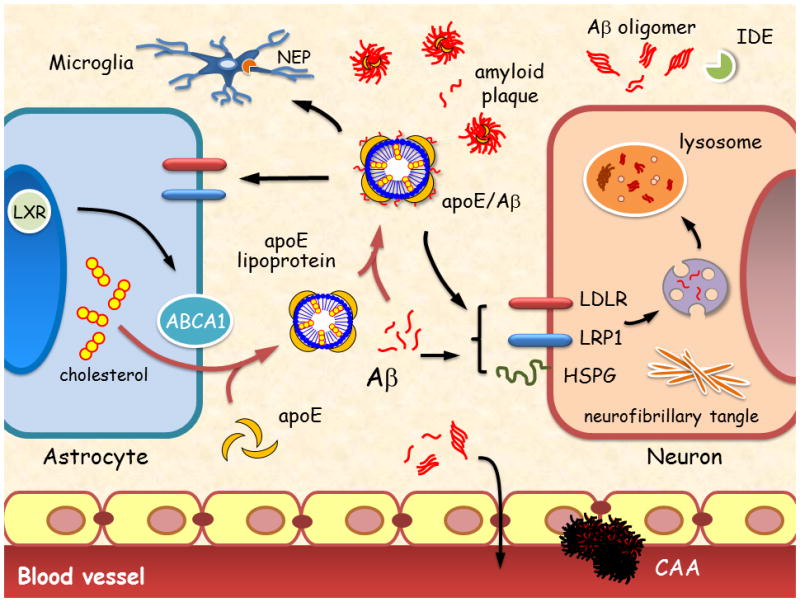
The main Aβ clearance pathways include receptor-mediated uptake by neurons and glia, drainage into interstitial fluid or through the BBB, and proteolytic degradation by IDE and neprilysin. Impaired clearance of Aβ can cause Aβ accumulation in brain parenchyma, leading to formation of neurotoxic Aβ oligomers and amyloid plaques. Aβ accumulation in the perivascular region leads to CAA, which disrupts blood vessel function. ApoE is primarily synthesized by astrocytes and microglia, and is lipidated by the ABCA1 transporter to form lipoprotein particles. Lipidated ApoE binds to soluble Aβ and facilitates Aβ uptake through cell surface receptors, including LRP1, LDLR, and HSPG175, 177 in a manner that probably depends on ApoE isoform and its level of lipidation. ApoE facilitates binding and internalization of soluble Aβ by glial cells, disrupts Aβ clearance at the BBB in an isoform-dependent manner (ApoE4 > ApoE3 > ApoE2) and influences CAA pathogenesis. Abbreviations: Aβ, amyloid-β; ABCA1, ATP-binding cassette A1; BBB, blood–brain barrier; CAA, cerebral amyloid angiopathy; HSPG, heparan sulphate proteoglycan; IDE, insulin-degrading enzyme; LDLR, low-density lipoprotein receptor; LRP1, low-density lipoprotein receptor-related protein 1; LXR, liver X receptor.
Fibrillar Aβ deposition is often detected in the brains of elderly, cognitively normal individuals in a manner that depends on the presence of APOE ε4, although such an association is weaker than that in patients with AD.30 In addition, APOE ε4 carriers have lower cerebrospinal fluid (CSF) Aβ42 levels and higher PiB-positive imaging, which reflect the presence of cerebral amyloid deposition and serve as potential biomarkers for AD.31, 32 Cognitively normal APOE ε4 carriers exhibit PiB-positive imaging about 56 years of age, compared with about 76 years of age in noncarriers.33 This difference suggests that APOE ε4 probably increases the risk of AD by initiating and accelerating Aβ accumulation, aggregation and deposition in the brain. Although APOE ε2 reduces the risk of dementia,34 in individuals older than 90 years, both the ε2 and ε4 alleles of APOE increase amyloid burden compared with APOE ε3, suggesting that the protective effects of APOE ε2 against AD might not be associated with Aβ deposition.
APOE ε4 also shows an association with CAA and CAA-related haemorrhages.35, 36 CAA refers to the pathological condition in which amyloid spreads and deposits throughout the cerebral blood vessel walls37 and is frequently detected in AD.23 Interestingly, although APOE ε2 is protective against AD, it is a risk factor for CAA-related haemorrhage, independently of AD, possibly by predisposing vessels to vasculopathic complications of CAA.36
Prediction of AD in MCI
MCI is a transitional stage between normal ageing and dementia, and is associated with increased risk of AD.38 The rate at which patients with amnesic MCI (aMCI) progress to clinically diagnosable AD is 10–15% per year, in contrast to a rate of 1–2% per year among healthy elderly individuals.39 The prevalence of APOE ε4 is substantially higher in both aMCI and dys-executive MCI than in control individuals.40 Patients with MCI who harbour APOE ε4 exhibit distinct cognitive profiles, which seem to resemble those of patients in the early stages of AD.41 A case–control study reported poorer memory performance among patients with MCI who were carriers of APOE ε4 compared with noncarriers.42 APOE ε4 is associated with impaired memory performance and increased risk of memory decline in middle-aged (40–59 years) and elderly (60–85 years) people with MCI.43, 44 Furthermore, patients with MCI who are carriers of APOE ε4 experience more-rapid decline in several cognitive and functional assessments, and severity of the deficits is strongly associated with the APOE ε4 gene dose.41, 45, 46 Importantly, the presence of APOE ε4 is associated with increased risk of progression from MCI to AD-type dementia.47–49 Among individuals with aMCI, APOE ε4 carriers tend to be younger than noncarriers, consistent with younger age of AD onset in individuals with APOE ε4.50 These findings indicate that the APOE ε4 genotype in patients with MCI can serve as a predictive factor for determination of clinical outcome and the risk of conversion to AD.
In patients with MCI, the adverse effects of APOE ε4 on cognitive functions correlate with the severity of neuronal pathology. Those who are carriers of APOE ε4 have lower CSF Aβ42 levels, higher tau levels and greater brain atrophy than do noncarriers.50 Furthermore, patients with MCI who are PiB-positive are more likely to be APOE ε4 carriers and exhibit worse memory performance than are PiB-negative patients.51 Other finding suggest, although not without controversy,52 that APOE ε4 has considerable deleterious effects on memory performance42 and might be used to predict disease progression in combination with AD biomarkers and neuroimaging approaches.53
Predicting cognitive decline in healthy cases
Healthy APOE ε4 carriers not diagnosed with MCI or AD show an accelerated longitudinal decline in memory tests, which starts around the age of 55–60 years, revealing a possible pre-MCI state in this genetic subset of individuals.54, 55 This memory decline, despite ongoing normal clinical status, suggests that pathological changes in AD might manifest in the brain as early as the sixth decade of life.56, 57 Thus, APOE ε4 is associated with cognitive decline many years before cognitive impairment becomes clinically apparent.56, 58 Interestingly, APOE ε4 has differential effects on memory performance depending on age. Some studies in young adults and children have found evidence of better cognitive performance in APOE ε4 carriers than in noncarriers, which could suggest antagonistic pleiotropy,59–61 in which APOE ε4 might offer benefits during development and early adulthood at the expense of more-rapid decline in cognitive function with ageing.62
Similar to the situation in patients with MCI, APOE ε4 is associated with enhanced amyloid pathology in cognitively normal people. The proportion of PiB-positive individuals follows a strong APOE allele-dependent pattern (ε4 > ε3 > ε2),25, 63, 64 and APOE ε4 increases the amount of amyloid deposition in a gene-dose-dependent manner.30
APOE ε4 and other AD risk factors
The APOE ε4 genotype combines synergistically with atherosclerosis, peripheral vascular disease, or type 2 diabetes in contributing to an increased risk of AD.65, 66 APOE ε4 is a risk factor for cardiovascular disease, suggesting that this allele and cerebrovascular disease might have compounding effects on cognitive decline in AD.67 Diabetes also increases the risk of AD, and the association is particularly strong among APOE ε4 carriers.66, 68, 69 Patients with diabetes who are carriers of APOE ε4 have more neuritic plaques, neurofibrillary tangles and CAA than do noncarriers.66 The combination of a diabetes-related factor—that is, hyperglycaemia, hyperinsulinaemia, and insulin resistance—and the APOE ε4 allele promotes neuritic plaque formation.69 APOE ε4 seems to modify the risk of AD in patients with diabetes—a disease that directly or indirectly causes vascular and neuronal damage and further exacerbates AD pathology. Furthermore, recent research demonstrated that, independently of Aβ, ApoE4 triggers inflammatory cascades that cause neurovascular dysfunction, including blood–brain barrier breakdown, leakage of blood-derived toxic proteins into the brain and reduction in the length of small vessels.70 This result suggests that ApoE4-associated damage to vascular systems in brain could have a key role in AD pathogenesis.
APOE and traumatic brain injury
Increasing evidence has shown that APOE ε4 is associated with poorer outcomes following traumatic brain injury (TBI), regardless of the severity of initial injury.71 A meta-analysis demonstrated that the outcome of TBI at 6 months after injury is worse in APOE ε4 carriers.72 TBI is associated with increased risk of AD,73 and such a risk is more evident in patients with APOE ε4.74 Only 10% of APOE ε4 noncarriers with TBI have Aβ plaque pathology, whereas 35% and 100% of TBI patients with one or two APOE ε4 alleles, respectively, possess Aβ pathology.75 The poorer outcomes associated with ApoE4 might relate to its reduced ability to repair and remodel synapses and protect neurons upon injury compared with ApoE3.8 These possibilities are currently under investigation.
APOE and vascular diseases
Vascular cognitive impairment, which comprises clinical conditions with cerebrovasculature-derived cognitive disturbances including vascular dementia, is observed in approximately 8–15% of aged individuals with cognitive dysfunction in Western clinic-based series.76 A recent meta-analysis has shown evidence of increased risk of vascular dementia in individuals with APOE ε4 compared with APOE ε3 (OR 1.72).77 Several studies suggest that the contribution of APOE ε4 to risk of vascular cognitive impairment is independent of other vascular risk factors including hypertension, dyslipidaemia and atherogenesis,78 whereas another report shows that age-related cognitive decline among APOE ε4 carriers is induced by brain damage owing to increased blood pressure.79 In addition, APOE ε4 is associated with poor outcome after subarachnoid haemorrhage,80 and is a strong risk factor for CAA-related intracranial haemorrhage.81 These results suggest that APOE ε4 is closely associated with neurovascular dysfunctions.
APOE and other types of dementia
Lewy body disease is thought to be the second most common kind of dementia comprised of a spectrum of diseases that includes Parkinson disease (PD), PD-associated dementia and dementia with Lewy bodies (DLB). Clinical and pathological features of PD and AD frequently overlap. Most studies, however, have failed to report associations between APOE ε4 and susceptibility to PD and PD-associated dementia.82, 83 DLB also shares clinical and pathological characteristics with AD and PD,84 and several reports have shown that APOE ε4 increases risk of DLB.85 Immunohistochemical analysis showed that deposition of Lewy bodies in patients with DLB who are APOE ε4 carriers is substantially more abundant than those who are noncarriers.86 As Lewy bodies are considerably increased in the cerebral cortex of DLB patients with Aβ deposition,87 the strong association between amyloid pathology and the pathology of Lewy body disease could explain why APOE ε4 increases risk of DLB. APOE ε4 might also be a risk factor for frontotemporal dementia,88 although the pathophysiological role of ApoE in this disease requires further investigation. The APOE genotypes do not seem to influence the risks of Huntington disease 89 nor amyotrophic lateral sclerosis.90
Mechanisms of ApoE isoforms in AD
APOE ε4 confers a gain of toxic functions, a loss of neuroprotective functions or both in the pathogenesis of AD (Figure 3).
Figure 3. The role of Apolipoprotein E4 in Alzheimer disease pathogenesis.
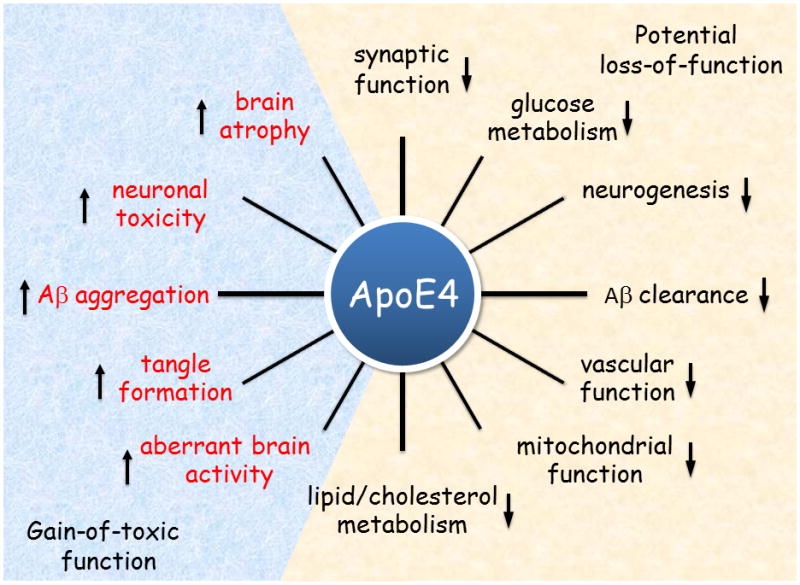
ApoE4 confers toxic gain of function, loss of neuroprotective function or both in the pathogenesis of Alzheimer disease. Key functional differences between ApoE4 and ApoE3 are illustrated. Abbreviations: Aβ, amyloid-β; ApoE, Apolipoprotein E.
Aβ metabolism and aggregation
Studies in humans and transgenic mice showed that brain Aβ levels and amyloid plaque loads are ApoE isoform-dependent (ε4 > ε3 > ε2),30, 63, 91 suggesting an important role of ApoE in modulating Aβ metabolism, aggregation, and deposition. ApoE4 is less efficient in Aβ clearance than is ApoE3 in young and old amyloid mouse models that express human ApoE isoforms.63 Additionally, ApoE isoforms differentially regulate cholesterol levels, which have been shown to modulate γ-secretase activity and Aβ production.92 Several studies reported an APOE genotype-dependent effect on CSF and brain ApoE levels (ε4 < ε3 < ε2) in ApoE-targeted replacement (ApoE-TR) mice, in which the mouse Apoe gene is replaced with human APOE isoforms.91, 93, 94 This result suggests that lower levels of total ApoE exhibited by APOE ε4 carriers might contribute to disease progression. However, whether human ApoE isoform status affects CSF and brain ApoE protein levels in healthy individuals and patients with AD remains to be established.95, 96
ApoE-knockout mice clear Aβ from the brain faster than do control mice.97 Stimulation of liver X receptors (LXRs)98, 99 or the retinoid X receptor (RXR)100 facilitates Aβ clearance, probably by increasing ApoE levels and lipidation. Further investigation is needed to determine whether ApoE levels are directly associated with Aβ clearance. In addition, a recent study showed that lack of one copy of ATP-binding cassette transporter A1 (ABCA1), which shuttles lipids to ApoE, impairs Aβ clearance and exacerbates amyloid deposition and memory deficits in ApoE4-TR mice, but not in ApoE3-TR mice.101 This result suggests that ApoE isoforms exhibit differential lipidation status, which affects Aβ clearance in an isoform-dependent manner. Alternatively, ApoE–lipoprotein particles may sequester Aβ and promote cellular uptake and degradation of ApoE–Aβ complexes.102
ApoE4–lipoproteins bind Aβ with lower affinity than do ApoE3–lipoproteins,103 suggesting that ApoE4 might be less efficient in mediating Aβ clearance. In addition, ApoE might modulate Aβ removal from the brain to the systemic circulation by transporting Aβ across the blood–brain barrier. In this respect, ApoE impedes Aβ clearance at the blood–brain barrier in an isoform-specific fashion (ApoE4 > ApoE3 and ApoE2).104 Finally, studies in microglia have shown that ApoE3 promotes enzyme-mediated degradation of Aβ more efficiently than does ApoE4.105 Together, these studies suggest that ApoE4 inhibits Aβ clearance and/or is less efficient in mediating Aβ clearance compared with ApoE3 and ApoE2.
ApoE also seems to regulate Aβ aggregation and deposition. An important study showed that deletion of the mouse Apoe gene essentially eliminates deposition of fibrillar Aβ in amyloid model mice.106 Given that ApoE is co-deposited with Aβ in human AD brains,24 it is possible that ApoE promotes Aβ aggregation and deposition in an isoform-dependent manner. The exact mechanisms by which ApoE isoforms differentially regulate Aβ aggregation and deposition require further investigation.
Brain activity and atrophy
AD is associated with both functional abnormalities of the hippocampus and cortical atrophy in the memory network.107, 108 Patients with AD or MCI who are APOE ε4 carriers exhibit greater medial temporal lobe atrophy, particularly in the hippocampal area.41, 109, 110 Structural MRI studies found that, compared with noncarriers, APOE ε4 carriers have accelerated age-related loss in cortical thickness and hippocampal volume that are tightly coupled to decline in cognitive performance.111–113
Functional MRI (fMRI) studies reported that ApoE4 disrupts resting state fMRI connectivity and the balance between brain networks, in the absence of amyloid pathology.114, 115 Furthermore, cognitively normal APOE ε4 carriers have elevated resting-state activity in the default mode network—a network that is preferentially affected early in AD—and higher hippocampal activation during memory tasks.116–118 Such changes have been hypothesized to represent a compensatory response by APOE ε4 carriers in which increased cognitive effort is required to achieve an equivalent level of performance to that of noncarriers.116, 118
Elevated baseline activity in brain networks of APOE ε4 carriers could potentially contribute to increased Aβ production, as Aβ levels are regulated by neuronal activity.119, 120 Interestingly, in adults who do not have dementia, increased hippocampal activity was associated with reduced cortical thickness in the medial temporal lobe and brain regions that are vulnerable to AD pathology.121 Studies suggested that hippocampal hyperactivity might represent impending synaptic dysfunction and incipient cognitive decline.122 Interestingly, another study showed a reduction of posterior default mode network connectivity in APOE ε4 carriers in cognitively normal elderly people, implying that APOE ε4 carriers may exhibit a more rapid decline in connectivity of this network than do noncarriers as they age.115
18F-fluorodeoxyglucose PET imaging, which measures cerebral metabolic rates of glucose as a proxy for neuronal activity, correlates with disease progression and predicts histopathological diagnosis in AD.123 Mounting evidence suggests that APOE ε4 carriers exhibit lower cerebral glucose metabolism.124–126 Healthy adults with APOE ε4 show altered patterns of brain metabolism both at rest and during cognitive challenges compared with noncarriers.126, 127 Representative studies illustrating the association of ApoE4 isoform with altered brain metabolism and activity, memory decline, and amyloid pathology in cognitively normal people are shown in Figure 4. Improved understanding of the mechanisms of ApoE4-related brain activity changes, brain atrophy and reduced metabolism should help to explain why ApoE4 is a risk factor for cognitive decline and AD.
Figure 4. Abnormal brain function and enhanced neuropathology and memory decline in cognitively normal APOE ε4 carriers.
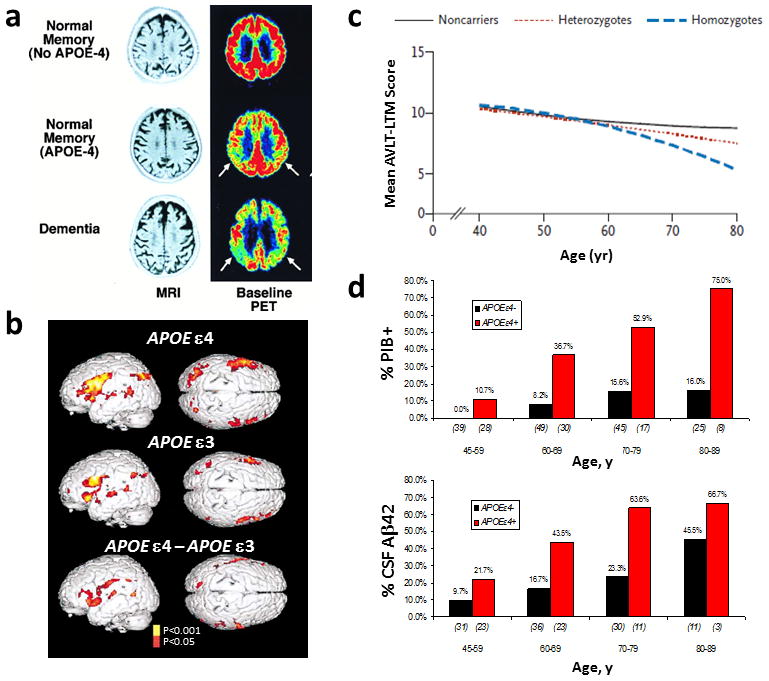
(a) 18F-fluorodeoxyglucose PET images show that cognitively normal APOE ε4 carriers have lower glucose metabolism than do noncarriers. (b) APOE ε4 carriers exhibit a greater increase in functional MRI signal in brain regions associated with task performance, and show increases in additional regions compared with APOE ε3 carriers. (c) Age-related memory decline occurs more rapidly in APOE ε4 carriers than noncarriers, starting from age 55–60 years. (d) APOE ε4 carriers show increased cerebral Aβ deposition which persists in greater frequencies with age compared with noncarriers. Increased PiB binding and reduced CSF Aβ42 levels reflect cerebral amyloid deposition. Abbreviations: Aβ, amyloid-β; APOE, apolipoprotein E; CSF, cerebrospinal fluid; PiB, Pittsburg compund B. Part a, is modified, with permission from the National Academy of Science, USA © Small, G. W. et al. Proc. Natl Acad. Sci. USA 97, 6037–6042 (2000). Part b, is modified, with permission from the Massachusetts Medical Society © Bookheimer, S. Y. et al. N. Engl. J. Med. 343, 450–456 (2000). Part c, is modified, with permission from the Massachusetts Medical Society © Caselli et al. N. Engl. J. Med. 361, 255–263 (2009). Part d is modified, with permission, from John Wiley and Sons © Morris, J. C. et al. Ann. Neurol. 67, 122–131 (2010).
Tau phosphorylation and neurotoxicity
ApoE is produced primarily by astrocytes and microglia. Neuronal ApoE expression can, however, be induced in response to stress or injury, probably for the purpose of neuronal repair and remodelling.128, 129 A truncated fragment of ApoE4, resulting from proteolytic cleavage of ApoE following stress or injury, increases tau hyperphosphorylation, cytoskeletal disruption and mitochondrial dysfunction.128, 130, 131 ApoE4 also exacerbates neurotoxicity triggered by Aβ and other insults.128, 131
A recent study showed that neurons in patients with temporal lobe epilepsy who harbour APOE ε4 are less resilient to the damaging hyperexcitability and more susceptible to Aβ toxicity than are those in APOE ε4 carriers,132 suggesting that ApoE3 might confer a neuroprotective advantage over ApoE4 against neuronal stress. Interestingly, astrocyte-derived ApoE4 has neuroprotective effects against excitotoxic injuries, whereas neuronal expression of ApoE4 promotes excitotoxic cell death. This result suggests that ApoE derived from various cellular sources might exhibit different physiological and pathological activity.133
Lipid metabolism
Abnormal lipid metabolism is strongly related to the pathogenesis of AD. In the CNS, ApoE mediates neuronal delivery of cholesterol, which is an essential component for axonal growth, synaptic formation and remodelling—events that are crucial for learning, memory formation and neuronal repair.134, 135 Brain cholesterol levels are substantially reduced in hippocampal and cortical areas in patients with AD compared with age-matched controls.136 Preferential degradation of ApoE4 relative to ApoE3 in astrocytes has been proposed to result in low levels of ApoE in the brain and CSF and reduced capacity for neuronal delivery of cholesterol, suggesting that low levels of total ApoE exhibited by APOE ε4 carriers may directly contribute to the disease progression.93 ApoE4 is also less efficient than ApoE3 in transporting brain cholesterol.137 Moreover, ApoE4-TR mice have abnormal cholesterol levels and impaired lipid metabolism.138 Insufficient levels of ApoE and/or impaired ApoE function in carriers of the ε4 allele might, therefore, lead to aberrant CNS cholesterol homeostasis and neuronal health, which contribute to AD risk.
Synaptic plasticity and spine integrity
Synaptic failure is an early pathological feature of AD.139, 140 Increasing evidence demonstrates that ApoE isoforms differentially regulate synaptic plasticity and repair.141, 142 In AD and healthy aged controls, APOE ε4 gene dosage correlates inversely with dendritic spine density in the hippocampus.143 ApoE4-TR mice also have lower dendritic spine density and length compared with ApoE3-TR mice.144, 145 ApoE3, but not ApoE4, prevents loss of synaptic networks induced by Aβ oligomers.146 ApoE isoforms also differentially regulate dendritic spines during ageing.143, 147 The age-dependence of these differences implies that the effects of ApoE isoforms on neuronal integrity might relate to increased risk of dementia in aged APOE ε4 carriers.
Reduced synaptic transmission was observed in 1-month-old ApoE4-TR mice compared with ApoE3-TR mice, suggesting that ApoE4 may also contribute to functional deficits early in development, which could account for alteration of neuronal circuitry that eventually results in cognitive disorders later in life.147 In addition, ApoE4 selectively impairs ApoE receptor trafficking and signalling, as well as glutamate receptor function and synaptic plasticity.141 Together, these findings suggest that the effect of APOE ε4 genotype on risk of AD might be mediated, at least in part, through direct effects on synaptic function.
Neuroinflammation
Neuroinflammation contributes to neuronal damage in the brain and is implicated in AD pathogenesis.148 ApoE colocalizes with plaque-associated amyloid and microglia, suggesting a role for ApoE in the innate immune response in AD. Lack of ApoE in mice is associated with increased inflammation in response to Aβ,149, 150 but ApoE isoforms might differently regulate the innate immune response.151 ApoE4 seems to have proinflammatory and/or reduced anti-inflammatory functions, which could further exacerbate AD pathology. For example, ApoE4-TR mice exhibit greater inflammatory responses to lipopolysaccharide compared with ApoE3-TR mice.152 In addition, young APOE ε4 carriers show an increased inflammatory response that may relate to AD risk later in life.153 Consistent with this notion, non-steroidal anti-inflammatory drugs were shown to reduce AD risk only in APOE ε4 carriers,154 suggesting that APOE genotype might determine the effect of anti-inflammatory medications for AD.
Neurogenesis
Hippocampal neurogenesis has an important role in structural plasticity and maintenance of brain networks. Dysfunctional neurogenesis resulting from early disease manifestations could, therefore, exacerbate neuronal vulnerability to AD and contribute to memory impairment.155 ApoE is required for maintenance of the neural stem or progenitor cell pool in the adult dentate gyrus region of the hippocampus.156 In ApoE-TR mice, ApoE4 inhibits hippocampal neurogenesis by impairing maturation of hilar γ-aminobutyric acid-containing interneurons, which contributes to learning and memory deficits.157, 158 These results demonstrate an important pathological role of ApoE4 in impairment of neurogenesis, which might contribute to AD pathogenesis.
ApoE as a therapeutic target for AD
Most therapeutic approaches for AD target the Aβ pathway. With the recent failure of clinical trials of drugs targeting solely Aβ, an urgent need exists to define new targets and develop alternative therapeutic strategies to treat AD. As APOE genotype determines AD risk, and ApoE has crucial roles in cognition, ApoE might offer an attractive alternative target for AD therapy. APOE genotype status could be included in clinical trial enrolment criteria, as some therapies might be effective only in specific APOE genotypes. Here, we briefly discuss several approaches that are currently being explored (Table 1).
Table 1.
ApoE-targeted strategies for treatment of Alzheimer disease
| Strategy | Rationale | Examples |
|---|---|---|
| Pharmacological approaches | ||
| Modulate ApoE levels | Promotes Aβ clearance, lipid homeostasis and synaptic function | LXR/RXR agonists, small molecules |
| Increase ABCA1 expression | Promotes ApoE lipidation and stabilizes ApoE, thereby decreasing amyloid deposition | LXR/RXR agonists, small molecules |
| Disrupt ApoE–Aβ interaction | Reduces Aβ aggregation and deposition | Aβ12–28P, small molecule inhibitors, ApoE-specific antibody |
| Conversion of ApoE4 to ApoE3 | Increases ApoE3-associated protective functions and decreases ApoE4-related toxic effects | Small compounds (i.e., disulphonate and (monosulphoalkyl) |
| Restore ApoE functions | Increases ApoE protective functions and decreases neuroinflammation | ApoE-mimetic peptide |
| Blockade of ApoE fragmentation | Decreases tau pathology, and toxicity to mitochondria | Inhibitors for specific proteinases involved in ApoE fragmentation |
| Increase LRP1 and/or LDLR levels | Enhances Aβ clearance, cholesterol transport and synaptic plasticity | Small molecules |
| Increase apoE receptor 2 and/or VLDLR levels | Increases ApoE signalling and synaptic plasticity | Small molecules |
| Restore brain vascular integrity | Eliminates ApoE4-mediated blood–brain barrier breakdown and leakage of blood-derived toxic molecules | Cyclosporine A |
| Nonpharmacological approaches | ||
| APOE genotyping prior to immunotherapy | Helps to predict clinical outcome for Aβ-targeted or other therapies | Aβ immunotherapy might be more effective in APOE ε4 carriers or noncarriers |
| Preventive care | Maintains healthy brain vasculature function | Physical exercise, intellectual activities (e.g., puzzles), social connections (e.g., calling family and friends), healthy diet |
Abbreviations: Aβ, amyloid-β; ABCA1, ATP-binding cassette transporter A1; AD, Alzheimer disease; ApoE, apolipoprotein E; LDLR, low-density lipoprotein receptor; LRP1, low-density lipoprotein receptor-related protein 1; LXR, liver X receptor; RXR, retinoid X receptor; VLDLR, very-low-density lipoprotein receptor.
APOE genotype and Aβ immunotherapy
Recent phase III clinical trials for immunotherapy have shown that bapineuzumab, an antibody that targets the N-terminus of Aβ, prevents Aβ deposition in the brains of APOE ε4 carriers with mild or moderate AD, but not noncarriers.159, 160 Bapineuzumab also lowers levels of phosphorylated tau in the CSF of both APOE ε4 carriers and noncarriers.159, 160 These reports suggest that Aβ immunotherapy is useful to eliminate Aβ from the brains of patients with AD and that its effect is likely to depend on ApoE isoforms. Major adverse effects of bapineuzumab—vasogenic cerebral oedema and microhaemorrhage—occur more frequently in APOE ε4 carriers than in noncarriers.159, 160 Although bapineuzumab failed to prevent cognitive and functional decline in these clinical trials, a combination of Aβ immunotherapy and an ApoE-targeted approach might lead to more effective therapeutic strategies.
Preventive of cognitive decline in APOE ε4 carriers
A prospective study of a cognitively normal cohort showed that risk of dementia in APOE ε4 carriers is negatively associated with high education, high level of leisure activities, and absence of vascular risk factors.161 A recent study demonstrated that physical exercise was strongly associated with reduced PiB-positivity in cognitively normal APOE ε4 carriers,31 indicating that a sedentary lifestyle in APOE ε4 carriers might increase the risk of amyloid deposition. Such studies indicate that high education, active leisure activities and exercise, and maintenance of vascular health could be beneficial in reducing the risk of AD and cognitive decline, particularly in APOE ε4 carriers.
Regulation of ApoE expression
ApoE levels in CSF and plasma tend to be lower in patients with AD than in healthy individuals, although such findings remain controversial.162, 163 Thus, increasing the expression of ApoE in all APOE genotypes may prevent or slow progression of AD through acceleration of Aβ metabolism and promotion of ApoE functions in lipid metabolism and synaptic support. Compounds that increase brain ApoE expression can be identified through comprehensive drug screening. Given that expression of ApoE is controlled by peroxisome proliferator-activated receptor-γ and LXRs, which form complexes with RXRs,100, 164 agonists or antagonists of these nuclear receptors are potential candidates as ApoE modulators. Indeed, recent work has demonstrated that oral administration of an RXR agonist, bexarotene, to an amyloid mouse model decreases Aβ plaque deposition and improves cognitive function in an ApoE-dependent manner.100 The LXR agonist TO901317 also increases ApoE levels in the brain, facilitates clearance of Aβ42, and reverses contextual memory deficit in amyloid mouse models.98, 99
In addition to ApoE, LXRs also regulate ABCA1, which promotes cholesterol efflux.165 Consequently, reduction of amyloid burden by the LXR agonist GW3965 depends on expression of ABCA1 in amyloid mouse models.166 These results suggest that upregulation of lipidated ApoE might be necessary to maximize therapeutic effects in AD. These studies did not, however, assess the effect of increasing human ApoE3 or ApoE4 specifically. Because Aβ deposition is greater in APP-transgenic mice expressing mouse ApoE than in those expressing human ApoE isoforms,167 further studies are needed to confirm the therapeutic effect of modulating the level of human ApoE isoforms. In addition, whether increasing ApoE4 is beneficial or harmful in AD brains remains unclear, and the effects might depend on age and disease status. Toxic functions associated with ApoE4 suggest that lowing ApoE4 expression might be beneficial in APOE ε4 carriers with cognitive decline during MCI and AD. Additional preclinical studies are needed to test potential beneficial or harmful effects of increasing or decreasing ApoE expression, particularly with regard to ApoE isoforms.
Blocking of ApoE-Aβ interaction
ApoE is required for deposition of Aβ fibrils in amyloid mouse models.106 Recent studies have demonstrated that haploinsufficiency of human APOE results in significantly decreased amyloid plaque deposition in amyloid mouse models regardless of APOE isoform status.168, 169 Thus, disruption of the interaction between ApoE and Aβ might reduce Aβ aggregation and deposition, and should be considered as a therapeutic approach. Aβ interacts with ApoE through amino acid residues 12–28. A synthetic peptide mimicking this sequence, Aβ12–28P, reduces Aβ deposition and ameliorates memory deficits in amyloid mouse models.170 Blocking the ApoE–Aβ interaction using Aβ-mimicking peptides could, therefore, be an effective approach for treatment of AD. Screening assays can also be used to identify compounds or ApoE-specific antibodies that block ApoE–Aβ interaction. These approaches should be assessed carefully because they could disrupt ApoE–lipid interactions and the associated beneficial functions of ApoE.
Other ApoE-based therapeutic approaches
ApoE4 is structurally different from ApoE2 and ApoE3 owing to different domain interactions,131 and this difference probably contributes to ApoE4 isoform-specific harmful effects. Modification of the structure of ApoE4 to form an ApoE3-like molecule might, therefore, be an interesting approach to ameliorate these harmful effects. Indeed, several molecules that bind to ApoE4 and interfere with domain interactions between N- and C-termini have been found. GIND–25 (disulphonate) and GIND–105 (monosulphoalkyl) are good candidates because they decrease Aβ production induced by ApoE4 to a similar level induced by ApoE3.131 CB9032258 (a phthalazinone analogue) and its derivatives disrupt ApoE4 domain interaction and restore functional activities of ApoE4 in neurons.171
An ApoE-mimetic peptide containing the receptor-binding region suppresses neuronal cell death and calcium influx associated with N-methyl-D-aspartate exposure in vitro.172 COG112, a chimeric peptide containing the receptor-binding region, is also reported to improve symptoms in mouse models of multiple sclerosis 173 and sciatic nerve crush174 through modulation of inflammatory responses. The effects of these peptides on AD pathogenesis are unknown, however, because they do not contain Aβ-interacting nor lipid-binding regions.13
ApoE receptors are also potential targets for AD therapy. For example, low-density lipoprotein receptor-related protein 1 and low-density lipoprotein receptor have crucial roles in brain lipid metabolism and Aβ clearance.175–177 ApoE receptor 2 and very low-density lipoprotein receptor are essential for reelin signalling, which is important for neuronal migration during development and synaptic plasticity in adult brains.178 Modulation of the expression of these ApoE receptors in AD brains might, therefore, restore lipid homeostasis and synaptic plasticity, and augment Aβ clearance.8, 178 Although ApoE-based therapies are still in the early stages of development, they offer great promises in the fight against AD. Clinical trials to further evaluate therapeutic potential of ApoE-based strategies are needed, with an eventual goal to develop curative and/or protective treatments for AD.
Conclusions
Work summarized in this Review highlights clinical evidence for the association between APOE ε4, AD and cognitive decline. Although the presence of APOE ε4 does not necessarily entail disease development, this genetic isoform probably accelerates the rate of disease conversion and progression. In particular, the effects of APOE ε4 on brain network connectivity, memory performance, and rate of cognitive decline are age-dependent in patients with AD and cognitively normal individuals. Thus, understanding the potential pathogenic link between APOE ε4 and cognitive function might allow for earlier identification of people at risk of developing AD. In combination with other putative AD biomarkers—such as MRI scans, PiB scans, and measurements of CSF Aβ and tau—APOE allele status could add predictive value to clinical diagnosis and evaluation of treatment efficacy.
Mechanistically, ApoE4 seems to increase risk of AD and cognitive decline through both Aβ-dependent and Aβ-independent pathways. ApoE isoforms differentially regulate Aβ production, aggregation and clearance. Independent of Aβ, ApoE4 might be less efficient than ApoE3 and ApoE2 in delivering cholesterol and essential lipids for the maintenance of synaptic integrity and plasticity. In addition, ApoE is a crucial regulator of the innate immune system, and in which ApoE4 promotes proinflammatory responses that could exacerbate AD pathogenesis. Finally, ApoE isoforms have differential roles in maintaining vascular health—roles that are crucial given that defects in vascular health are strongly associated with AD. Elucidating the contribution of ApoE4 to AD pathogenesis is a considerable challenge, but one that affords the potential to assist in combating AD.
Key Points.
The ε4 allele of the apolipoprotein E (APOE) gene is the main genetic risk factor for Alzheimer disease (AD)
APOE ε4 carriers have enhanced AD pathology, accelerated age-dependent cognitive decline and worse memory performance than do noncarriers.
Numerous structural and functional brain changes associated with AD pathogenesis are detected in APOE ε4 carriers before clinical symptoms become evident.
ApoE affects amyloid-β (Aβ) clearance, aggregation and deposition in an isoform-dependent manner.
ApoE4 also contributes to AD pathogenesis by Aβ-independent mechanisms that involve synaptic plasticity, cholesterol homeostasis, neurovascular functions, and neuroinflammation.
ApoE-targeted AD therapy should focus on restoration of the physiological function of ApoE through increased expression and lipidation, and inhibition of the detrimental effects of ApoE4.
Review criteria.
This Review was based on searches of the PubMed database using the following terms: “apolipoprotein E”, “cognitive decline”, “Alzheimer disease”, “amyloid beta”, “synaptic plasticity”, “cerebral amyloid angiopathy”, “mild cognitive impairment”, “cholesterol”, “brain activity”, “cerebrovascular diseases”, “brain metabolism”, “neurogenesis”, “brain atrophy”, “neuroinflammation”, “tau” and “traumatic brain injury”. Only articles published in English were retrieved. Full-text papers were available for most of the articles that were chosen for review, and the references of these articles were searched for further relevant material.
Acknowledgments
Works in authors’ laboratories are supported by the NIH, the Alzheimer’s Association, the American Health Assistance Foundation, and Xiamen University Research Funds. We thank Caroline Stetler and Owen Ross for critical reading of the manuscript before submission.
Biographies
Chia-Chen Liu is a Postdoctoral Research fellow in the Department of Neuroscience at Mayo Clinic College of Medicine, Jacksonville, FL, USA. She is also a visiting scientist in the Institute of Neuroscience at Xiamen University in Xiamen, China. She gained her Ph.D. from Washington University School of Medicine, St. Louis, MO, USA. Her current research interests focus on apolipoprotein E and its receptors in Alzheimer diseases and vascular dementia.
Takahisa Kanekiyo is an Instructor in the Department of Neuroscience at Mayo Clinic College of Medicine, Jacksonville, FL, USA. He gained his M.D. and Ph.D. from Osaka University Graduate School of Medicine, Japan, and engaged in postdoctoral training at Washington University School of Medicine, St. Louis, MO, USA. His research interests focus on apolipoprotein E and its receptors in neurovascular diseases including Alzheimer diseases and cerebral amyloid angiopathy.
Huaxi Xu is an adjunct Professor in the Institute of Neuroscience at Xiamen University in Xiamen, China. His research focuses on the molecular and cellular mechanisms underlying the pathogenesis of Alzheimer disease. His major contributions include dissecting the cellular trafficking pathways for amyloid precursor protein (APP) and presenilins, and identifying cellular factors that regulate APP processing and neuronal apoptosis. He serves as Co-Editor-in-Chief of Molecular Neurodegeneration.
Guojun Bu is a Professor of Neuroscience at Mayo Clinic College of Medicine, Jacksonville, FL, USA. He is also an adjunct Professor in the Institute of Neuroscience at Xiamen University in Xiamen, China. Since 1994, he has led a research programme studying the biological and pathological functions of Apolipoprotein E (ApoE) and ApoE receptors using cellular and animal models, with a specific focus on the pathogenesis of Alzheimer disease. His group has defined a critical role for the ApoE receptor lipoprotein receptor-related protein 1 in brain ApoE metabolism and in the clearance of amyloid peptides. He founded and serves as the Editor-in-Chief of Molecular Neurodegeneration.
Footnotes
Author contributions
All authors contributed to researching data for the article, discussion of the content, writing the article, and to review and/or editing of the manuscript before submission.
References
- 1.Alzheimer’s Association. 2012 Alzheimer’s disease facts and figures. Alzheimer’s dement. 2012;8:131–168. doi: 10.1016/j.jalz.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 3.Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 4.Roberson ED, et al. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 5.Zheng H, Koo E. Biology and pathophysiology of the amyloid precursor protein. Mol Neurodegener. 2011;6:27. doi: 10.1186/1750-1326-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mawuenyega KG, et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corder EH, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 8.Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10:333–44. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–22. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farrer LaCLHJL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: A meta-analysis. JAMA. 1997;278:1349–1356. [PubMed] [Google Scholar]
- 11.Mahley RW, Rall SC., Jr Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–37. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 12.Lahoz C, et al. Apolipoprotein E genotype and cardiovascular disease in the Framingham Heart Study. Atherosclerosis. 2001;154:529–537. doi: 10.1016/s0021-9150(00)00570-0. [DOI] [PubMed] [Google Scholar]
- 13.Frieden C, Garai K. Structural differences between apoE3 and apoE4 may be useful in developing therapeutic agents for Alzheimer’s disease. Proc Natl Acad Sci USA. 2012;109:8913–8. doi: 10.1073/pnas.1207022109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen J, Li Q, Wang J. Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc Natl Acad Sci USA. 2011;108:14813–8. doi: 10.1073/pnas.1106420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong N, Weisgraber KH. Understanding the association of apolipoprotein E4 with Alzheimer disease: clues from its structure. J Biol Chem. 2009;284:6027–31. doi: 10.1074/jbc.R800009200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harold D, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–93. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lambert JC, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–9. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 18.Chartier-Harlin MC, et al. Apolipoprotein E, epsilon 4 allele as a major risk factor for sporadic early and late-onset forms of Alzheimer’s disease: analysis of the 19q13.2 chromosomal region. Hum Mol Genet. 1994;3:569–74. doi: 10.1093/hmg/3.4.569. [DOI] [PubMed] [Google Scholar]
- 19.Houlden H, et al. ApoE genotype is a risk factor in nonpresenilin early-onset Alzheimer’s disease families. Am J Med Genet. 1998;81:117–21. doi: 10.1002/(sici)1096-8628(19980207)81:1<117::aid-ajmg19>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 20.William Rebeck G, Reiter JS, Strickland DK, Hyman BT. Apolipoprotein E in sporadic Alzheimer’s disease: Allelic variation and receptor interactions. Neuron. 1993;11:575–580. doi: 10.1016/0896-6273(93)90070-8. [DOI] [PubMed] [Google Scholar]
- 21.Roses AD, et al. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J. 2010;10:375–84. doi: 10.1038/tpj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cruchaga C, et al. Association and expression analyses with single-nucleotide polymorphisms in TOMM40 in Alzheimer disease. Arch Neurol. 2011;68:1013–9. doi: 10.1001/archneurol.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ellis RJ, et al. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: the CERAD experience, Part XV. Neurology. 1996;46:1592–6. doi: 10.1212/wnl.46.6.1592. [DOI] [PubMed] [Google Scholar]
- 24.Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991;541:163–6. doi: 10.1016/0006-8993(91)91092-f. [DOI] [PubMed] [Google Scholar]
- 25.Kok E, et al. Apolipoprotein E–dependent accumulation of Alzheimer disease–related lesions begins in middle age. Ann Neurol. 2009;65:650–657. doi: 10.1002/ana.21696. [DOI] [PubMed] [Google Scholar]
- 26.Polvikoski T, et al. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med. 1995;333:1242–7. doi: 10.1056/NEJM199511093331902. [DOI] [PubMed] [Google Scholar]
- 27.Schmechel DE, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:9649–53. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klunk WE, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 29.Barthel H, et al. Cerebral amyloid-beta PET with florbetaben (18F) in patients with Alzheimer’s disease and healthy controls: a multicentre phase 2 diagnostic study. Lancet Neurol. 2011;10:424–35. doi: 10.1016/S1474-4422(11)70077-1. [DOI] [PubMed] [Google Scholar]
- 30.Reiman EM, et al. Fibrillar amyloid-β burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc Natl Acad Sci USA. 2009;106:6820–6825. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Head D, et al. Exercise engagement as a moderator of the effects of APOE genotype on amyloid deposition. Arch Neurol. 2012 doi: 10.1001/archneurol.2011.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prince JA, Zetterberg H, Andreasen N, Marcusson J, Blennow K. APOE ε4 allele is associated with reduced cerebrospinal fluid levels of Aβ42. Neurology. 2004;62:2116–2118. doi: 10.1212/01.wnl.0000128088.08695.05. [DOI] [PubMed] [Google Scholar]
- 33.Fleisher AS, et al. Apolipoprotein E ε4 and age effects on florbetapir positron emission tomography in healthy aging and Alzheimer disease. Neurobiol Aging. 2012 doi: 10.1016/j.neurobiolaging.2012.04.017. [DOI] [PubMed] [Google Scholar]
- 34.Berlau DJ, Corrada MM, Head E, Kawas CH. APOE epsilon2 is associated with intact cognition but increased Alzheimer pathology in the oldest old. Neurology. 2009;72:829–34. doi: 10.1212/01.wnl.0000343853.00346.a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greenberg SM, et al. Apolipoprotein E epsilon 4 is associated with the presence and earlier onset of hemorrhage in cerebral amyloid angiopathy. Stroke. 1996;27:1333–7. doi: 10.1161/01.str.27.8.1333. [DOI] [PubMed] [Google Scholar]
- 36.Biffi A, et al. Variants at APOE influence risk of deep and lobar intracerebral hemorrhage. Ann Neurol. 2010;68:934–43. doi: 10.1002/ana.22134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vinters HV. Cerebral amyloid angiopathy. A critical review. Stroke. 1987;18:311–24. doi: 10.1161/01.str.18.2.311. [DOI] [PubMed] [Google Scholar]
- 38.Morris JC, et al. Mild cognitive impairment represents early-stage Alzheimer disease. Arch Neurol. 2001;58:397–405. doi: 10.1001/archneur.58.3.397. [DOI] [PubMed] [Google Scholar]
- 39.Petersen RC, et al. Mild cognitive impairment clinical characterization and outcome. Arch Neurol. 1999;56:303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- 40.Pa J, et al. Clinical-neuroimaging characteristics of dysexecutive mild cognitive impairment. Ann Neurol. 2009;65:414–23. doi: 10.1002/ana.21591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Farlow MR, et al. Impact of APOE in mild cognitive impairment. Neurology. 2004;63:1898–1901. doi: 10.1212/01.wnl.0000144279.21502.b7. [DOI] [PubMed] [Google Scholar]
- 42.Smith GE, et al. Apolipoprotein E genotype influences cognitive ‘phenotype’ in patients with Alzheimer’s disease but not in healthy control subjects. Neurology. 1998;50:355–362. doi: 10.1212/wnl.50.2.355. [DOI] [PubMed] [Google Scholar]
- 43.Ramakers IHGB, et al. The association between APOE genotype and memory dysfunction in subjects with mild cognitive impairment is related to age and Alzheimer pathology. Dement Geriatr Cogn Disord. 2008;26:101–108. doi: 10.1159/000144072. [DOI] [PubMed] [Google Scholar]
- 44.Dik MG, et al. APOE-ε4 is associated with memory decline in cognitively impaired elderly. Neurology. 2000;54:1492–1497. doi: 10.1212/wnl.54.7.1492. [DOI] [PubMed] [Google Scholar]
- 45.Whitehair DC, et al. Influence of apolipoprotein E ε4 on rates of cognitive and functional decline in mild cognitive impairment. Alzheimer’s dement. 2010;6:412–419. doi: 10.1016/j.jalz.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cosentino S, et al. APOE ε4 allele predicts faster cognitive decline in mild Alzheimer disease. Neurology. 2008;70:1842–1849. doi: 10.1212/01.wnl.0000304038.37421.cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fleisher AS, et al. Clinical predictors of progression to Alzheimer disease in amnestic mild cognitive impairment. Neurology. 2007;68:1588–1595. doi: 10.1212/01.wnl.0000258542.58725.4c. [DOI] [PubMed] [Google Scholar]
- 48.Elias-Sonnenschein LS, Viechtbauer W, Ramakers IH, Verhey FR, Visser PJ. Predictive value of APOE-epsilon4 allele for progression from MCI to AD-type dementia: a meta-analysis. J Neurol Neurosurg Psychiatry. 2011;82:1149–56. doi: 10.1136/jnnp.2010.231555. [DOI] [PubMed] [Google Scholar]
- 49.Petersen RC, et al. Apolipoprotein E status as a predictor of the development of Alzheimer’s disease in memory-impaired individuals. JAMA. 1995;273:1274–1278. [PubMed] [Google Scholar]
- 50.Vemuri P, et al. Effect of apolipoprotein E on biomarkers of amyloid load and neuronal pathology in Alzheimer disease. Ann Neurol. 2010;67:308–316. doi: 10.1002/ana.21953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pike KE, et al. β-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain. 2007;130:2837–2844. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- 52.Bunce D, Fratiglioni L, Small BJ, Winblad B, Bäckman L. APOE and cognitive decline in preclinical Alzheimer disease and non-demented aging. Neurology. 2004;63:816–821. doi: 10.1212/01.wnl.0000137041.86153.42. [DOI] [PubMed] [Google Scholar]
- 53.Lin AL, Laird AR, Fox PT, Gao JH. Multimodal MRI neuroimaging biomarkers for cognitive normal adults, amnestic mild cognitive impairment, and Alzheimer’s disease. Neurology research international. 2012;2012:907409. doi: 10.1155/2012/907409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caselli RJ, et al. Cognitive domain decline in healthy apolipoprotein E epsilon4 homozygotes before the diagnosis of mild cognitive impairment. Arch Neurol. 2007;64:1306–11. doi: 10.1001/archneur.64.9.1306. [DOI] [PubMed] [Google Scholar]
- 55.Caselli RJ, et al. Longitudinal changes in cognition and behavior in asymptomatic carriers of the APOE e4 allele. Neurology. 2004;62:1990–1995. doi: 10.1212/01.wnl.0000129533.26544.bf. [DOI] [PubMed] [Google Scholar]
- 56.Caselli RJ, et al. Longitudinal modeling of age-related memory decline and the APOE ε4 effect. N Engl J Med. 2009;361:255–263. doi: 10.1056/NEJMoa0809437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caselli RJ, et al. Longitudinal modeling of frontal cognition in APOE epsilon4 homozygotes, heterozygotes, and noncarriers. Neurology. 2011;76:1383–8. doi: 10.1212/WNL.0b013e3182167147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Izaks GJ, et al. The association of APOE genotype with cognitive function in persons aged 35 years or older. PLoS ONE. 2011;6:e27415. doi: 10.1371/journal.pone.0027415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mondadori CRA, et al. Better memory and neural efficiency in young apolipoprotein E ε4 carriers. Cerebral Cortex. 2007;17:1934–1947. doi: 10.1093/cercor/bhl103. [DOI] [PubMed] [Google Scholar]
- 60.Jochemsen HM, Muller M, van der Graaf Y, Geerlings MI. APOE epsilon4 differentially influences change in memory performance depending on age. The SMART-MR study. Neurobiol Aging. 2012;33:832 e15–22. doi: 10.1016/j.neurobiolaging.2011.07.016. [DOI] [PubMed] [Google Scholar]
- 61.Bloss CS, Delis DC, Salmon DP, Bondi MW. Decreased cognition in children with risk factors for Alzheimer’s disease. Biol Psychiatry. 2008;64:904–906. doi: 10.1016/j.biopsych.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tuminello ER, Han SD. The Apolipoprotein E antagonistic pleiotropy hypothesis: review and recommendations. Int J Alzheimers Dis. 2011 doi: 10.4061/2011/726197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Castellano JM, et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morris JC, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–131. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haan MN, Shemanski L, Jagust WJ, Manolio TA, Kuller L. The role of APOE epsilon4 in modulating effects of other risk factors for cognitive decline in elderly persons. JAMA. 1999;282:40–6. doi: 10.1001/jama.282.1.40. [DOI] [PubMed] [Google Scholar]
- 66.Peila R, Rodriguez BL, Launer LJ. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes. 2002;51:1256–62. doi: 10.2337/diabetes.51.4.1256. [DOI] [PubMed] [Google Scholar]
- 67.Kalmijn S, Feskens EJ, Launer LJ, Kromhout D. Cerebrovascular disease, the apolipoprotein e4 allele, and cognitive decline in a community-based study of elderly men. Stroke. 1996;27:2230–5. doi: 10.1161/01.str.27.12.2230. [DOI] [PubMed] [Google Scholar]
- 68.Irie F, et al. Enhanced risk for Alzheimer disease in persons with type 2 diabetes and APOE epsilon4: the Cardiovascular Health Study Cognition Study. Arch Neurol. 2008;65:89–93. doi: 10.1001/archneurol.2007.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matsuzaki T, et al. Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology. 2010;75:764–70. doi: 10.1212/WNL.0b013e3181eee25f. [DOI] [PubMed] [Google Scholar]
- 70.Bell RD, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 2011;10:241–52. doi: 10.1016/S1474-4422(10)70325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou W, et al. Meta-analysis of APOE4 allele and outcome after traumatic brain injury. J Neurotrauma. 2008;25:279–90. doi: 10.1089/neu.2007.0489. [DOI] [PubMed] [Google Scholar]
- 73.Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A. Head injury as a risk factor for Alzheimer’s disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry. 2003;74:857–62. doi: 10.1136/jnnp.74.7.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Magnoni S, Brody DL. New perspectives on amyloid-beta dynamics after acute brain injury: moving between experimental approaches and studies in the human brain. Arch Neurol. 2010;67:1068–73. doi: 10.1001/archneurol.2010.214. [DOI] [PubMed] [Google Scholar]
- 75.Nicoll JA, Roberts GW, Graham DI. Apolipoprotein E epsilon 4 allele is associated with deposition of amyloid beta-protein following head injury. Nat Med. 1995;1:135–7. doi: 10.1038/nm0295-135. [DOI] [PubMed] [Google Scholar]
- 76.Jellinger KA. Morphologic diagnosis of “vascular dementia” - a critical update. J Neurol Sci. 2008;270:1–12. doi: 10.1016/j.jns.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 77.Yin YW, et al. Association between apolipoprotein E gene polymorphism and the risk of vascular dementia: a meta-analysis. Neurosci Lett. 2012;514:6–11. doi: 10.1016/j.neulet.2012.02.031. [DOI] [PubMed] [Google Scholar]
- 78.Prince M, et al. The association between APOE and dementia does not seem to be mediated by vascular factors. Neurology. 2000;54:397–402. doi: 10.1212/wnl.54.2.397. [DOI] [PubMed] [Google Scholar]
- 79.Bender AR, Raz N. Age-related differences in memory and executive functions in healthy APOE varepsilon4 carriers: the contribution of individual differences in prefrontal volumes and systolic blood pressure. Neuropsychologia. 2012;50:704–14. doi: 10.1016/j.neuropsychologia.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lanterna LA, et al. Meta-analysis of APOE genotype and subarachnoid hemorrhage. Neurology. 2007;69:766–775. doi: 10.1212/01.wnl.0000267640.03300.6b. [DOI] [PubMed] [Google Scholar]
- 81.Biffi A, et al. APOE genotype and extent of bleeding and outcome in lobar intracerebral haemorrhage: a genetic association study. Lancet Neurol. 2011;10:702–9. doi: 10.1016/S1474-4422(11)70148-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Federoff M, Jimenez-Rolando B, Nalls MA, Singleton AB. A large study reveals no association between APOE and Parkinson’s disease. Neurobiol Dis. 2012;46:389–392. doi: 10.1016/j.nbd.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ezquerra M, et al. Lack of association of APOE and tau polymorphisms with dementia in Parkinson’s disease. Neurosci Lett. 2008;448:20–3. doi: 10.1016/j.neulet.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 84.McKeith IG, et al. Dementia with Lewy bodies. Semin Clin Neuropsychiatry. 2003;8:46–57. doi: 10.1053/scnp.2003.50006. [DOI] [PubMed] [Google Scholar]
- 85.Harrington CR, et al. Influence of apolipoprotein E genotype on senile dementia of the Alzheimer and Lewy body types. Significance for etiological theories of Alzheimer’s disease. Am J Pathol. 1994;145:1472–84. [PMC free article] [PubMed] [Google Scholar]
- 86.Mann DM, Brown SM, Owen F, Baba M, Iwatsubo T. Amyloid beta protein (abeta) deposition in dementia with Lewy bodies: predominance of A beta 42(43) and paucity of A beta 40 compared with sporadic Alzheimer’s disease. Neuropathol Appl Neurobiol. 1998;24:187–94. doi: 10.1046/j.1365-2990.1998.00112.x. [DOI] [PubMed] [Google Scholar]
- 87.Pletnikova O, et al. Abeta deposition is associated with enhanced cortical alpha-synuclein lesions in Lewy body diseases. Neurobiol Aging. 2005;26:1183–92. doi: 10.1016/j.neurobiolaging.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 88.Seripa D, et al. The APOE gene locus in frontotemporal dementia and primary progressive aphasia. Arch Neurol. 2011;68:622–8. doi: 10.1001/archneurol.2011.90. [DOI] [PubMed] [Google Scholar]
- 89.Saft C, et al. Apolipoprotein E genotypes do not influence the age of onset in Huntington’s disease. J Neurol Neurosurg Psychiatry. 2004;75:1692–6. doi: 10.1136/jnnp.2003.022756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jawaid A, et al. Does apolipoprotein E genotype modify the clinical expression of ALS? Eur J Neurol. 2011;18:618–24. doi: 10.1111/j.1468-1331.2010.03225.x. [DOI] [PubMed] [Google Scholar]
- 91.Bales KR, et al. Human APOE isoform-dependent effects on brain β-amyloid levels in PDAPP transgenic mice. J Neurosci. 2009;29:6771–6779. doi: 10.1523/JNEUROSCI.0887-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Osenkowski P, Ye W, Wang R, Wolfe MS, Selkoe DJ. Direct and potent regulation of gamma-secretase by its lipid microenvironment. J Biol Chem. 2008;283:22529–40. doi: 10.1074/jbc.M801925200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Riddell DR, et al. Impact of apolipoprotein E (apoE) polymorphism on brain apoE levels. J Neurosci. 2008;28:11445–11453. doi: 10.1523/JNEUROSCI.1972-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sullivan PM, et al. Reduced levels of human apoE4 protein in an animal model of cognitive impairment. Neurobiol Aging. 2011;32:791–801. doi: 10.1016/j.neurobiolaging.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 95.Wahrle SE, et al. Apolipoprotein E levels in cerebrospinal fluid and the effects of ABCA1 polymorphisms. Mol Neurodegener. 2007;2:7. doi: 10.1186/1750-1326-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Beffert U, et al. Apolipoprotein E and beta-amyloid levels in the hippocampus and frontal cortex of Alzheimer’s disease subjects are disease-related and apolipoprotein E genotype dependent. Brain Res. 1999;843:87–94. doi: 10.1016/s0006-8993(99)01894-6. [DOI] [PubMed] [Google Scholar]
- 97.DeMattos RB, et al. ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron. 2004;41:193–202. doi: 10.1016/s0896-6273(03)00850-x. [DOI] [PubMed] [Google Scholar]
- 98.Riddell DR, et al. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol Cell Neurosci. 2007;34:621–8. doi: 10.1016/j.mcn.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 99.Vanmierlo T, et al. Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol Aging. 2011;32:1262–72. doi: 10.1016/j.neurobiolaging.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 100.Cramer PE, et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–6. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fitz NF, et al. Abca1 Deficiency Affects Alzheimer’s Disease-Like Phenotype in Human ApoE4 But Not in ApoE3-Targeted Replacement Mice. J Neurosci. 2012;32:13125–36. doi: 10.1523/JNEUROSCI.1937-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.LaDu MJ, et al. Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem. 1994;269:23403–23406. [PubMed] [Google Scholar]
- 104.Deane R, et al. ApoE isoform–specific disruption of amyloid β peptide clearance from mouse brain. J Clin Invest. 2008;118:4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jiang Q, et al. ApoE promotes the proteolytic degradation of Aβ. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bales KR, et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet. 1997;17:263–4. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- 107.Ries ML, et al. Magnetic resonance imaging characterization of brain structure and function in mild cognitive impairment: a review. J Am Geriatr Soc. 2008;56:920–934. doi: 10.1111/j.1532-5415.2008.01684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nestor PJ, Scheltens P, Hodges JR. Advances in the early detection of Alzheimer’s disease. Nat Med. 2004;10 (Suppl):S34–41. doi: 10.1038/nrn1433. [DOI] [PubMed] [Google Scholar]
- 109.Korf ESC, Wahlund LO, Visser PJ, Scheltens P. Medial temporal lobe atrophy on MRI predicts dementia in patients with mild cognitive impairment. Neurology. 2004;63:94–100. doi: 10.1212/01.wnl.0000133114.92694.93. [DOI] [PubMed] [Google Scholar]
- 110.Hashimoto M, et al. Apolipoprotein E ε4 and the pattern of regional brain atrophy in Alzheimer’s disease. Neurology. 2001;57:1461–1466. doi: 10.1212/wnl.57.8.1461. [DOI] [PubMed] [Google Scholar]
- 111.Espeseth T, et al. Accelerated age-related cortical thinning in healthy carriers of apolipoprotein E ε4. Neurobiol Aging. 2008;29:329–340. doi: 10.1016/j.neurobiolaging.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 112.Reiman EM, et al. Hippocampal volumes in cognitively normal persons at genetic risk for Alzheimer’s disease. Ann Neurol. 1998;44:288–291. doi: 10.1002/ana.410440226. [DOI] [PubMed] [Google Scholar]
- 113.Fennema-Notestine C, et al. Presence of APOE ε4 allele associated with thinner frontal cortex in middle age. J Alzheimers Dis. 2011;26:49–60. doi: 10.3233/JAD-2011-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sheline YI, et al. APOE4 allele disrupts resting state fMRI connectivity in the absence of amyloid plaques or decreased CSF Abeta42. J Neurosci. 2010;30:17035–40. doi: 10.1523/JNEUROSCI.3987-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Machulda Mm, JDTVP, et al. Effect of apoe ε4 status on intrinsic network connectivity in cognitively normal elderly subjects. Arch Neurol. 2011;68:1131–1136. doi: 10.1001/archneurol.2011.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bookheimer SY, et al. Patterns of brain activation in people at risk for Alzheimer’s disease. N Engl J Med. 2000;343:450–456. doi: 10.1056/NEJM200008173430701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Filippini N, et al. Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc Natl Acad Sci USA. 2009;106:7209–14. doi: 10.1073/pnas.0811879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bondi MW, Houston WS, Eyler LT, Brown GG. fMRI evidence of compensatory mechanisms in older adults at genetic risk for Alzheimer disease. Neurology. 2005;64:501–508. doi: 10.1212/01.WNL.0000150885.00929.7E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bero AW, et al. Neuronal activity regulates the regional vulnerability to amyloid-[beta] deposition. Nat Neurosci. 2011;14:750–756. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cirrito JR, et al. Synaptic activity regulates interstitial fluid Amyloid-β levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 121.Putcha D, et al. Hippocampal hyperactivation associated with cortical thinning in Alzheimer’s disease signature regions in non-demented elderly adults. J Neurosci. 2011;31:17680–17688. doi: 10.1523/JNEUROSCI.4740-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.O’Brien JL, et al. Longitudinal fMRI in elderly reveals loss of hippocampal activation with clinical decline. Neurology. 2010;74:1969–1976. doi: 10.1212/WNL.0b013e3181e3966e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mosconi L, et al. Pre-clinical detection of Alzheimer’s disease using FDG-PET, with or without amyloid imaging. J Alzheimers Dis. 2010;20:843–854. doi: 10.3233/JAD-2010-091504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Small GW, et al. Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA. 1995;273:942–7. [PubMed] [Google Scholar]
- 125.Reiman EM, et al. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med. 1996;334:752–8. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- 126.Small GW, et al. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc Natl Acad Sci USA. 2000;97:6037–6042. doi: 10.1073/pnas.090106797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Reiman EM, et al. Correlations between apolipoprotein E ε4 gene dose and brain-imaging measurements of regional hypometabolism. Proc Natl Acad Sci USA. 2005;102:8299–8302. doi: 10.1073/pnas.0500579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Huang Y. Aβ-independent roles of apolipoprotein E4 in the pathogenesis of Alzheimer’s disease. Trends Mol Med. 2010;16:287–294. doi: 10.1016/j.molmed.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 129.Aoki K, et al. Increased expression of neuronal apolipoprotein E in human brain with cerebral infarction. Stroke. 2003;34:875–880. doi: 10.1161/01.STR.0000064320.73388.C6. [DOI] [PubMed] [Google Scholar]
- 130.Brecht WJ, et al. Neuron-specific apolipoprotein E4 proteolysis is associated with increased Tau phosphorylation in brains of transgenic mice. J Neurosci. 2004;24:2527–2534. doi: 10.1523/JNEUROSCI.4315-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci USA. 2006;103:5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Aboud O, Mrak RE, Boop F, Griffin ST. Apolipoprotein epsilon 3 alleles are associated with indicators of neuronal resilience. BMC medicine. 2012;10:35. doi: 10.1186/1741-7015-10-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Buttini M, et al. Cellular source of apolipoprotein E4 determines neuronal susceptibility to excitotoxic injury in transgenic mice. Am J Pathol. 2010;177:563–9. doi: 10.2353/ajpath.2010.090973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mauch DH, et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- 135.Pfrieger FW. Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol Life Sci. 2003;60:1158–1171. doi: 10.1007/s00018-003-3018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Svennerholm L, Gottfries CG. Membrane lipids, selectively diminished in Alzheimer brains, suggest synapse loss as a primary event in early-onset form (Type I) and demyelination in late-onset form (Type II) J Neurochem. 1994;62:1039–1047. doi: 10.1046/j.1471-4159.1994.62031039.x. [DOI] [PubMed] [Google Scholar]
- 137.Rapp A, Gmeiner B, Hüttinger M. Implication of apoE isoforms in cholesterol metabolism by primary rat hippocampal neurons and astrocytes. Biochimie. 2006;88:473–483. doi: 10.1016/j.biochi.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 138.Hamanaka H, et al. Altered cholesterol metabolism in human apolipoprotein E4 knock-in mice. Hum Mol Genet. 2000;9:353–361. doi: 10.1093/hmg/9.3.353. [DOI] [PubMed] [Google Scholar]
- 139.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 140.Koffie R, Hyman B, Spires-Jones T. Alzheimer’s disease: synapses gone cold. Mol Neurodegener. 2011;6:63. doi: 10.1186/1750-1326-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen Y, Durakoglugil MS, Xian X, Herz J. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci USA. 2010 doi: 10.1073/pnas.0914984107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Buttini M, et al. Modulation of alzheimer-like synaptic and cholinergic deficits in transgenic mice by human apolipoprotein E depends on isoform, aging, and overexpression of amyloid β peptides but not on plaque formation. J Neurosci. 2002;22:10539–10548. doi: 10.1523/JNEUROSCI.22-24-10539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ji Y, et al. Apolipoprotein E isoform-specific regulation of dendritic spine morphology in apolipoprotein E transgenic mice and Alzheimer’s disease patients. Neuroscience. 2003;122:305–315. doi: 10.1016/j.neuroscience.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 144.Dumanis SB, et al. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J Neurosci. 2009;29:15317–15322. doi: 10.1523/JNEUROSCI.4026-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wang C, et al. Human apoE4-targeted replacement mice display synaptic deficits in the absence of neuropathology. Neurobiol Dis. 2005;18:390–398. doi: 10.1016/j.nbd.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 146.Sen A, Alkon DL, Nelson TJ. Apolipoprotein E3 (ApoE3) but not ApoE4 protects against synaptic loss through increased expression of protein kinase Cε. J Biol Chem. 2012;287:15947–15958. doi: 10.1074/jbc.M111.312710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Klein RC, Mace BE, Moore SD, Sullivan PM. Progressive loss of synaptic integrity in human apolipoprotein E4 targeted replacement mice and attenuation by apolipoprotein E2. Neuroscience. 2010;171:1265–1272. doi: 10.1016/j.neuroscience.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bales KR, Du Y, Holtzman D, Cordell B, Paul SM. Neuroinflammation and Alzheimer’s disease: critical roles for cytokine/Aβ-induced glial activation, NF-κB, and apolipoprotein E. Neurobiol Aging. 2000;21:427–432. doi: 10.1016/s0197-4580(00)00143-3. [DOI] [PubMed] [Google Scholar]
- 149.LaDu MJ, et al. Apolipoprotein E and apolipoprotein E receptors modulate Aβ-induced glial neuroinflammatory responses. Neurochem Int. 2001;39:427–434. doi: 10.1016/s0197-0186(01)00050-x. [DOI] [PubMed] [Google Scholar]
- 150.Lynch JR, Morgan D, Mance J, Matthew WD, Laskowitz DT. Apolipoprotein E modulates glial activation and the endogenous central nervous system inflammatory response. J Neuroimmunol. 2001;114:107–113. doi: 10.1016/s0165-5728(00)00459-8. [DOI] [PubMed] [Google Scholar]
- 151.Keene CD, Cudaback E, Li X, Montine KS, Montine TJ. Apolipoprotein E isoforms and regulation of the innate immune response in brain of patients with Alzheimer’s disease. Curr Opin Neurobiol. 2011;21:920–8. doi: 10.1016/j.conb.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lynch JR, et al. APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J Biol Chem. 2003;278:48529–48533. doi: 10.1074/jbc.M306923200. [DOI] [PubMed] [Google Scholar]
- 153.Ringman Jm, EDGDH, et al. Plasma signaling proteins in persons at genetic risk for alzheimer disease: Influence of apoe genotype. Arch Neurol. 2012;69:757–764. doi: 10.1001/archneurol.2012.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Szekely CA, et al. NSAID use and dementia risk in the Cardiovascular Health Study: role of APOE and NSAID type. Neurology. 2008;70:17–24. doi: 10.1212/01.wnl.0000284596.95156.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mu Y, Gage F. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener. 2011;6:85. doi: 10.1186/1750-1326-6-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yang CP, Gilley JA, Zhang G, Kernie SG. ApoE is required for maintenance of the dentate gyrus neural progenitor pool. Development. 2011;138:4351–4362. doi: 10.1242/dev.065540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Andrews-Zwilling Y, et al. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J Neurosci. 2010;30:13707–17. doi: 10.1523/JNEUROSCI.4040-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Li G, et al. GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell Stem Cell. 2009;5:634–45. doi: 10.1016/j.stem.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Salloway S. European Federation of Neurological Societies. Stockholm; Sweden: 2012. [Google Scholar]
- 160.Sperling R. European Federation of Neurological Societies. Stockholm; Sweden: 2012. [Google Scholar]
- 161.Ferrari C, et al. How can elderly apolipoprotein E epsilon4 carriers remain free from dementia? Neurobiol Aging. 2012 doi: 10.1016/j.neurobiolaging.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 162.Hesse C, et al. Measurement of apolipoprotein E (apoE) in cerebrospinal fluid. Neurochem Res. 2000;25:511–7. doi: 10.1023/a:1007516210548. [DOI] [PubMed] [Google Scholar]
- 163.Gupta VB, et al. Plasma apolipoprotein E and Alzheimer disease risk. Neurology. 2011;76:1091–1098. doi: 10.1212/WNL.0b013e318211c352. [DOI] [PubMed] [Google Scholar]
- 164.Chawla A, et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7:161–71. doi: 10.1016/s1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- 165.Vaya J, Schipper HM. Oxysterols, cholesterol homeostasis, and Alzheimer disease. J Neurochem. 2007;102:1727–37. doi: 10.1111/j.1471-4159.2007.04689.x. [DOI] [PubMed] [Google Scholar]
- 166.Donkin JJ, et al. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem. 2010;285:34144–54. doi: 10.1074/jbc.M110.108100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Fryer JD, et al. Human apolipoprotein E4 alters the amyloid-beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci. 2005;25:2803–10. doi: 10.1523/JNEUROSCI.5170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kim J, et al. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-beta amyloidosis. J Neurosci. 2011;31:18007–12. doi: 10.1523/JNEUROSCI.3773-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bien-Ly N, Gillespie AK, Walker D, Yoon SY, Huang Y. Reducing human apolipoprotein E levels attenuates age-dependent Abeta accumulation in mutant human amyloid precursor protein transgenic mice. J Neurosci. 2012;32:4803–11. doi: 10.1523/JNEUROSCI.0033-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sadowski MJ, et al. Blocking the apolipoprotein E/amyloid-beta interaction as a potential therapeutic approach for Alzheimer’s disease. Proc Natl Acad Sci USA. 2006;103:18787–92. doi: 10.1073/pnas.0604011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Chen HK, et al. Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J Biol Chem. 2012;287:5253–66. doi: 10.1074/jbc.M111.276162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Aono M, et al. Protective effect of apolipoprotein E-mimetic peptides on N-methyl-D-aspartate excitotoxicity in primary rat neuronal-glial cell cultures. Neuroscience. 2003;116:437–45. doi: 10.1016/s0306-4522(02)00709-1. [DOI] [PubMed] [Google Scholar]
- 173.Li FQ, et al. Apolipoprotein E-derived peptides ameliorate clinical disability and inflammatory infiltrates into the spinal cord in a murine model of multiple sclerosis. J Pharmacol Exp Ther. 2006;318:956–65. doi: 10.1124/jpet.106.103671. [DOI] [PubMed] [Google Scholar]
- 174.Li FQ, Fowler KA, Neil JE, Colton CA, Vitek MP. An apolipoprotein E-mimetic stimulates axonal regeneration and remyelination after peripheral nerve injury. J Pharmacol Exp Ther. 2010;334:106–15. doi: 10.1124/jpet.110.167882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kanekiyo T, et al. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-β uptake. J Neurosci. 2011;31:1644–1651. doi: 10.1523/JNEUROSCI.5491-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Liu Q, et al. Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J Neurosci. 2010;30:17068–17078. doi: 10.1523/JNEUROSCI.4067-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kim J, et al. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid feposition and increases extracellular abeta clearance. Neuron. 2009;64:632–644. doi: 10.1016/j.neuron.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Herz J, Chen Y. Reelin, lipoprotein receptors and synaptic plasticity. Nat Rev Neurosci. 2006;7:850–859. doi: 10.1038/nrn2009. [DOI] [PubMed] [Google Scholar]