

Abstract
c-Met, the receptor tyrosine kinase whose natural ligand is hepatocyte growth factor, is known to have a key role in cell motility. We have previously shown that lysophosphatidic acid (LPA) induced a decrease in c-Met activation via serine phosphorylation of c-Met at cell-cell contacts. Here, we demonstrate that lipopolysaccharide (LPS) treatment of human bronchial epithelial cells induced internalization of c-Met via phosphorylation at its tyrosine residue 1003. In addition, it induced epithelial barrier dysfunction as evidenced by a decrease in transepithelial resistance (TER) in a time-dependent manner. Pretreatment with a c-Met inhibitor (PHA-665752) or inhibition of protein kinase C (PKC)-α attenuated the LPS-mediated phosphorylation of c-Met and its internalization. LPS-induced c-Met tyrosine 1003 phosphorylation, activation of PKCα, and c-Met internalization were, however, reversed by pretreatment of cells with LPA, which increased c-Met accumulation at cell-cell contacts. Inhibition of LPS-mediated c-Met tyrosine (Y1003) phosphorylation and internalization by prior treatment with PHA-665752, inhibition of PKCα, or overexpression of c-MetY1003A mutant attenuated LPS-induced reduction of TER. Furthermore, we found that c-Met accumulation at cell-cell contacts contributed to LPA-enhanced epithelial barrier integrity, since downregulation of c-Met by specific small-interfering RNA attenuated LPA-increased TER. The data reveal a novel biological function of c-Met in the regulation of lung epithelial barrier integrity.
Keywords: tyrosine phosphorylation, lysophosphatidic acid, protein kinase C
plasma membrane receptor trafficking is important for regulating cell-surface receptor expression, intracellular signaling, and its ensuing biological functions such as cell proliferation and cell migration (10, 21). c-Met is a receptor tyrosine kinase that is predominantly expressed on epithelial cells regulating cell motility upon binding of its natural ligand hepatocyte growth factor (HGF) (20). HGF treatment rapidly induces c-Met tyrosine phosphorylation, internalization, and subsequent proteasomal degradation (17, 23). Based on the specific site of c-Met phosphorylation, the fate of c-Met is decided. For instance, phosphorylation of the tyrosine residues Y1230, Y1234, and Y1235 forms docking sites for the recruitment of key intracellular signaling molecules that transduce downstream signals (20, 23). Phosphorylation at the tyrosine residue Y1003, which is in the juxtamembrane domain, induces c-Met internalization via its interaction with c-cbl (22). Phosphorylation at serine residue S985 negatively regulates HGF-mediated c-Met kinase activity (9, 11) and is therefore considered inhibitory. Protein kinase C (PKC)-δ and -ε phosphorylate S985 of c-Met (11). Overall, these studies suggest that tyrosine phosphorylation at Y1003 mediates c-Met internalization, whereas serine phosphorylation at S985 induces c-Met inactivation and trafficking to the cell-cell contacts. The involvement of PKC isoforms in tyrosine phosphorylation of epidermal growth factor receptor (EGF-R) has been reported (35), whereas the role of PKC in c-Met Y1003 phosphorylation has not been studied.
The contribution of c-Met toward maintaining cell-cell junctions in the endothelium has been reported (19, 26). Activation of c-Met promotes increased human endothelial barrier function via activation of glycogen synthesis kinase 3β (19) and hyaluronan receptor, CD44 (26), and sphingosine 1-phosphate receptor 1 (8). A major function of lung epithelium is to protect against the inhaled substances such as particle matter, pathogen, and smoke via maintaining epithelial barrier integrity. Recent studies have shown that c-Met interacts with E-cadherin, a major constituent of adherens junctions of epithelia (6, 18, 26); however, the role of c-Met in maintaining lung epithelial barrier integrity has not been investigated.
Lysophosphatidic acid (LPA) is a natural-occurring bioactive phospholipid that has been detected in plasma and bronchoalveolar lavage fluids. LPA regulates release of both pro- and anti-inflammatory factors, such as interleukin-8, PGE2, IL-13Rα2, and IL-33 decoy receptor, in lung epithelial cells (29, 37, 38). Intratracheal injection of LPA attenuated lipopolysaccharide (LPS)-induced lung inflammation in a murine model of acute lung injury (12), indicating a protective role of LPA in lung inflammatory diseases. In addition to its effects on regulation of inflammatory responses, the role of LPA in cytoskeleton rearrangement has been well demonstrated. LPA treatment induces cell migration, including lung epithelial cells (31, 32). Our recent study has shown that LPA regulates lung epithelial integrity by inducing E-cadherin and c-Met serine phosphorylation and accumulation at cell-cell contacts (12, 36). LPA induced serine phosphorylation of c-Met, leading to the inactivation of c-Met. HGF-induced c-Met activation and internalization was reversed by LPA treatment. The underlying mechanism involved serine phosphorylation of c-Met by PKCδ (36).
Endotoxin-induced acute lung injury is characterized by increased cytokine release, neutrophil infiltration, and alveolar-capillary barrier dysfunction (2, 12, 34). Our laboratory and others have shown that LPS induced lung endothelial and epithelial barrier disruption as evidenced by reduction of transepithelial and transendothelial resistance (TER) as well as increased dextran leak in a Transwell permeability assay (2, 12, 34). LPS treatment induced E-cadherin internalization in lung epithelial cells (12); however, its effect on c-Met is not known. Here, we show that PKCα regulates LPS-induced tyrosine phosphorylation of c-Met at Y1003 and c-Met internalization, which is associated with lung epithelial barrier dysfunction. Furthermore, LPA treatment attenuates LPS-induced activation of PKCα, Y1003 c-Met phosphorylation, and epithelial barrier dysfunction. This study is the first report demonstrating that c-Met trafficking in lung epithelial barrier function is regulated by LPS and LPA.
MATERIALS AND METHODS
Cell culture and reagents.
Human bronchial epithelial cells (HBEpCs) were purchased from Lonza (Rockville, MD). The passage 1 HBEpCs were cultured in serum-free basal essential growth medium (BEGM) supplemented with growth factors (Lonza). Cells were incubated in a 5% CO2 incubator at 37°C to ∼80% confluence and subsequently propagated in six-well cell culture dishes. All experiments were carried out between passages 1 and 4. 1-Oleoyl (18:1) LPA and LPS were purchased from Sigma-Aldrich (St. Louis, MO). Myr-PKCζ peptide inhibitor was from Biomol (Plymouth Meeting, PA). Antibodies to PKCα and E-cadherin (K20) were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies to phospho (p)-PKCα, c-Met, p-Y1003-c-Met, and p-Y1230/1234/1235-c-Met were from Cell Signaling Technology (Danvers, MA). Horseradish peroxidase-conjugated goat anti-rabbit and anti-mouse secondary antibodies were purchased from Molecular Probes (Eugene, OR). ECL kit for detection of proteins by Western blotting was obtained from Amersham Pharmacia (Piscataway, NJ). Bronchial epithelial cell basal medium (BEBM) and a supplement kit were purchased from Lonza. All other reagents were of analytical grade.
Preparation of cell lysates and Western blotting.
After the indicated treatments, cells were rinsed with ice-cold PBS and lysed in 150 μl of lysis buffer containing 20 mM Tris·HCl (pH 7.4), 150 mM NaCl, 2 mM EGTA, 5 mM β-glycerophosphate, 1 mM MgCl2, 1% Triton X-100, 1 mM sodium orthovanadate, 10 μg/ml protease inhibitors, 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 μg/ml pepstatin. Cell lysates were sonicated on ice for 12 s and centrifuged at 500 g for 5 min at 4°C in a microfuge. Protein concentrations were determined with a BCA protein assay kit (Thermo Fisher Scientific, Rockford, IL) using BSA as standard. Equal amounts of cell lysates (20 μg) were subjected to 10% SDS-PAGE analysis, transferred to polyvinylidene difluoride membranes, blocked with 5% (wt/vol) BSA in 25 mM Tris·HCl, pH 7.4, 137 mM NaCl, and 0.1% Tween 20 (TBST) for 1 h, and incubated with primary antibodies in 5% (wt/vol) BSA in TBST for 1–2 h. The membranes were washed at least three times with TBST at 15-min intervals and then incubated with either mouse, rabbit, or goat horseradish peroxidase-conjugated secondary antibody (1:2,000) for 1 h. They were then developed with the enhanced chemiluminescence detection system according to the manufacturer's instructions.
Cell surface protein isolation.
HBEpCs grown in D100 dishes were treated with LPS for 16 h. Cell surface proteins were isolated by the Pierce Cell Surface Protein Isolation Kit (Thermo Fisher Scientific) according to the manufacturer's instruction. Briefly, cell surface proteins were labeled with a cell-impermeable, cleavable biotinylation reagent, Sulfo-NHS-SS-Biotin, for 30 min at 4°C, followed by column purification. The isolated cell surface proteins were analyzed by Western blotting with a c-Met antibody.
Immunofluorescence staining.
HBEpCs were grown in a glass chamber until ∼80–90% confluence. After treatment, cells were fixed with 3.7% formaldehyde for 20 min and then immunostained with E-cadherin (K20) (12), c-Met, or V5 tag antibody followed by three washes and incubated with the fluorescent probe-conjugated secondary antibody. Images were captured by a Nikon ECLIPSE TE 300 inverted microscope.
Construction of c-Met wild-type and Y1003A mutant plasmids.
Human c-Met cDNA was synthesized by RT-PCR using HBEpCs total RNA as a template. The primers are 5′-CACCATGAAGGCCCCCGCTGTG-3′ and 5′-TGATGTCTCCCAGAAGGAGGC-3′. The resulting PCR product was purified, followed by one-step cloning into a pcDNA3.1D/V5-His vector. The PCR conditions were as follows: 98°C for 15 s and 35 cycles of 98°C for 15 s, 58°C for 15 s, and 72°C for 60 s. The Y100A mutant was generated by a Site Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) according the manufacturer's instructions (31, 33). Human V5-tagged c-Met plasmid was used a template. The primers are 5′- ATCTTCTGGAAAAGTAGCTCGGGCGTCTACAGATTCATTTGAAACC-3′ and 5′-GGTTTCAAATGAATCTGTAGACGCCCGAGCTACTTTTCCAGAAGA T-3′.
Transfection of small-interfering RNA of c-Met.
Smartpool RNA duplexes corresponding to c-Met and scrambled control small-interfering RNA (siRNA) were purchased from Santa Cruz Biotechnology. Transient transfection of siRNA was carried out using Transmessenger Transfection Reagent (Qiagen, Chatsworth, CA). Briefly, siRNA (50 nM) was condensed with Enhancer R and formulated with Transmessenger reagent, according to the manufacturer's instruction. The transfection complex was diluted into 900 μl of BEBM medium and added directly to the cells. The medium was replaced with complete BEGM medium after 3 h. Cells were cultured in the BEGM medium for 72 h.
Transfection of c-Met, c-Met mutant, and PKCα short-hairpin RNA plasmids.
HBEpCs grown on six-well plates (60–70% confluence) were transfected with V5-tagged-c-Met, c-MetY1003A, and PKCα short-hairpin RNA (shRNA) plasmids (2 μg) using FuGENE HD transfection reagent according to the manufacturer's protocol. c-Met or c-Met Y1003A mutant transfected cells were analyzed after 48 h. PKCα shRNA transfected cells were analyzed after 72 h.
Measurement of transepithelial resistance by electrical cell impedance sensor.
HBEpCs were grown to 100% confluence over gold microelectrodes. Transepithelial resistance (TER) was measured in an electrical cell-substrate impedance sensing system (Applied BioPhysics, Foster City, CA). The total TER measured dynamically across the epithelial monolayer was determined as the combined resistance between the basal surface of the cell and the electrode, reflecting alterations in cell-cell adhesion (12).
Statistical analyses.
All results were subjected to statistical analysis using Microsoft Excel, and, wherever appropriate, the data were also analyzed by Student's t-test and expressed as means ± SD. Data were collected from at least three independent experiments, and P < 0.05 was considered significant.
RESULTS
LPS induces phosphorylation of c-Met at Y1003.
To investigate if LPS regulates c-Met phosphorylation, HBEpCs were treated with LPS (5 μg/ml) for 0–6 h. The cell lysates were analyzed by immunoblottings with antibodies to phospho-Y1003-c-Met, phospho-Y1230/1234/1235-c-Met, and total c-Met. Tyrosine phosphorylation of c-Met at all the tyrosine residues was extremely low in untreated HBEpCs, whereas a strong band of total c-Met (∼140 kDa) was detected (Fig. 1A). LPS treatment induced significant Y1003 phosphorylation of c-Met in a time-dependent manner after 3 h, without inducing phosphorylation at Y1230/1234/1235 and altering c-Met expression (Fig. 1, A and B). Pretreatment with a specific c-Met tyrosine kinase inhibitor, PHA-665752 (1–100 nM), attenuated the LPS-induced Y1003 phosphorylation in a dose-dependent manner (Fig. 1, C and D). These data demonstrate that LPS induced c-Met phosphorylation at Y1003 without affecting c-Met expression and other tyrosine residues.
Fig. 1.

Lipopolysaccharide (LPS) induces phosphorylation of c-Met at Y1003. A: human bronchial epithelial cells (HBEpCs) were treated with LPS (5 μg/ml) for 0–6 h. Cell lysates were analyzed by immunoblotting with antibodies to phospho-Y1003 c-Met (p-Y1003-c-Met), phospho (p)-Y1230/1234/1235-c-Met, and c-Met. Shown are representative images from three independent experiments. B: quantitative analysis from three independent experiments in A (mean ± SD). *P < 0.01 vs. 0 h of LPS treatment. C: HBEpCs were treated with PHA-665752 (PHA, 1, 20, and 100 nM, 1 h) before LPS (5 μg/ml) challenge for 6 h. Cell lysates were analyzed by immunoblotting with antibodies to p-Y1003-c-Met and c-Met. Shown are representative images from three independent experiments. D: quantitative analysis from three independent experiments in C (mean ± SD). *P < 0.01 vs. untreated cells; **P < 0.01 vs. LPS alone-treated cells.
PKCα mediates LPS-induced Y1003 phosphorylation of c-Met.
Tyrosine phosphorylation is negatively controlled by the activity of protein tyrosine phosphatases, which is subsequently regulated by PKC (25, 27). We have previously shown that LPS induced activation of PKCα in lung epithelial cells (29). To further investigate the role of PKCα in LPS-induced Y1003 phosphorylation of c-Met, cells were pretreated with Go-6976, a commonly used PKCα inhibitor, before LPS treatment. Figure 2, A and B, shows that LPS (5 μg/ml, 6 h) induced phosphorylation of c-Met at Y1003, and this effect was attenuated by Go-6976 in a dose-dependent manner. Furthermore, PKCα was downregulated by PKCα shRNA transfection. As shown in Fig. 2, C and D, PKCα shRNA attenuated LPS-induced c-Met Y1003 phosphorylation as well as PKCα expression. The data suggest that PKCα regulates LPS-induced Y1003 phosphorylation of c-Met.
Fig. 2.

Protein kinase C (PKC)-α regulates LPS-induced Y1003 phosphorylation of c-Met. A: HBEpCs were treated with Go-6976 (1, 5, and 10 μM, 1 h) before LPS (5 μg/ml) challenge for 6 h. Cell lysates were analyzed by immunoblotting with antibodies to p-Y1003-c-Met and c-Met. Shown are representative images from three independent experiments. B: quantitative analysis from three independent experiments in A (mean ± SD). *P < 0.01 vs. untreated cells; **P < 0.05 vs. LPS alone-treated cells. C: HBEpCs were transfected with PKCα short-hairpin RNA (shRNA, 2 μg, 3 days) before the LPS (5 μg/ml) challenge for 6 h. Cell lysates were analyzed by immunoblotting with antibodies to p-Y1003-c-Met, c-Met, PKCα, and β-actin. Shown are representative images from three independent experiments. D: quantitative analysis from three independent experiments in C (mean ± SD). *P < 0.01 vs. untreated cells; **P < 0.05 vs. LPS alone-treated cells.
LPS-induced Y1003 phosphorylation of c-Met regulates c-Met internalization.
Y1003 of c-Met is localized in the juxtamembrane domain, and its phosphorylation is known to regulate c-Met internalization and degradation (6). We have shown that LPS did not reduce c-Met expression, although it induced phosphorylation of c-Met at Y1003 (Fig. 1). We next investigated whether Y1003 phosphorylation of c-Met by LPS regulates c-Met internalization. HBEpCs were treated with LPS (5 μg/ml) for 16 h, and then cell surface proteins were isolated by using Sulfo-NHS-SS-Biotin labeling. As shown in Fig. 3, A and B, c-Met expression on the cell surface was significantly reduced by LPS treatment. Furthermore, HBEpC cells were cultured in a glass bottom chamber and treated with LPS (5 μg/ml) for 16 h. c-Met localization was then examined by immunostaining with a c-Met antibody. Figure 3, C and D, shows that, in untreated cells, c-Met is predominantly expressed on the cell membrane, especially at the cell-cell contacts. LPS treatment reduced c-Met content on the cell membrane and increased its levels in the cytoplasm. We also treated cells with PHA-665752 or Go-6976 before LPS challenge. c-Met and PKC inhibitors both suppressed LPS-induced c-Met internalization (Fig. 3, C and D). To directly determine the role of Y1003 phosphorylation in LPS-induced c-Met internalization, V5-tagged human c-Met wild-type and c-MetY1003A mutant were overexpressed in HBEpCs before LPS treatment. As shown in Fig. 4, A and B, LPS induced c-Met wild-type internalization without altering c-MetY1003A expression on the cell surface. The data clearly exhibit that Y1003 phosphorylation of c-Met is the key to LPS-induced c-Met internalization.
Fig. 3.

LPS-induced Y1003 phosphorylation of c-Met regulates LPS-mediated c-Met internalization. A: HBEpCs grown on D100 dishes were treated with LPS (5 μg/ml) for 16 h. Cell surface proteins were isolated. The expression of c-Met on cell surface was analyzed by immunoblotting with an antibody to c-Met. The total cell lysates were analyzed by immunoblotting with an antibody to β-actin. Shown are representative images from three independent experiments. B: quantitative analysis from three independent experiments in A (mean ± SD). *P < 0.01 vs. untreated cells. C: HBEpCs grown on glass bottom slides were treated with PHA-665752 (20 nM) or Go-6976 (Go, 10 μM) for 1 h before LPS (5 μg/ml) challenge for a further 16 h. Cells were fixed, and c-Met localization was detected by immunostaining with a c-Met antibody (Ab) followed by incubation with a fluorescent probe-conjugated second antibody. Shown are representative images from three independent experiments. D: the pixel intensities of c-Met at cell-cell contacts were measured by MetaVue software. *P < 0.01 vs. untreated cells; **P < 0.01 vs. LPS alone-treated cells.
Fig. 4.
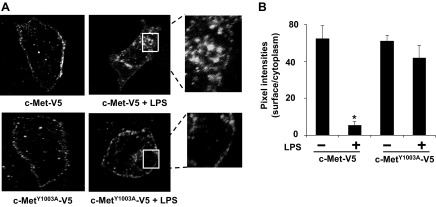
c-MetY1003A is resistant to LPS-induced internalization. A: HBEpCs grown on glass bottom slides were transfected with V5-tagged c-Met or c-MetY1003A plasmid. Cells were treated with LPS (5 μg/ml) for 16 h, and then cells were fixed. The localization of the overexpressed c-Met and mutant was detected by immunostaining with a V5 tag antibody followed by incubation with a fluorescent probe-conjugated second antibody. Shown are representative images from three independent experiments. B: the pixel intensities of overexpressed c-Met or mutant at cell surfaces and cytoplasm were measured by MetaVue software. *P < 0.01 vs. untreated cells.
Y1003 phosphorylation of c-Met regulates LPS-reduced TER.
We have earlier shown that LPS induced E-cadherin internalization, causing a reduction of TER (22). The role of c-Met in the regulation of endothelial barrier integrity has been well studied (12, 36); however, the role of c-Met trafficking in the regulation of lung epithelial barrier integrity remains unclear. To investigate the role of Y1003 phosphorylation of c-Met in LPS-induced reduction of TER, cells grown on electrical cell impedance sensor chambers were treated with PHA-665752 or Go-6976 before the LPS challenge. Go-6976 slightly increased the TER, whereas PHA-665752 rapidly reduced the TER, which returned back to normal at 4 h. In agreement with our previous study (12), LPS reduced TER in a time-dependent manner, whereas PHA-665752 or Go-6976 pretreatment partly rescued it in 8–16 h (Fig. 5, A and B). Furthermore, we found that overexpression of c-Met wild type attenuated the LPS-induced decrease in TER; the effect was further enhanced by overexpression of c-MetY1003A. These results indicate that LPS-induced Y1003 phosphorylation of c-Met and c-Met internalization is involved in LPS-reduced lung epithelial barrier disruption.
Fig. 5.
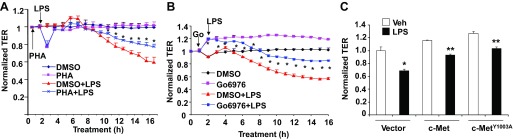
LPS-induced Y1003 phosphorylation of c-Met regulates LPS-reduced transepithelial resistance (TER). HBEpCs grown on golden microelectrodes were treated with DMSO, PHA-665752 (20 nM) (A), or Go-6976 (Go, 10 μM) (B) for 1 h before LPS (5 μg/ml) challenge for 16 h. The changes in TER were measured by electrical cell impedance sensor (ECIS). *P < 0.01 vs. LPS-treated cells. C: overexpressing c-Met or c-MetY1003A HBEpCs were treated with LPS (5 μg/ml) for 16 h, and the changes in TER were measured. *P < 0.01 vs. control cells; **P < 0.05 vs. LPS-treated empty vector transfected cells.
c-Met is involved in LPA-induced lung epithelial barrier enhancement.
Previous studies have shown that c-Met is associated with E-cadherin at cell-cell contacts (29). We have observed earlier that LPA rapidly increased TER in lung epithelial cells, which is dependent on E-cadherin accumulation at cell-cell contacts (22). Here, we examined the role of c-Met in LPA-induced lung epithelial barrier enhancement. c-Met expression was downregulated by the transfection of HBEpCs with a specific c-Met siRNA before LPA treatment. LPA (1 μM, 15 min) enhanced E-cadherin accumulation at cell-cell contacts and enhanced TER, whereas the effects were partly attenuated by c-Met siRNA (Fig. 6, A and B). The effect of c-Met siRNA on loss of c-Met expression was confirmed by Western blotting (Fig. 6C). The data demonstrate that LPA-induced c-Met accumulation at cell-cell contacts contributes to the lung epithelial barrier integrity. We have shown that posttreatment of LPA reversed LPS-induced lung epithelial barrier dysfunction (12). Furthermore, to test if LPA pretreatment could attenuate the LPS-induced decrease in TER, cells were treated with LPA (1 μM, 1 h) before LPS (5 μg/ml) for 16 h. As shown in Fig. 6D, the pretreatment of LPA significantly reversed the effect of LPS on lung epithelial barrier dysfunction.
Fig. 6.

c-Met is involved in lysophosphatidic acid (LPA)-induced lung epithelial barrier enhancement. A: HBEpCs grown on glass bottom slides were transfected with scrambled small-interfering RNA (siRNA) or c-Met siRNA (50 ng, 72 h) before LPA (1 μM, 15 min) treatment. E-cadherin localization was detected by immunostaining with an E-cadherin antibody followed by incubation with a fluorescence-labeled second antibody. Shown are representative images from three independent experiments. B: HBEpCs grown on golden microelectrodes were transfected with scrambled siRNA or c-Met siRNA (50 ng, 72 h) before LPA (1 μM, 30 min) treatment. The changes in TER were measured by ECIS. *P < 0.01 vs. untreated scrambled siRNA transfected cells; **P < 0.05 vs. LPA-treated scrambled siRNA transfected cells. C: the effect of c-Met siRNA on c-Met expression was analyzed by immunoblotting with antibodies to c-Met and E-cadherin. Shown are representative images from three independent experiments. D: HBEpCs were pretreated with LPA (1 μM, 1h) before LPS challenge (5 μg/ml, 16 h). TER changes were normalized to LPS treatment alone. *P < 0.01 vs. LPS alone treatment.
LPA reversed LPS-induced c-Met internalization, Y1003 phosphorylation, and activation of PKCα.
We have previously shown that LPA treatment induced serine phosphorylation of c-Met and c-Met accumulation at cell-cell contacts (29). To investigate if LPA treatment reverses LPS-induced c-Met internalization, cells were treated with LPS for 16 h, followed by LPA treatment for a further 1 h. LPS treatment reduced plasma membrane c-Met content, whereas LPA posttreatment reversed the trend (Fig. 7, A and B). Furthermore, we found that pretreatment with LPA attenuated LPS-induced phosphorylation of c-Met and PKCα (Fig. 7, C and D). These results suggest that LPA reversed LPS-induced lung epithelial barrier dysfunction by reducing LPS-mediated activation of PKCα, Y1003 c-Met phosphorylation, and c-Met internalization.
Fig. 7.

LPA reverses LPS-induced c-Met internalization, Y1003 phosphorylation, and phosphorylation of PKCα. A: HBEpCs grown on glass bottom slides were treated with LPS (5 μg/ml) for 16 h before adding LPA (1 μM) for 1 h. Cells were fixed, and c-Met localization was detected by immunostaining with a c-Met antibody followed by incubation with a fluorescence-labeled second antibody. Shown are representative images from three independent experiments. B: the pixel intensities of c-Met at cell-cell contacts were measured by MetaVue software. *P < 0.01 vs. untreated cells; **P < 0.05 vs. LPS-treated cells. C: HBEpCs were pretreated with LPA (1 μM) for 1 h before the LPS (5 μg/ml, 6 h) challenge. Cell lysates were analyzed by immunoblotting with antibodies to p-Y1003-c-Met, c-Met, p-PKCα, PKCα, and β-actin. Shown are representative images from three independent experiments. D: quantitative analysis from three independent experiments in C (mean ± SD). *P < 0.01 vs. untreated cells; **P < 0.01 vs. LPS alone-treated cells.
DISCUSSION
Overexpression of c-Met has been detected in a variety of human tumors, including lung cancers. c-Met activation regulates cell scattering, which has been implicated in cancer metastasis (5, 20). Ligand-induced plasma membrane receptor internalization is a common process for controlling receptor stability and signaling. The elucidation of c-Met trafficking regulation therefore will provide the basis for devising interventions that impact cell injury, repair, and cellular homeostasis. Here, we demonstrate that the bacterial endotoxin, LPS, induces Y1003 c-Met phosphorylation, triggering c-Met internalization and the disruption of lung epithelial barrier. The process is facilitated by PKCα and is impeded by an epithelial barrier enhancer, LPA. This is the first report to reveal the role of c-Met trafficking in the regulation of lung epithelial barrier function, providing a new therapeutic target to attenuate endotoxin-induced lung injury.
Receptor tyrosine kinase internalization is regulated by phosphorylation at certain tyrosine residues, such as Y1068 and Y1086 in case of EGF-R internalization (13). c-Met contains several tyrosine residues that are phosphorylated, such as Y1003 in the juxtamembrane domain, Y1231/1234/1235 in the kinase domain, and Y1349/1356/1365 in the COOH-terminal domain (20, 23). The present study reveals that LPS induces tyrosine phosphorylation of c-Met at Y1003 that is known to be involved in the c-Met internalization. LPS, however, did not induce phosphorylation of any other known tyrosine residues in c-Met and c-Met expression. Phosphorylation of Y1003 c-Met by HGF is known to recruit c-Cbl, the E3 ubiquitin ligase that targets c-Met to proteasomal degradation (22). Both HGF and LPS induce Y1003 phosphorylation of c-Met; however, only HGF promotes c-Met degradation. Unlike HGF (16, 36), LPS failed to induce lung epithelial cell scattering (data not shown). The discrepancy between the LPS and HGF effects might be due to the fact that HGF phosphorylates several tyrosine residues of c-Met, whereas LPS appears to phosphorylate only Y1003. LPS is the ligand for Toll-like receptor 4 (TLR4), which plays an important role in innate immunity (1). Cross talk between TLR4 and other cell surface receptors such as proteinase inhibitor receptor-2 (4), FcγReceptorIII (CD16) (24), and LPA receptor 1 (30) regulate immune responses. The present study demonstrates that the cross talk between LPS receptor and c-Met regulates c-Met internalization, but not its degradation. Interestingly, LPS-induced Y1003 c-Met phosphorylation and internalization plays a critical role in LPS-mediated lung epithelial barrier disruption, since inhibition of Y1003 c-Met phosphorylation attenuated LPS-induced c-Met internalization and epithelial barrier dysfunction. Future studies will examine if LPS induces c-cbl interaction with c-Met and the role of c-cbl in LPS-induced c-Met internalization.
Growing evidence suggests that PKC isotypes mediate c-Met trafficking and activation (14, 15, 36). Inhibition of PKCα blocked HGF-induced c-Met trafficking toward the perinuclear compartment, whereas PKCε activation contributed to c-Met trafficking back to the cell surface (14). In lung epithelial cells, LPS treatment induced PKCα activation (3, 29). We show here that PKCα modulates LPS-induced c-Met phosphorylation at Y1003 and internalization. Because PKCα is a serine/threonine kinase, the effect on Y1003 phosphorylation of c-Met must be indirect and might be mediated by the regulation of tyrosine phosphatase activity. Indeed, it has been known that PKC regulates tyrosine phosphatase activity, therefore mediating protein tyrosine phosphorylation. For example, PKCδ associates with SHPTP1 tyrosine phosphatase, thus inducing phosphorylation and inhibition of SHPTP1 (25). Receptor-type protein tyrosine phosphatase β (RPTP-β) is responsible for c-Met dephosphorylation (28). Although the role of PKC in regulation of RPTP-β has not been reported, the phosphorylation of its isoform, RPTP-α, has been known to be regulated by PKC (27). We hypothesize that PKC regulates RPTP-β phosphorylation and activation, thus altering Y1003 c-Met phosphorylation. Future studies may investigate if the RPTP-β mediates LPS-induced Y1003 c-Met phosphorylation and internalization.
In addition to inducing cell scattering, c-Met activation has been implicated in strengthening cell-cell junctions (19, 26). HGF activates c-Met, thus increasing lung endothelial barrier function (19, 26); however, it had no effect on TER in lung epithelial cells (data not shown). HGF-mediated increase in endothelial barrier integrity is dependent on the cross talk between c-Met and sphingosine 1-phosphate receptor 1 (7); however, sphingosine 1-phosphate receptor 1 expression is very low in HBEpCs (data not shown). Here, we show that the cross talk between LPS signaling and c-Met regulates lung epithelial barrier function. This is the first report to demonstrate that LPS induces Y1003 phosphorylation of c-Met at the juxtamembrane domain therefore increasing c-Met internalization and disrupting lung epithelial barrier integrity through PKCα. This study provides a new target to prevent endotoxin-induced lung injury by attenuating c-Met internalization.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grants RO1-HL-01916, HL-112791 (to Y. Zhao), and R37-079396 (to V. Natarajan) and American Heart Association Award 12SDG9050005 (J. Zhao).
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: Y.Z. and V.N. conception and design of research; Y.Z., J.Z., R.K.M., and J.W. performed experiments; Y.Z., J.Z., and J.W. analyzed data; Y.Z., J.Z., E.W.S., and R.S. interpreted results of experiments; Y.Z. prepared figures; Y.Z. drafted manuscript; Y.Z., J.Z., R.K.M., and V.N. edited and revised manuscript; Y.Z., J.Z., R.K.M., J.W., E.W.S., and V.N. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Andrew Khoo for manuscript editing.
REFERENCES
- 1. Beutler B. Tlr4: central component of the sole mammalian LPS sensor. Curr Opin Immunol 12: 20–26, 2000 [DOI] [PubMed] [Google Scholar]
- 2. Birukova AA, Wu T, Tian Y, Meliton A, Sarich N, Tian X, Leff A, Birukov KG. Iloprost improves endothelial barrier function in LPS-induced lung injury. Eur Respir J 41: 165–176, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boncoeur E, Tardif V, Tessier MC, Morneau F, Lavoie J, Gendreau-Berthiaume E, Grygorczyk R, Dagenais A, Berthiaume Y. Modulation of epithelial sodium channel activity by lipopolysaccharide in alveolar type II cells: involvement of purinergic signaling. Am J Physiol Lung Cell Mol Physiol 298: L417–L426, 2010 [DOI] [PubMed] [Google Scholar]
- 4. Bucci M, Vellecco V, Harrington L, Brancaleone V, Roviezzo F, Mattace Raso G, Ianaro A, Lungarella G, De Palma R, Meli R, Cirino G. Cross talk between Toll-Like Receptor 4 (TLR4) and Proteinase Activated Receptor 2 (PAR-2) is involved in vascular function. Br J Pharmacol 168: 411–420, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cecchi F, Rabe DC, Bottaro DP. Targeting the HGF/Met signaling pathway in cancer therapy. Expert Opin Ther Targets 16: 553–572, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davies G, Jiang WG, Mason MD. HGF/SF modifies the interaction between its receptor c-Met, and the E-cadherin/catenin complex in prostate cancer cells. Int J Mol Med 7: 385–388, 2001 [DOI] [PubMed] [Google Scholar]
- 7. Ephstein Y, Singleton PA, Chen W, Wang L, Salgia R, Kanteti P, Dudek SM, Garcia JG, Jacobson JR. Critical role of S1PR1 and integrin beta4 in HGF/c-Met-mediated increases in vascular integrity. J Biol Chem 288: 2191–2200, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ephstein Y, Singleton PA, Chen W, Wang L, Salgia R, Kanteti P, Dudek SM, Garcia JG, Jacobson JR. Critical role of S1PR1 and integrin beta4 in HGF/c-Met-mediated increases in vascular integrity. J Biol Chem 288: 2191–2200, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gandino L, Longati P, Medico E, Prat M, Comoglio PM. Phosphorylation of serine 985 negatively regulates the hepatocyte growth factor receptor kinase. J Biol Chem 269: 1815–1820, 1994 [PubMed] [Google Scholar]
- 10. Grandal MV, Madshus IH. Epidermal growth factor receptor and cancer: control of oncogenic signalling by endocytosis. J Cell Mol Med 12: 1527–1534, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hashigasako A, Machide M, Nakamura T, Matsumoto K. Bi-directional regulation of Ser-985 phosphorylation of c-met via protein kinase C and protein phosphatase 2A involves c-Met activation and cellular responsiveness to hepatocyte growth factor. J Biol Chem 279: 26445–26452, 2004 [DOI] [PubMed] [Google Scholar]
- 12. He D, Su Y, Usatyuk PV, Spannhake EW, Kogut P, Solway J, Natarajan V, Zhao Y. Lysophosphatidic acid enhances pulmonary epithelial barrier integrity and protects endotoxin-induced epithelial barrier disruption and lung injury. J Biol Chem 284: 24123–24132, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiang X, Huang F, Marusyk A, Sorkin A. Grb2 regulates internalization of EGF receptors through clathrin-coated pits. Mol Biol Cell 14: 858–870, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kermorgant S, Zicha D, Parker PJ. PKC controls HGF-dependent c-Met traffic, signalling and cell migration. Embo J 23: 3721–3734, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kermorgant S, Zicha D, Parker PJ. Protein kinase C controls microtubule-based traffic but not proteasomal degradation of c-Met. J Biol Chem 278: 28921–28929, 2003 [DOI] [PubMed] [Google Scholar]
- 16. Kim HJ, Sammak PJ, Ingbar DH. Hepatocyte growth factor stimulates migration of type II alveolar epithelial cells on the provisional matrix proteins fibronectin and fibrinogen. Chest 116: 94S–95S, 1999 [DOI] [PubMed] [Google Scholar]
- 17. Lefebvre J, Ancot F, Leroy C, Muharram G, Lemiere A, Tulasne D. Met degradation: more than one stone to shoot a receptor down. FASEB J 26: 1387–1399, 2012 [DOI] [PubMed] [Google Scholar]
- 18. Li G, Schaider H, Satyamoorthy K, Hanakawa Y, Hashimoto K, Herlyn M. Downregulation of E-cadherin and Desmoglein 1 by autocrine hepatocyte growth factor during melanoma development. Oncogene 20: 8125–8135, 2001 [DOI] [PubMed] [Google Scholar]
- 19. Liu F, Schaphorst KL, Verin AD, Jacobs K, Birukova A, Day RM, Bogatcheva N, Bottaro DP, Garcia JG. Hepatocyte growth factor enhances endothelial cell barrier function and cortical cytoskeletal rearrangement: potential role of glycogen synthase kinase-3beta. FABEB J 16: 950–962, 2002 [DOI] [PubMed] [Google Scholar]
- 20. Ma PC, Maulik G, Christensen J, Salgia R. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev 22: 309–325, 2003 [DOI] [PubMed] [Google Scholar]
- 21. Marmor MD, Yarden Y. Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene 23: 2057–2070, 2004 [DOI] [PubMed] [Google Scholar]
- 22. Maulik G, Madhiwala P, Brooks S, Ma PC, Kijima T, Tibaldi EV, Schaefer E, Parmar K, Salgia R. Activated c-Met signals through PI3K with dramatic effects on cytoskeletal functions in small cell lung cancer. J Cell Mol Med 6: 539–553, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Niemann HH. Structural insights into Met receptor activation. Eur J Cell Biol 90: 972–981, 2011 [DOI] [PubMed] [Google Scholar]
- 24. Rittirsch D, Flierl MA, Day DE, Nadeau BA, Zetoune FS, Sarma JV, Werner CM, Wanner GA, Simmen HP, Huber-Lang MS, Ward PA. Cross-talk between TLR4 and FcgammaReceptorIII (CD16) pathways. PLoS Pathog 5: e1000464, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rosdahl JA, Mourton TL, Brady-Kalnay SM. Protein kinase C delta (PKCdelta) is required for protein tyrosine phosphatase mu (PTPmu)-dependent neurite outgrowth. Mol Cell Neurosci 19: 292–306, 2002 [DOI] [PubMed] [Google Scholar]
- 26. Singleton PA, Salgia R, Moreno-Vinasco L, Moitra J, Sammani S, Mirzapoiazova T, Garcia JG. CD44 regulates hepatocyte growth factor-mediated vascular integrity. Role of c-Met, Tiam1/Rac1, dynamin 2, and cortactin. J Biol Chem 282: 30643–30657, 2007 [DOI] [PubMed] [Google Scholar]
- 27. Tracy S, van der Geer P, Hunter T. The receptor-like protein-tyrosine phosphatase, RPTP alpha, is phosphorylated by protein kinase C on two serines close to the inner face of the plasma membrane. J Biol Chem 270: 10587–10594, 1995 [DOI] [PubMed] [Google Scholar]
- 28. Xu Y, Zhou J, Carey TE, McHugh JB, Voorhees JJ, Fisher GJ. Receptor-type Protein tyrosine phosphatase beta regulates met phosphorylation and function in head and neck squamous cell carcinoma. Neoplasia New York 14: 1015–1022, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhao J, Chen Q, Li H, Myerburg M, Spannhake EW, Natarajan V, Zhao Y. Lysophosphatidic acid increases soluble ST2 expression in mouse lung and human bronchial epithelial cells. Cell Signal 24: 77–85, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao J, He D, Su Y, Berdyshev E, Chun J, Natarajan V, Zhao Y. Lysophosphatidic acid receptor 1 modulates lipopolysaccharide-induced inflammation in alveolar epithelial cells and murine lungs. Am J Physiol Lung Cell Mol Physiol 301: L547–L556, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao J, Mialki RK, Wei J, Coon TA, Zou C, Chen BB, Mallampalli RK, Zhao Y. SCF E3 ligase F-box protein complex SCFFBXL19 regulates cell migration by mediating Rac1 ubiquitination and degradation. FASEB J In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao J, Wei J, Mialki R, Zou C, Mallampalli RK, Zhao Y. Extracellular signal-regulated kinase (ERK) regulates cortactin ubiquitination and degradation in lung epithelial cells. J Biol Chem 287: 19105–19114, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao J, Wei J, Mialki RK, Mallampalli DF, Chen BB, Coon T, Zou C, Mallampalli RK, Zhao Y. F-box protein FBXL19-mediated ubiquitination and degradation of the receptor for IL-33 limits pulmonary inflammation. Nat Immunol 13: 651–658, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao Y, Gorshkova IA, Berdyshev E, He D, Fu P, Ma W, Su Y, Usatyuk PV, Pendyala S, Oskouian B, Saba JD, Garcia JG, Natarajan V. Protection of LPS-induced murine acute lung injury by sphingosine-1-phosphate lyase suppression. Am J Respir Cell Mol Biol 45: 426–435, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhao Y, He D, Saatian B, Watkins T, Spannhake EW, Pyne NJ, Natarajan V. Regulation of lysophosphatidic acid-induced epidermal growth factor receptor transactivation and interleukin-8 secretion in human bronchial epithelial cells by protein kinase Cdelta, Lyn kinase, and matrix metalloproteinases. J Biol Chem 281: 19501–19511, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao Y, He D, Stern R, Usatyuk PV, Spannhake EW, Salgia R, Natarajan V. Lysophosphatidic acid modulates c-Met redistribution and hepatocyte growth factor/c-Met signaling in human bronchial epithelial cells through PKC delta and E-cadherin. Cell Signal 19: 2329–2338, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao Y, Natarajan V. Lysophosphatidic acid (LPA) and its receptors: Role in airway inflammation and remodeling. Biochim Biophys Acta 1831: 86–92, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhao Y, Natarajan V. Lysophosphatidic acid signaling in airway epithelium: role in airway inflammation and remodeling. Cell Signal 21: 367–377, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]