

Abstract
Hypoxia-induced pulmonary hypertension (HPH) is characterized by sustained pulmonary vasoconstriction and vascular remodeling, both of which are mediated by pulmonary artery smooth muscle cell (PASMC) contraction and proliferation, respectively. An increase in cytosolic Ca2+ concentration ([Ca2+]cyt) is a major trigger for pulmonary vasoconstriction and an important stimulus for cell proliferation in PASMCs. Ca2+ influx through voltage-dependent Ca2+ channels (VDCC) is an important pathway for the regulation of [Ca2+]cyt. The potential role for L- and T-type VDCC in the development of HPH is still unclear. Using a hypoxic-induced pulmonary hypertension mouse model, we undertook this study to identify if VDCC in pulmonary artery (PA) are functionally upregulated and determine which type of VDCC are altered in HPH. Mice subjected to chronic hypoxia developed pulmonary hypertension within 4 wk, and high-K+- and U-46619-induced contraction of PA was greater in chronic hypoxic mice than that in normoxic control mice. Additionally, we demonstrate that high-K+- and U-46619-induced Ca2+ influx in PASMC is significantly increased in the hypoxic group. The VDCC activator, Bay K8864, induced greater contraction of the PA of hypoxic mice than in that of normoxic mice in isometric force measurements. L-type and T-type VDCC blockers significantly attenuated absolute contraction of the PA in hypoxic mice. Chronic hypoxia did not increase high-K+- and U-46619-induced contraction of mesenteric artery (MA). Compared with MA, PA displayed higher expression of calcium channel voltage-dependent L-type α1C-subunit (Cav1.2) and T-type α1H-subunit (Cav3.2) upon exposure to chronic hypoxia. In conclusion, both L-type and T-type VDCC were functionally upregulated in PA, but not MA, in HPH mice, which could result from selectively increased expression of Cav1.2 and Cav3.2.
Keywords: hypoxia, voltage-dependent calcium ion channel, pulmonary artery, mouse, calcium channel voltage-dependent L-type α1C-subunit, calcium channel voltage-dependent T-type α1H-subunit
persistent hypoxia induces vasoconstriction of small pulmonary arteries (PA) and vascular remodeling, including smooth muscle cell hypertrophy and hyperplasia, eventually resulting in hypoxic pulmonary hypertension (HPH). Pulmonary hypertension associated with hypoxia belongs to the third group in the classification of pulmonary hypertension according to the proceedings of the Fourth World Symposium on Pulmonary Hypertension at Dana Point 2008 (17). Over time, HPH causes right ventricle hypertrophy, right ventricular failure, and death (1, 37). Pulmonary hypertension related to chronic obstructive pulmonary disease (COPD) is one of the most common forms of HPH and is significantly associated with increased mortality (8, 36). Currently, there is no specific therapy for pulmonary hypertension associated with COPD, which provides motivation for researchers to understand the pathogenic mechanisms of HPH.
Sustained pulmonary vasoconstriction and vascular remodeling are the predominant features of HPH, both of which are respectively mediated by pulmonary artery smooth muscle cell (PASMC) contraction and proliferation (27). A rise of cytosolic Ca2+ concentration ([Ca2+]cyt) is a major trigger for pulmonary vasoconstriction and an important stimulus for PASMC proliferation. Chronic hypoxia increases the resting [Ca2+]cyt and alters the electromechanical coupling in PASMC, and normoxic recovery reverses the alterations in the electrophysiological properties of PASMC and causes normalization of pulmonary arterial pressure (5, 6).
In PASMC, [Ca2+]cyt is regulated by two pathways: voltage-dependent Ca2+ influx and voltage-independent Ca2+ influx. The latter includes Ca2+ entry through receptor-operated Ca2+ channels (ROC) and store-operated Ca2+ channels (SOC). Lin et al. showed that both ROC and SOC of PASMC were upregulated by chronic hypoxia and contributed to the enhanced vascular tone in HPH (29). In PASMC when the membrane is depolarized, Ca2+ influx through voltage-dependent Ca2+ channels (VDCC) contributes to another important pathway to regulate [Ca2+]cyt.
VDCC are organized into six subtypes by their functional characteristics and further divided into groups based on their sensitivity to membrane depolarization (high voltage: L, P, Q, R, and N type and low voltage: T type). L-, T- and P/Q-type channels have been identified in vascular smooth muscle cells (18, 44); however, expression of P- and Q-type channels has not been investigated in the pulmonary vasculature. L-type VDCC are activated at more depolarized potentials and display slower inactivation and faster deactivation times compared with T-type VDCC. The L-type VDCC antagonists nifedipine or verapamil can prevent hypoxic pulmonary vasoconstriction (HPV), inhibit PASMC smooth muscle cell proliferation, and attenuate HPH. These data imply that Ca2+ influx through L-type VDCC is one of the important [Ca2+]cyt regulatory pathways in HPH (6, 12, 13, 23, 24, 47). T-type VDCC have been identified in human PASMC and are required for cell cycle progression and proliferation (44). Currently, the potential role for T-type VDCC in the development of HPH is still not understood.
Hypoxia affects all systems of the body, and not only causes pulmonary vasoconstriction and HPH, but also induces systemic vasodilation (28, 32). Previous data have shown that acute hypoxia reduces K+ currents in PASMC but not in mesenteric arterial smooth muscle cells (MASMC) and that chronic hypoxia downregulates expression of voltage-gated K+ (KV) channels in PASMC but not in MASMC (40, 63). Suppression of KV channel expression reduces KV current, causes membrane depolarization, which activates VDCC, and increases Ca2+ influx, ultimately leading to a rise in [Ca2+]cyt in PASMC (40, 63). In MASMC, hypoxia negligibly affects KV channel expression, but increases ATP-sensitive K+ current, and induces hyperpolarization (40, 63, 64). Based on these different responses to hypoxia, comparative research on pulmonary and systemic circulation could be helpful in exploring the mechanisms of HPH to facilitate the development of treatments for HPH. In this regard, we used the HPH mouse model to examine whether VDCC in the PA are functionally upregulated by chronic hypoxia and determine which type of VDCC are altered compared with VDCC from the mesenteric artery (MA).
MATERIALS AND METHODS
HPH mouse model.
HPH was induced by exposure of male mice (8 wk old C57BL/6) to chronic hypoxia (10% O2) in a normobaric ventilated chamber (46). Briefly, adult age-matched male mice were kept in hypoxic conditions for 4 wk to develop pulmonary hypertension. All studies were approved by the University of Illinois at Chicago Institutional Animal Care and Use Committee and were performed according to the guidelines of the University of Illinois at Chicago that comply with national and international regulations.
Hemodynamic and right ventricular hypertrophy.
Pulmonary hemodynamic and right ventricular hypertrophy measurements were done as described (46). Briefly, right ventricular systolic pressure (RVSP) was measured by a catheter (Millar, Houston, TX) inserted in the right ventricle (RV) via the external right jugular vein. To determine RV hypertrophy, the RV was separated from the left ventricle (LV) and septum (S). The Fulton index or the ratio of RV weight to LV + S weight [RV/(LV + S)] was determined and calculated as a measurement for RV hypertrophy.
Histology and pulmonary vascular morphometry.
Before removal, the lungs were perfused with 10 ml of saline through the RV. The lung tissue or vessels were then fixed in 10% formalin and paraffin embedded. Sections (6 μm thick) were cut, and hematoxylin and eosin (H&E) staining was performed to analyze the thickness of pulmonary artery (PA).
Isolation of PA and mesentery artery.
After anesthesia using a procedure approved by the Institutional Animal Care and Use Committee, the lungs and mesentery were quickly removed from the mouse, washed with cold saline (to remove blood from the lung tissue), and placed in a dissection plate. Under a stereomicroscope, the right and left branches of the intrapulmonary arteries (250∼400 μm diameter) and MA (250∼400 μm diameter) were isolated from the mice. Adipose and connective tissues were carefully removed with fine forceps and ophthalmological scissors, and the remaining arteries were cut into 2- to 3-mm-long rings.
Isometric tension measurements.
Two tungsten hooks (125 μm diameter) were inserted through the lumen of the rings. One hook was mounted on the bottom of a perfusion chamber, and the other was connected to an isometric force transducer (Harvard Apparatus, Holliston, MA). Isometric tension was continuously monitored and acquired using DATAQ software (DATAQ Instruments, Akron, OH). Optimal resting tension for mouse PA and MA preparations has been previously reported to be ∼300 mg (57, 58). Unless indicated otherwise, resting tension was set at 300 mg. All the rings were equilibrated for 1 h at resting tension and then challenged three times with 40 mM K+ (40K) perfusate to obtain a stable contractile response (57). Isolated rings were superfused with modified Krebs solution (MKS; at 37°C) consisting of the following (in mM): 138 NaCl, 1.8 CaCl2, 4.7 KCl, 1.2 MgSO4, 1.2 NaH2PO4, 5 HEPES, and 10 glucose (pH 7.4 with NaOH). For Ca2+-free solution, CaCl2 was replaced by equimolar MgCl2, and 1 mM EGTA was added to chelate residual Ca2+. In the high-K+ solution, NaCl was replaced by equimolar KCl to maintain osmolarity. The active tension induced by agonists was normalized by the basal tension and expressed as net increase in tension (g).
Drugs.
Mibefradil and ω-agatoxin were prepared as concentrated stock solutions in distilled water. Nifedipine, Bay K8864, and U-46619 were prepared as stocks in dimethyl sulfoxide. All stock solutions were aliquoted and kept frozen at −20°C until use. On the day of experiments, aliquots of the stock solutions were dissolved in MKS to the proper concentrations. The pH values of all solutions were measured after addition of the drugs and adjusted to 7.4. All drugs were from Sigma Chemical, unless otherwise indicated.
Mouse PASMC isolation.
PASMC were isolated from mouse lungs as described previously, using a modification of the Marshall et al. (33) method. A mixture of 5 ml of medium 199 (M199) growth medium containing 5 g/l low-melting-point agarose type VII (Sigma, St. Louis, MO), 5 g/l iron beads (diameter <10 μM; Sigma), and antibiotics (penicillin and streptomycin) was slowly injected over a period of 60 s through the RV, thereby perfusing the PA. M199 growth medium (1 ml) containing 5 g/l agarose type VII was injected in airways through the trachea. The lungs were plunged in cold PBS to cause the agarose to gel. Because of the rapidly solidifying nature of the agarose and the size of the iron particles, the likelihood of traversing the capillary space is minimized. All the lobes were then isolated and finely minced in a petri dish. The tissue was further disrupted by passing through a 16-gauge followed by an 18-gauge needle approximately five times. The suspension was then mixed in M199 growth medium containing 80 U/ml type IV collagenase (Sigma) and incubated at 37°C for 90 min. With the use of a magnetic column (Invitrogen), the arteries containing the iron beads were collected on the sides. The supernatant was aspirated, and, following three washes, the arteries were suspended in 5 ml M199 containing 20% FBS. Aliquots of the suspension were transferred to T25 culture flasks. Cells from the hypoxic group were incubated at 3% O2, whereas cells from the normoxic control were cultured in air. Smooth muscle cell purity was determined by immunostaining with smooth muscle specific actin antibody.
[Ca2+]cyt measurement.
Cells were cultured on 25-mm cover slips (Fisher Scientific, Waltham, MA) and were then placed in a recording chamber on the stage of an invert fluorescent microscope (Eclipse Ti-E; Nikon, Tokyo, Japan) equipped with an objective lens (S Plan Fluor 20×/0.45 ELWD; Nikon) and an EM-CCD camera (Evolve; Photometrics, Tucson, AZ). [Ca2+]cyt was monitored using a membrane-permeable Ca2+-sensitive fluorescent indicator, fura 2-acetoxymethyl ester (fura 2-AM; Invitrogen-Molecular Probes, Eugene, OR), and imaged with NIS Elements 3.2 software (Nikon). These cells were incubated in HEPES-buffered solution containing 4 μM fura 2-AM for 60 min at room temperature (25°C). The loaded cells were then washed with HEPES-buffered solution for 10 min to remove excess extracellular indicator. Fura 2 was excited with 340- and 380-nm wavelengths (D340×v2 and D380×v2 filters, respectively; Chroma Technology, Bellows Falls, VT) by a xenon arc lamp (Lambda LS; Sutter Instrument, Novato, CA) and an optical filter changer (Lambda 10-B). Emission of fura 2 was collected through a dichroic mirror (400DCLP) and a wide band emission filter (D510/80m). [Ca2+]cyt within a region of interest (5 × 5 μm), which was placed at the peripheral region of each cell, was measured as the ratio of fluorescence intensities (F340/F380) every 2 s. The recording chamber was continuously perfused at a flow rate of 2 ml/min using a mini-pump (model 3385; Control, Friendswood, TX). [Ca2+]cyt measurements were carried out at 32°C using an automatic temperature controller (TC-344B; Warner Instruments, Hamden, CT), since increased fura 2 compartmentalization in organelles and cell dye leakage have been reported to occur at physiological temperature (45).
Real-time RT-PCR analysis.
The total RNA from dissected pulmonary arteries of either normoxic control mice or hypoxic mice was purified using Trizol reagent (Invitrogen, Grand Island, NY) and quantified with NanoDrop 2000c (Thermo Scientific, Waltham, MA). Equal amounts of RNA were reverse-transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Grand Island, NY). The quantitative Real-Time PCR analysis was performed with SYBR Green Master Mix (Roche Applied Science, Indianapolis, IN) and gene-specific primers for calcium channel voltage-dependent L-type α1C-subunit (Cav1.2, forward: 5′-TTGAGCAACCTTGTGGCATCCTTG-3′; reverse: 5′-ACGGGTCTGCATCTCATCGAAGTT-3′), Cav2.1 (forward: 5′-TGT GTT TCA GAT CCT GAC TGG CGA-3′; reverse: 5′-AGA ACA CGT TCA GCA GGG TGT AGT-3′), Cav3.1 (forward: 5′-AGA ATG TCA GCT TCT CCG CAG TCA-3′; reverse: 5′-GCT GAA CAC CAA CGA TGC CAA AGA-3′), calcium channel voltage-dependent L-type α1H-subunit (Cav3.2, forward: 5′-TGA GGA TGT TGA GTG CCG TTC AGA-3′; reverse: 5′-CAT GCC GGC CAT GAC AAT GAA GAA-3′), and 18S RNA control (forward: 5′-TTA GAG TGT TCA AAG CAG GCC CGA-3′; reverse: 5′-TCT TGG CAA ATG CTT TCG CTC TGG-3′) using a Bio-Rad CFX384 Real-Time System C1000 Thermal Cycler (Bio-Rad, Hercules, CA). The specificity of amplification of intended targets was evaluated by melting curve analysis. The cycle threshold Ct values were normalized using 18S rRNA as an internal standard and analyzed by the ΔΔCt method for relative quantification using the Bio-Rad CFX Manager Software. The change in ΔΔCt with respect to control was calculated by making normoxic values equal to one and adjusting corresponding hypoxic values proportionally.
Immunohistochemistry.
For immunohistochemistry, sections of isolated PA and MA were incubated with the following primary antibodies: rabbit anti-Cav1.2 (Sigma) and rabbit anti-Cav3.2 (Santa Cruz). Alexa Fluor 488-labeled chicken anti-rabbit IgG (Molecular Probes) was used as secondary antibody.
Western blotting.
Protein samples were prepared by homogenizing mouse lung tissue using a glass Dounce tissue grinder, followed by brief sonication, in 1× RIPA lysis buffer (Millipore) containing 2% n-dodecyl-β-d-maltoside (Thermo Scientific) and protease inhibitor cocktail (Roche). Insoluble tissue was removed by centrifugation (5,000 g, for 5 min). Protein concentrations were determined by the BCA Protein Assay Kit (Pierce). Proteins were loaded equally (30 μg) into separate wells on 4–20% SDS gradient gels (Mini-PROTEAN TGX; Bio-Rad), separated at 120 V for 2 h, and transferred onto polyvinylidene difluoride membranes. Membranes were then stained with Ponceau S and cut in half at 75-kDa standard marker, with the top used for Cav1.2 or Cav3.2 analysis (∼250 kDa) and the bottom for β-actin loading controls (∼44 kDa). Primary antibodies used for analysis included: monoclonal mouse anti-Cav1.2 (Millipore) and anti-Cav3.2 at 1:200 (Alomone); monoclonal β-actin at 1:1,000 (Santa Cruz Biotechnology). Intensity of Western blot bands was determined with ImageJ software.
Statistics.
The composite data are expressed as means ± SE. Statistical analysis was performed using paired or unpaired Student's t-test or ANOVA and post hoc tests (Student-Newman-Keuls) where appropriate. Differences were considered to be significant at P < 0.05.
RESULTS
Chronic hypoxia induces pulmonary hypertension in mice.
To investigate the role of VDCC in the development of HPH, we initially begin by examining the development of HPH in the mouse model. Mice exposed to sustained hypoxia (10% O2) developed pulmonary hypertension within 4 wk as reflected by a significant increase in RVSP: 34.256 ± 1.413 mmHg in hypoxic mice vs. 22.354 ± 1.522 mmHg in normoxic mice (Fig. 1A). Lung tissue sections with H&E staining showed that the wall thickness of the vessels was greater in HPH mice than in the normoxic group (Fig. 1B). Additionally, a significant increase in the Fulton index [RV/(LV + S)] was found in the hypoxic group (0.225 ± 0.0113) compared with that in the normoxic control (0.334 ± 0.0344, Fig. 1C). These results demonstrate that chronic hypoxia induces pulmonary hypertension in mice.
Fig. 1.

Chronic hypoxia induces pulmonary hypertension in mice. A: representative records of right ventricular pressure (RVP, left) and summarized data (mean ± SE) of right ventricular systolic pressure (RVSP, right) in normoxic mice (Nor, n = 6) and mice exposed to hypoxia for 4 wk (Hyp, n = 13). ***P < 0.001 vs. Nor. B: hematoxylin and eosin (H&E) staining of lung sections (left, horizontal bar = 10 μm) and calculated pulmonary arterial (PA) wall thickness (determined by the percentage ratio of vascular wall area to total area; right) in Nor (n = 10) and Hyp (n = 10) mice. **P < 0.01 vs. Nor. C: summarized data (mean ± SE) of the ratio of right ventricle (RV) to left ventricle (LV) plus septum (S) weight [RV/(LV + S)] in Nor (n = 7) and Hyp (n = 7) mice. **P < 0.01 vs. Nor.
High-K+- and U-46619-induced contraction of PA is enhanced in HPH mice.
Pulmonary hypertension is due in part to sustained pulmonary vasoconstriction and vascular remodeling. To investigate the effect of hypoxia on pulmonary vasoconstriction, we measured the contraction of PA from normoxic and hypoxic mice in response to high K+ and U-46619. Mounted PA rings were tested for endothelial function by preconstricting vessels with phenylephrine (PE, 100 nM), followed by a vasodilation challenge with acetylcholine (ACh, 10 μM). Assessment of intact endothelial layer function in mouse PA vessels has been previously reported by our group (25). ACh treatment of endothelium-denuded PA preparations produced a slight relaxation in active tension (0.1768 ± 0.0654 g) that was not significantly different from PE-induced active tension (0.2050 ± 0.0819 g) or washout (0.2295 ± 0.0936 g). These data indicate the endothelial layer was functionally disrupted (Fig. 2).
Fig. 2.

Assessment of endothelium function in PA rings. A: representative tracing showing changes in phenylephrine (PE, 100 nM)-induced contraction before, during, and after application of acetylcholine (ACh, 10 μM) in pulmonary artery (PA) ring preparations. B: summarized data (means ± SE) showing change in absolute tension in the presence or absence of ACh. Application of ACh did not significantly decrease PE-induced contraction in PA rings (n = 4, not significant).
As we have previously shown (57), both high-K+ solution and U-46619, a thromboxane A2 analog, induced vasoconstriction in pulmonary arteries from normoxic mice (Fig. 3, A and B, left). The vasoconstriction observed in response to high-K+ solution (10–120 mM K+) and U-46619 (0.3–300 nM) was concentration dependent (Fig. 3, A-C). However, the vasoconstriction observed in pulmonary arteries of HPH mice in response to high K+ and U-46619 was significantly greater than in normoxic mice (Fig. 3, A-C, right). These data indicate that high-K+- and U-46619-induced vasoconstriction in HPH mice is significantly enhanced compared with normoxic mice.
Fig. 3.

Chronic hypoxia enhances active tension induced by high K+ or U-46619 in pulmonary arteries. A and B: representative tracings showing changes in tension before, during, and after application of various concentrations of extracellular K+ (10–120 mM; A) or U-46619 (0.3–300 nM; B) in isolated PA from normoxic mice (Normoxia) and mice exposed to chronic hypoxia for 4 wk (Hypoxia). Horizontal bars depict 5 min. C: summarized data (means ± SE) showing the dose-response curves of high-K+-induced absolute active tension (left) and U-46619-induced absolute active tension (right) in isolated pulmonary arterial rings from Nor (n = 8, open circles) and Hyp (n = 8, closed squares) mice. **P < 0.01, Nor vs. Hyp.
Ca2+ influx through VDCC is increased in the PA of HPH mice.
Increased [Ca2+]cyt is a major trigger for pulmonary vasoconstriction, and Ca2+ influx through VDCC is an important mechanism for the increased [Ca2+]cyt. To determine the effect of hypoxia on Ca2+ influx through VDCC, we measured [Ca2+]cyt in PASMC from normoxic and hypoxic mice. Raising the extracellular K+ concentration causes membrane depolarization, which subsequently increases open probability of VDCC and promotes Ca2+ influx (51, 57). In primary PASMC isolated from normoxic mice, application of a high-K+ (40 mM) solution had a small effect on the [Ca2+]cyt (Fig. 4, A, left, and C, left). However, in primary PASMC from hypoxic mice, high K+ significantly increased the [Ca2+]cyt (Fig. 4A, right, and C, left). Similarly, U-46619 had minimal effect on the [Ca2+]cyt in normoxic PASMC (Fig. 4, B, left, and C, right), but U-46619 significantly increased the [Ca2+]cyt in primary PASMC isolated from hypoxic mice (Fig. 4, B, right, and C, right). These data suggest that the activity of VDCC is enhanced in hypoxic mice, leading to significantly increased [Ca2+]cyt.
Fig. 4.

Chronic hypoxia enhances 40 mM K+ (40K)-induced Ca2+ influx in mouse pulmonary arterial smooth muscle cells (PASMC). A and B: representative records showing changes in cytosolic Ca2+ concentration ([Ca2+]cyt) before, during, and after application of 40K (A) or 100 nM U-46619 (B) in freshly dissociated PASMC from mice exposed to normoxia or hypoxia for 4 wk. C: summarized data (means ± SE) showing the amplitudes of 40K-mediated increase in [Ca2+]cyt (left) and U-46619-mediated increase in [Ca2+]cyt (right) in PASMC from Nor (n = 14 for 40K and n = 37 for U-46619) and Hyp (n = 23 for 40K and n = 69 for U-46619) mice. **P < 0.01 vs. Nor.
Activation of VDCC in HPH mice enhances pulmonary vasoconstriction.
Increased [Ca2+]cyt in PASMC can lead to contraction. Given that [Ca2+]cyt and VDCC activity is enhanced in PASMC from hypoxic mice, we investigated the effect of activation of VDCC on PA contraction. Bay K8864 selectively activates VDCC and induces vascular contraction. Using isometric force measurements, we show that treatment of normoxic PA rings with increasing concentrations of Bay K8864 results in vasoconstriction (Fig. 5, A, left, and B). Treatment of hypoxic PA rings with Bay K8864 results in a significantly stronger contraction than that of normoxic PA rings (Fig. 5, A, right, and B). These data further demonstrate the enhanced activity of VDCC can contribute to HPH in mice.
Fig. 5.

Chronic hypoxia enhances Bay K8644-mediated pulmonary vasoconstriction. A: representative tracings showing isometric tension before, during, and after application of the voltage-dependent Ca2+ channels (VDCC) agonist Bay K8644 (0.1–10 μM) in isolated pulmonary arterial rings from mice exposed to normoxia and hypoxia (for 4 wk). B: summarized data (means ± SE) showing the dose-response curves of Bay K8644-induced active tension (depicted as increase in absolute active tension, left, and tension increase normalized to the maximal tension) in PA rings isolated from Nor (open circles, n = 4) and Hyp (solid squares, n = 8) mice. **P < 0.01, vs. Nor.
L-type and T-type VDCC blockers significantly attenuate contraction of the PA in HPH mice.
To determine which VDCC are involved in the enhanced contraction of the PA in HPH mice, we examined the high-K+- and U-46619-induced vasoconstriction of PA rings after treatment with VDCC blockers. Nifedipine, mibefradil, and ω-agatoxin are L-type, T-type, and P/Q-type VDCC blockers, respectively. In PA rings from normoxic mice, both nifedepine and mibefradil caused dose-dependent inhibition of high-K+ (40K)-induced PA contraction (Fig. 6, Aa, left, and Ab). Nifedepine and mibefradil inhibited the high-K+-induced contraction of PA rings from hypoxic mice to a significantly greater degree than in normoxic mice (Fig. 6, Aa, right, and Ab). Interestingly, when normalized to the initial contraction, a decrease in sensitivity to nifedipine but not miberadil is observed in hypoxic PA preparations (Fig. 6, Ac and Bc). In contrast, ω-agatoxin did not inhibit the high-K+-induced contraction in PA rings from normoxic or hypoxic mice (Fig. 6, Ca–c).
Fig. 6.

L- and T-type VDCC blockers inhibit high-K+-induced active tension in PA rings from hypoxic mice to a greater extent than in PA rings from normoxic mice. A–C: representative tracings (a) and summarized data expressed as absolute decrease (b) and percent decrease (c) to 40K-mediated active tension in the presence of various concentrations of nifedipine (0.03–3 μM; A), mibefradil (0.03–3 μM; B), and ω-agatoxin (0.1–30 nM; C) in PA rings isolated from Nor (left, and open circles, n = 4) and Hyp (right, and solid circles, n = 4) mice. *P < 0.05 and **P < 0.01 vs. Nor. The inhibitory effects of nifedipine, a L-type VDCC blocker, and mibefradil, a T-type VDCC blocker, on 40K-induced vasoconstriction is greater in PA rings from hypoxic mice than in PA rings from normoxic mice.
We also investigated the effect of VDCC blockers on U-46619-induced vasoconstriction. Similarly to the results from high-K+-induced contraction, nifedipine and mibefradil achieved greater inhibition of U-46619-induced vasoconstriction in PA rings from hypoxic mice than in those isolated from normoxic mice (Fig. 7, A and B). Again, the percent decrease in U-46619-induced contractions revealed that hypoxic PA rings were less sensitive to nifedipine while sensitivity to mibefradil remained the same compared with normoxia (Fig. 7, Ac and Bc). The U-46619-induced contraction was not inhibited by ω-agatoxin in either normoxic or hypoxic PA rings (Fig. 7C). Together, these results suggest that nifedipine-sensitive L-type VDCC and mibefradil-sensitive T-type VDCC contribute to the enhanced pulmonary vasoconstriction in HPH mice.
Fig. 7.
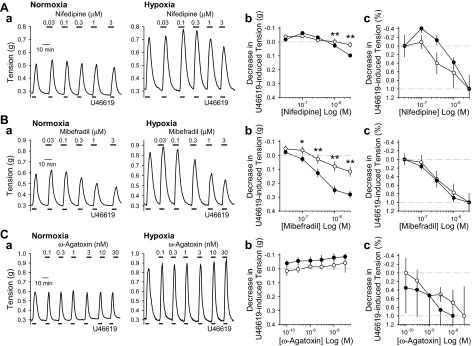
L- and T-type VDCC blockers inhibit U-46619-induced active tension in PA rings from hypoxic mice to a greater extent than in PA rings from normoxic mice. A–C: representative tracings (a) and summarized data expressed as absolute decrease (b) and percentage decrease (c) to 100 nM U-46619-mediated active tension in the presence of various concentrations of nifedipine (0.03–3 μM; A), mibefradil (0.03–3 μM; B), or ω-agatoxin (0.1–30 nM; C) in PA rings isolated from Nor (left, and open circles, n = 4) and Hyp (right, and solid circles, n = 4) mice. *P < 0.05 and **P < 0.01 vs. Nor. The inhibitory effects of high-dose nifedipine, an L-type VDCC blocker, on U-46619-induced vasoconstriction is greater in PA rings from hypoxic mice than in PA rings from normoxic mice.
Chronic hypoxia does not enhance high-K+- or U-46619-induced vasoconstriction or VDCC activity in MA.
It is known that hypoxia causes constriction of the pulmonary, but not systemic, arteries (15), and chronic hypoxia induces pulmonary hypertension but not systemic hypertension (28). We therefore investigated the high-K+- and U-46619-induced vasoconstriction of MA isolated from normoxic and hypoxic mice. Treatment with high K+ (e.g., 40 mM K+) induced contraction of MA rings with no significant difference between normoxic and hypoxic mice (Fig. 8, A and B). Additionally, U-46619 dose-dependently induced similar vasoconstriction in MA from normoxic and hypoxic mice (Fig. 8, A and C). These results demonstrate that hypoxia does not enhance vasoconstriction in MA (Fig. 8), and hypoxia selectively enhances vasoconstriction in PA (Figs. 3 and 5–7).
Fig. 8.

Chronic hypoxia does not affect 40K- and U46619-induced contraction of mesenteric arteries (MA) in mice. A: representative tracings showing isometric tension before, during, and after application of 40K and various concentrations (1–100 nM) of U-46619 (a thromboxane A2 analog) in isolated MA rings from mice exposed to normoxia (top) and hypoxia (bottom). B: summarized data (means ± SE) showing the amplitudes of 40K-induced active tension in MA rings isolated from Nor (n = 4) and Hyp (n = 4) mice. C: summarized data showing the dose-response curves of U-46619-induced active tension (depicted as increase in absolute active tension, left, and tension increase normalized to the maximal tension) in PA rings isolated from Nor (open circles, n = 4) and Hyp (solid squares, n = 4) mice. D: representative tracings showing isometric tension before, during, and after application of various concentrations (0.1–10 μM) of Bay K8644 (a VDCC activator) in MA rings isolated from Nor (left) and Hyp (right) mice. E: summarized data showing the dose-response curves of Bay K8644-induced changes in active tension (depicted as changes in absolute active tension, left, and tension change normalized to the maximal tension) in MA rings isolated from Nor (open circles, n = 4) and Hyp (solid squares, n = 4) mice.
We also investigated the activity of VDCC in MA from normoxic and hypoxic mice. Treatment with Bay K8644 (0.1–10 μM) did not induce contraction of MA rings isolated either from normoxic or hypoxic mice (Fig. 8, D and E). These data indicate that VDCC activity is not enhanced in the MA of hypoxic mice. Together, these results show that pulmonary vasoconstriction due to Ca2+ influx through nifedipine- and mibefradil-sensitive VDCC is enhanced in PA but not MA of HPH mice.
Chronic hypoxia upregulates expression of Cav1.2 and Cav3.2 in the PA.
To determine whether hypoxia altered the expression of VDCC, we examined the mRNA expression of L-type (Cav1.2), T-type (Cav3.1 and Cav3.2), and P/Q-type (Cav2.1) VDCC by real-time RT-PCR. In the PA, exposure to chronic hypoxia resulted in increased expression of Cav1.2, Cav3.1, and Cav3.2 compared with normoxic controls (Fig. 9Aa). In the MA, chronic hypoxia resulted in increased expression of Cav3.1 (Fig. 9Ab). The increase in expression of Cav3.1 after hypoxia is similar in PA and MA (Fig. 9Ac). However, the increase in expression of Cav1.2 and Cav3.2 after hypoxia is only seen in the PA (Fig. 9Ac). In addition to the upregulated mRNA expression, immunohistochemistry showed increased expression of Cav1.2 and Cav3.2 in PA from hypoxic mice compared with MA in which there was no significant change due to hypoxia (Fig. 9B). Western blot analysis of mouse lungs from normoxic and hypoxic animals indicates that hypoxia upregulates expression of Cav1.2 by ∼10-fold, whereas protein expression of Cav3.2 increases only slightly, by ∼40% with hypoxia (Fig. 9C). These results demonstrate that hypoxia selectively upregulates protein expression of L-type (Cav1.2) and T-type (Cav3.2) VDCC in the mouse PA and that hypoxia upregulates protein expression of another subtype of T-type VDCC, Cav3.1, in both PA and MA.
Fig. 9.

Chronic hypoxia upregulates mRNA and protein expression of calcium channel voltage-dependent L-type α1C-subunit (Cav1.2) and T-type α1H-subunit (Cav3.2) in PA but not in MA from mice. A: relative mRNA expression levels of Cav1.2, Cav2.1, Cav3.1, and Cav3.2 in PA (a) and MA (b) isolated from Nor (open bars, n = 6) and Hyp (solid bars, n = 5) mice. **P < 0.01 vs. Nor. c, Comparison of hypoxia-mediated changes in mRNA expression of Cav1.2, Cav2.1, Cav3.1, and Cav3.2 in MA (open bars) and PA (solid bars). **P < 0.01 vs. MA. B: a, immunohistochemical images (×60) showing Cav1.2 and Cav3.2 in PA (left) and MA (right) isolated from Nor (top) and Hyp (bottom) mice. b, Summarized data (means ± SE) showing mRNA expression level of Cav1.2 and Cav3.2 in PA and MA isolated from Nor (open bars; n = 8) and Hyp (solid bars; n = 8) mice. *P < 0.05 and **P < 0.01 vs. Nor. C: a, representative Western blot image of protein lysates from normoxic and hypoxic mouse lung tissues probed for Cav1.2 (top) and Cav3.2 (bottom). Normoxic and hypoxic samples were loaded on the same membrane. Brain tissues where probed as a positive control. b, Summarized data (means ± SE) showing protein expression level normalized to β-actin for Cav1.2 and Cav3.2 in normoxic and hypoxic mouse lung homogenates (n = 3, *P < 0.05 vs. Nor).
DISCUSSION
In the present study, our findings demonstrate that HPH mice have 1) increased high-K+- and U-46619-induced contraction of the PA, 2) increased high-K+- and U-46619-induced Ca2+ influx in primary PASMC, and 3) enhanced pulmonary vasoconstriction due to Ca2+ influx through VDCC compared with normoxic mice. In addition, the data from this study show that 1) L-type and T-type, but not P/Q-type, channels contribute to the enhanced high-K+- and U-46619-induced pulmonary vasoconstriction and 2) Cav1.2 and Cav3.2 are significantly upregulated in PA (but not in MA) from hypoxic mice compared with normoxic mice. We also show that the hypoxia-mediated changes are specific to the PA by demonstrating that there is no difference in high-K+- and U-46619-induced vasoconstriction or VDCC function and expression in MA from normoxic and hypoxic mice. Together, these data suggest that chronic hypoxia functionally enhances pulmonary vasoconstriction associated with Ca2+ influx through L-type and T-type VDCC by selectively upregulating Cav1.2 and Cav3.2 channels in PASMC. To determine if chronic hypoxia affected VDCC in the systemic circulation, we challenged the MA with high K+ and U-46619 but did not find significant difference between normoxic and hypoxic mice, demonstrating the effects of hypoxia on VDCC are specific to the pulmonary vasculature and not the systemic (or mesenteric) vasculature.
HPV is an important mechanism for maintaining a proper ventilation-to-perfusion ratio by diverting blood away from poorly ventilated areas to areas of the lung with relatively high ventilation. Persistent hypoxia or exposure to chronic hypoxia, however, causes sustained HPV and pulmonary arterial medial hypertrophy leading to increased pulmonary vascular resistance and pulmonary arterial pressure. Studies have demonstrated that acute hypoxia causes pulmonary vasoconstriction via changes in membrane potential (Em) and [Ca2+]cyt in PASMC (19, 31, 35). The resting Em is primarily determined by K+ permeability and the K+ concentration gradient across the plasma membrane (14). Inhibition of K+ channels causes membrane depolarization, increases open probability of VDCC, promotes Ca2+ influx, increases [Ca2+]cyt, and triggers PASMC contraction. Our group and others have demonstrated that chronic hypoxia results in decreased expression and function of Kv channels (11, 39, 41, 42, 53, 54, 61, 62, 65). In the present study, we show that, in addition to inhibiting Kv channels, chronic hypoxia selectively enhances VDCC function and expression in PA and PASMC (but not in MA). The hypoxic-mediated downregulation of Kv channels (leading to membrane depolarization and VDCC activation) and upregulation of L- and T-type VDCC (leading to increased availability of VDCC for membrane depolarization-mediated Ca2+ influx through VDCC) both contribute to enhanced pulmonary vasoconstriction and potentially to the development of HPH.
Previous studies have demonstrated enhanced high-K+-induced contraction after exposure to chronic hypoxia (9, 23). Hirenallur et al. (23) demonstrated that enhanced high-K+-induced pulmonary vasoconstriction is associated with upregulation of Cav1.2 (CACNA1C) channels and increase in whole cell voltage-dependent Ca2+ currents in PASMC from hypoxic newborn piglets, suggesting abnormal Ca2+-dependent tone is an important mechanism in the pathogenesis of neonatal pulmonary arterial hypertension (PAH). In our study, we have demonstrated that hypoxia leads to an increase in [Ca2+]cyt in mouse PASMC potentially because of enhanced VDCC activity. However, it should be mentioned that Ca2+ measurements were taken at subphysiological temperature. Although fura 2 is a widely used fluorescent probe for examining dynamic changes of intracellular Ca2+ in live cells, there are several limitations, namely organelle compartmentalization and dye leakage from loaded cells that prevent accurate Ca2+ measurements at 37°C (45). One method used for circumventing these potential artifacts is performing experiments at lower temperatures. Whether these data reflect actual Ca2+ responses to hypoxia at physiological temperature is unknown and will require further study.
In addition to enhanced high-K+-induced PA contraction, in the present study, we provide evidence of enhanced agonist-induced contraction in HPH mice. We demonstrate that PA contraction induced by the thromboxane A2 analog U-46619 was increased in HPH mice. Thromboxane A2, an arachidonic acid metabolite, is an endothelium-derived constricting factor that binds specifically to Gq/11 protein-coupled receptors (e.g., TP receptor) and causes increased [Ca2+]cyt and sensitization of the contractile proteins to Ca2+ in PASMC (38, 48, 51, 60). Inhibition of thromboxane synthase by furegrelate sodium attenuates the development of HPH in neonatal piglet (22). Additionally, thromboxane A2 inhibits KV channels (along with other K+ channels), which causes membrane depolarization and opens VDCC, resulting in pulmonary vasoconstriction (10). The thromboxane A2-mediated membrane depolarization is also associated with its activating effect on nonselective cation channels (60). In rat PASMC, U-46619 induces a nonselective cation current that is sensitive to ruthenium red, a blocker of transient potential vanilloid-related channels. The results of our study demonstrate that enhanced VDCC expression and function also contribute to the enhanced agonist-induced contraction in HPH mice.
In PASMC, L- and T-type VDCC are the main pathways for voltage-mediated Ca2+ influx involved in excitation-contraction coupling and proliferation (16, 26, 50). In the present study, we show that L-type (Cav1.2) and T-type (Cav3.2) channels are specifically upregulated in the PA (but not in the MA) of hypoxic mice, suggesting a role for these channels in the pathogenesis of HPH. Inhibition of L- and T-type channels with nifedipine and mibefradil, respectfully, significantly attenuated the enhanced high-K+- and U-46619-induced vasoconstriction to a greater extent in hypoxic mice than in normoxic mice. Paradoxically, hypoxic tissues experience a decrease in sensitivity to nifedipine as indicated by a rightward shift in the dose-response curve, whereas sensitivity to mibefradil remains unchanged compared with normoxic preparations (Figs. 6 and 7). Although the diminished effect of nifedipine on HPH mice was unexpected, it was not surprising, since we have also shown that Cav1.2 expression is significantly increased in hypoxic lung mouse tissue (Fig. 8). Therefore, the diminished effect of nifedipine on L-type VDCC is most likely due to enhanced expression and activity of VDCC in the chronically hypoxic pulmonary vasculature.
Recently, we have shown that the dihydropyridine Ca2+ channel blockers, including nifedipine, can activate the Ca2+-sensing receptor, which we have also shown is upregulated in PASMC isolated from idiopathic PAH patients (59), leading to enhanced extracellular Ca2+-induced Ca2+ influx. Clinical use of nifedipine has also proven to be problematic. Inhibition of L-type channels improves the pulmonary arterial pressure in ∼40% of pediatric patients with PAH (4) and only 10–15% of adults with PAH (43). This may also partially explain the less than robust effectiveness of inhibition of L-type channels seen in our hypoxic preparations, as well as in adult PAH patients.
Unlike the rat model, where HPH is caused almost entirely by sustained vasoconstriction, two mechanisms are thought to contribute equally for generating HPH in the mouse: 1) prolonged vasoconstriction and 2) structural remodeling of the vasculature, leading to reduction of the lumen (7). Inhibition of VDCC with nifedipine and other VDCC blockers significantly attenuates smooth muscle cell proliferation cultured in media containing growth factors and serum (30, 49, 55), which would indicate that Ca2+ influx through L-type and T-type VDCC is required for smooth muscle cell proliferation. Silenced expression of T-type VDCC in human PASMC was also shown to significantly attenuate proliferation (44), providing further evidence that T-type channels are essential in controlling proliferation. Future studies examining contributions from both L- and T-type VDCC to vascular remodeling in HPH mice are necessary.
Although nifedipine and mibefradil are highly recognized pharmacological tools for studying L- and T-type channels, respectively, there is evidence of nonspecific inhibition at higher concentrations in several cell types. Nifedipine has been shown to potently inhibit T-type Ca2+ currents in mouse spermatocytes (3), rat hypothalamic neurons (2), and rat clonal pituitary cells (21) (IC50 ∼ 0.4–50 μM), among others. Typically mibefradil is more selective for blocking T-type Ca2+ currents over L-type Ca2+ currents (∼10–30 more) (20, 34); however, in cultured rat spinal motoneurons, mibefradil was equally effective at blocking L-type Ca2+ currents (52). Additionally, a metabolized form of mibefadil has been reported to preferentially block L-type over T-type Ca2+ currents in insulin-secreting cells (56). To our knowledge, the overlapping pharmacology of these two inhibitors has not been extensively studied in vascular myocytes; however, we must always be cognizant of the limitations these pharmacological tools bring. Therefore, the use of more highly specific inhibitors and knockout mouse studies will be required to elucidate the role of L- and T-type Ca2+ channels in the vasculature, particularly pertaining to their contribution in the development of PAH. The molecular mechanism involved in hypoxia-mediated selective upregulation of Cav1.2 and Cav3.2 in PASMC and PA is unclear. The results from this study demonstrate that chronic hypoxia significantly upregulated the mRNA expression level of Cav1.2 and Cav3.2 only in the PA but not in the MA. Hypoxia also upregulates Cav3.1, a T-type VDCC found in neurons and cardiac tissue; however, the augmenting effect occurred to the same extent in the PA and MA. These data indicate that the augmenting effect of chronic hypoxia on the mRNA expression level of Cav1.2 and Cav3.2 is specific to only PA, but the augmenting effect of hypoxia on the Cav3.2 expression level seems to be a nonspecific phenomenon that occurs in both the PA and MA. The data from immunohistochemistry experiments confirmed that chronic hypoxia selectively upregulated the protein expression of Cav1.2 and Cav3.2 in the PA but not the MA.
In conclusion, our study demonstrates that chronic hypoxia enhances the high-K+- and U-46619-induced contraction in the PA, but not in the MA, because of selective functional upregulation of L-type (Cav1.2) and T-type (Cav3.2) VDCC in the PA. These studies provide insight into the role of Ca2+-dependent tone in the development of HPH.
GRANTS
This work was supported in parts by grants from the National Heart, Lung, and Blood Institute of the National Institutes of Health (HL-115014, HL-066012, and HL-098053), and funds from the Natural Science Foundation of China (30810103904, 81228001, and 81270117).
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: J. Wan, K.A.S., J. Wang, C.W., A.M., and J.X.-J.Y. conception and design of research; J. Wan, A.Y., A.M.Z., G.V., H.T., M.S.C., and E.A.K. performed experiments; J. Wan, A.Y., A.M.Z., G.V., K.A.S., H.T., R.J.A., M.S.C., E.A.K., A.M., and J.X.-J.Y. analyzed data; J. Wan, A.M.Z., G.V., K.A.S., H.T., R.J.A., M.S.C., E.A.K., J. Wang, C.W., A.M., and J.X.-J.Y. interpreted results of experiments; J. Wan, A.Y., A.M.Z., G.V., K.A.S., H.T., R.J.A., E.A.K., A.M., and J.X.-J.Y. prepared figures; J. Wan, K.A.S., and J.X.-J.Y. drafted manuscript; J. Wan, A.Y., A.M.Z., K.A.S., R.J.A., M.S.C., E.A.K., J. Wang, C.W., A.M., and J.X.-J.Y. edited and revised manuscript; J. Wan, A.Y., A.M.Z., G.V., K.A.S., H.T., R.J.A., M.S.C., E.A.K., J. Wang, C.W., A.M., and J.X.-J.Y. approved final version of manuscript.
REFERENCES
- 1. Abud EM, Maylor J, Undem C, Punjabi A, Zaiman AL, Myers AC, Sylvester JT, Semenza GL, Shimoda LA. Digoxin inhibits development of hypoxic pulmonary hypertension in mice. Proc Natl Acad Sci USA 109: 1239–1244, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Akaike N, Kostyuk P, Osipchuk Y. Dihydropyridine-sensitive low-threshold calcium channels in isolated rat hypothalamic neurones. J Physiol 412: 181–195, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arnoult C, Villaz M, Florman H. Pharmacological properties of the T-type Ca2+ current of mouse spermatogenic cells. Mol Pharmacol 53: 1104–1111, 1998 [PubMed] [Google Scholar]
- 4. Barst RJ. Recent advances in the treatment of pediatric pulmonary artery hypertension. Pediatr Clin North Am 46: 331–345, 1999 [DOI] [PubMed] [Google Scholar]
- 5. Bonnet S, Dubuis E, Vandier C, Martin S, Marthan R, Savineau JP. Reversal of chronic hypoxia-induced alterations in pulmonary artery smooth muscle electromechanical coupling upon air breathing. Cardiovasc Res 53: 1019–1028, 2002 [DOI] [PubMed] [Google Scholar]
- 6. Bonnet S, Hyvelin JM, Bonnet P, Marthan R, Savineau JP. Chronic hypoxia-induced spontaneous and rhythmic contractions in the rat main pulmonary artery. Am J Physiol Lung Cell Mol Physiol 281: L183–L192, 2001 [DOI] [PubMed] [Google Scholar]
- 7. Cahill E, Rowan S, Sands M, Banahan M, Ryan D, Howell K, McLoughlin P. The pathophysiological basis of chronic hypoxic pulmonary hypertension in the mouse: vasoconstrictor and structural mechanisms contribute equally. Exp Physiol 97: 796–806, 2012 [DOI] [PubMed] [Google Scholar]
- 8. Chaouat A, Bugnet AS, Kadaoui N, Schott R, Enache I, Ducolone A, Ehrhart M, Kessler R, Weitzenblum E. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 172: 189–194, 2005 [DOI] [PubMed] [Google Scholar]
- 9. Chen WS, Li XQ, Cao W, Xiao X, Dong L, Zhang JZ. Vardenafil ameliorates calcium mobilization in pulmonary artery smooth muscle cells from hypoxic pulmonary hypertensive mice. Arch Med Res 43: 265–273, 2012 [DOI] [PubMed] [Google Scholar]
- 10. Cogolludo A, Moreno L, Bosca L, Tamargo J, Perez-Vizcaino F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction. Circ Res 93: 656–663, 2003 [DOI] [PubMed] [Google Scholar]
- 11. Coppock EA, Martens JR, Tamkun MM. Molecular basis of hypoxia-induced pulmonary vasoconstriction: role of voltage-gated K+ channels. Am J Physiol Lung Cell Mol Physiol 281: L1–L12, 2001 [DOI] [PubMed] [Google Scholar]
- 12. Davidson A, Bossuyt A, Dab I. Acute effects of oxygen, nifedipine, and diltiazem in patients with cystic fibrosis and mild pulmonary hypertension. Pediatr Pulmonol 6: 53–59, 1989 [DOI] [PubMed] [Google Scholar]
- 13. Fike CD, Kaplowitz MR. Nifedipine inhibits pulmonary hypertension but does not prevent decreased lung eNOS in hypoxic newborn pigs. Am J Physiol Lung Cell Mol Physiol 277: L449–L456, 1999 [DOI] [PubMed] [Google Scholar]
- 14. Firth A, Remillard C, Platoshyn O, Fantozzi I, Ko E, Yuan J. Functional ion channels in human pulmonary artery smooth muscle cells: Voltage-dependent cation channels. Pulm Circ 1: 48–71, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fishman AP. Hypoxia on the pulmonary circulation. How and where it acts. Circ Res 38: 221–231, 1976 [DOI] [PubMed] [Google Scholar]
- 16. Fleischmann BK, Murray RK, Kotlikoff MI. Voltage window for sustained elevation of cytosolic calcium in smooth muscle cells. Proc Natl Acad Sci USA 91: 11914–11918, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Galie N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, Beghetti M, Corris P, Gaine S, Gibbs JS, Gomez-Sanchez MA, Jondeau G, Klepetko W, Opitz C, Peacock A, Rubin L, Zellweger M, Simonneau G. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 34: 1219–1263, 2009 [DOI] [PubMed] [Google Scholar]
- 18. Hansen PB, Poulsen CB, Walter S, Marcussen N, Cribbs LL, Skott O, Jensen BL. Functional importance of L- and P/Q-type voltage-gated calcium channels in human renal vasculature. Hypertension 58: 464–470, 2011 [DOI] [PubMed] [Google Scholar]
- 19. Harder DR, Madden JA, Dawson C. Hypoxic induction of Ca2+-dependent action potentials in small pulmonary arteries of the cat. J Appl Physiol 59: 1389–1393, 1985 [DOI] [PubMed] [Google Scholar]
- 20. Heady T, Gomora J, Macdonald T, Perez-Reyes E. Molecular pharmacology of T-type Ca2+ channels. Jpn J Pharmacol 85: 339–350, 2001 [DOI] [PubMed] [Google Scholar]
- 21. Herrington J, Lingle C. Kinetic and pharmacological properties of low voltage-activated Ca2+ current in rat clonal (GH3) pituitary cells. J Neurophysiol 68: 213–232, 1992 [DOI] [PubMed] [Google Scholar]
- 22. Hirenallur SD, Detweiler N, Haworth S, Leming J, Gordon J, Rusch N. Furegrelate, a thromboxane synthase inhibitor, blunts the development of pulmonary arterial hypertension in neonatal piglets. Pulm Circ 2: 193–200, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hirenallur SDK, Haworth ST, Leming JT, Chang J, Hernandez G, Gordon JB, Rusch NJ. Upregulation of vascular calcium channels in neonatal piglets with hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 295: L915–L924, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kennedy TP, Michael JR, Summer W. Calcium channel blockers in hypoxic pulmonary hypertension. Am J Med 78: 18–26, 1985 [DOI] [PubMed] [Google Scholar]
- 25. Ko EA, Wan J, Yamamura A, Zimnicka AM, Yamamura H, Yoo HY, Tang H, Smith KA, Sundivakkam PC, Zeifman A, Ayon RJ, Makino A, Yuan JXJ. Functional characterization of voltage-dependent Ca2+ channels in mouse pulmonary arterial smooth muscle cells: divergent effect of ROS. Am J Physiol Cell Physiol. 10.1152/ajpcell.00304.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kuga T, Kobayashi S, Hirakawa Y, Kanaide H, Takeshita A. Cell cycle-dependent expression of L- and T-type Ca2+ currents in rat aortic smooth muscle cells in primary culture. Circ Res 79: 14–19, 1996 [DOI] [PubMed] [Google Scholar]
- 27. Kuhr FK, Smith KA, Song MY, Levitan I, Yuan JX. New mechanisms of pulmonary arterial hypertension: role of Ca2+ signaling. Am J Physiol Heart Circ Physiol 302: H1546–H1562, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leach RM, Robertson TP, Twort CH, Ward JP. Hypoxic vasoconstriction in rat pulmonary and mesenteric arteries. Am J Physiol Lung Cell Mol Physiol 266: L223–L231, 1994 [DOI] [PubMed] [Google Scholar]
- 29. Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, Sham JS. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res 95: 496–505, 2004 [DOI] [PubMed] [Google Scholar]
- 30. Maci'a M, Lozano R, Villarroya M, Garci'a AG, Ruiz-Torres A. On the antiatherogenity of calcium channel blockers: studies in proliferating vascular smooth muscle cells on age sensitivity, dose dependent inhibitory effect, and time of action. Arch Gerontol Geriatr 35: 51–57, 2002 [DOI] [PubMed] [Google Scholar]
- 31. Madden JA, Dawson CA, Harder DR. Hypoxia-induced activation in small isolated pulmonary arteries from the cat. J Appl Physiol 59: 113–118, 1985 [DOI] [PubMed] [Google Scholar]
- 32. Madden JA, Vadula MS, Kurup VP. Effects of hypoxia and other vasoactive agents on pulmonary and cerebral artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 263: L384–L393, 1992 [DOI] [PubMed] [Google Scholar]
- 33. Marshall C, Mamary AJ, Verhoeven AJ, Marshall BE. Pulmonary artery NADPH-oxidase is activated in hypoxic pulmonary vasoconstriction. Am J Respir Cell Mol Biol 15: 633–644, 1996 [DOI] [PubMed] [Google Scholar]
- 34. Mishra S, Hermsmeyer K. Selective inhibition of T-type Ca2+ channels by Ro 40–5967. Circ Res 75: 144–148, 1994 [DOI] [PubMed] [Google Scholar]
- 35. Murray TR, Chen L, Marshall BE, Macarak EJ. Hypoxic contraction of cultured pulmonary vascular smooth muscle cells. Am J Respir Cell Mol Biol 3: 457–465, 1990 [DOI] [PubMed] [Google Scholar]
- 36. Oswald-Mammosser M, Weitzenblum E, Quoix E, Moser G, Chaouat A, Charpentier C, Kessler R. Prognostic factors in COPD patients receiving long-term oxygen therapy. Importance of pulmonary artery pressure. Chest 107: 1193–1198, 1995 [DOI] [PubMed] [Google Scholar]
- 37. Peinado VI, Pizarro S, Barbera JA. Pulmonary vascular involvement in COPD. Chest 134: 808–814, 2008 [DOI] [PubMed] [Google Scholar]
- 38. Perez-Vizcaino F, Cogolludo AL, Ibarra M, Fajardo S, Tamargo J. Pulmonary artery vasoconstriction but not [Ca2+]i signal stimulated by thromboxane A2 is partially resistant to NO. Pediatr Res 50: 508–514, 2001 [DOI] [PubMed] [Google Scholar]
- 39. Platoshyn O, Brevnova EE, Burg ED, Yu Y, Remillard CV, Yuan JX. Acute hypoxia selectively inhibits KCNA5 channels in pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol 290: C907–C916, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Platoshyn O, Yu Y, Golovina VA, McDaniel SS, Krick S, Li L, Wang JY, Rubin LJ, Yuan JX. Chronic hypoxia decreases KV channel expression and function in pulmonary artery myocytes. Am J Physiol Lung Cell Mol Physiol 280: L801–L812, 2001 [DOI] [PubMed] [Google Scholar]
- 41. Post JM, Gelband CH, Hume JR. [Ca2+]i inhibition of K+ channels in canine pulmonary artery. Novel mechanism for hypoxia-induced membrane depolarization. Circ Res 77: 131–139, 1995 [DOI] [PubMed] [Google Scholar]
- 42. Post JM, Hume JR, Archer SL, Weir EK. Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. Am J Physiol Cell Physiol 262: C882–C890, 1992 [DOI] [PubMed] [Google Scholar]
- 43. Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med 327: 76–81, 1992 [DOI] [PubMed] [Google Scholar]
- 44. Rodman D, Reese K, Harral J, Fouty B, Wu S, West J, Hoedt-Miller M, Tada Y, Li KX, Cool C, Fagan K, Cribbs L. Low-voltage-activated (T-type) calcium channels control proliferation of human pulmonary artery myocytes. Circ Res 96: 864–872, 2005 [DOI] [PubMed] [Google Scholar]
- 45. Roe M, Lemasters J, Herman B. Assessment of Fura-2 for measurements of cytosolic free calcium. Cell Calcium 11: 63–73, 1990 [DOI] [PubMed] [Google Scholar]
- 46. Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, Grimminger F. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest 115: 2811–2821, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shimoda LA, Sham JS, Shimoda TH, Sylvester JT. L-type Ca2+ channels, resting [Ca2+]i, and ET-1-induced responses in chronically hypoxic pulmonary myocytes. Am J Physiol Lung Cell Mol Physiol 279: L884–L894, 2000 [DOI] [PubMed] [Google Scholar]
- 48. Somlyo AP, Somlyo AV. Signal transduction by G-proteins, Rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J Physiol 522: 177–185, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sung JY, Choi HC. Nifedipine inhibits vascular smooth muscle cell proliferation and reactive oxygen species production through AMP-activated protein kinase signaling pathway. Vasc Pharmacol 56: 1–8, 2012 [DOI] [PubMed] [Google Scholar]
- 50. Thakali KM, Kharade SV, Sonkusare SK, Rhee SW, Stimers JR, Rusch NJ. Intracellular Ca2+ silences L-type Ca2+ channels in mesenteric veins: mechanism of venous smooth muscle resistance to calcium channel blockers. Circ Res 106: 739–747, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tosun M, Paul RJ, Rapoport RM. Role of extracellular Ca2+ influx via L-type and non-L-type Ca2+ channels in thromboxane A2 receptor-mediated contraction in rat aorta. J Pharmacol Exp Ther 284: 921–928, 1998 [PubMed] [Google Scholar]
- 52. Viana F, Van den Bosch L, Missiaen L, Vandenberghe W, Droogmans G, Nilius B, Robberecht W. Mibefradil (Ro 40-5967) blocks multiple types of voltage-gated calcium channels in cultured rat spinal motoneurones. Cell Calcium 22: 299–311, 1997 [DOI] [PubMed] [Google Scholar]
- 53. Wang J, Juhaszova M, Rubin LJ, Yuan XJ. Hypoxia inhibits gene expression of voltage-gated K+ channel alpha subunits in pulmonary artery smooth muscle cells. J Clin Invest 100: 2347–2353, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang J, Weigand L, Wang W, Sylvester JT, Shimoda LA. Chronic hypoxia inhibits Kv channel gene expression in rat distal pulmonary artery. Am J Physiol Lung Cell Mol Physiol 288: L1049–L1058, 2005 [DOI] [PubMed] [Google Scholar]
- 55. Wu JR, Liou SF, Lin SW, Chai CY, Dai ZK, Liang JC, Chen IJ, Yeh JL. Lercanidipine inhibits vascular smooth muscle cell proliferation and neointimal formation via reducing intracellular reactive oxygen species and inactivating Ras-ERK1/2 signaling. Pharmacol Res 59: 48–56, 2009 [DOI] [PubMed] [Google Scholar]
- 56. Wu S, Zhang M, Vest P, Bhattacharjee A, Liu L, Li M. A mibefradil metabolite is a potent intracellular blocker of L-type Ca2+ currents in pancreatic beta-cells. J Pharmacol Exp Ther 292: 939–943, 2000 [PubMed] [Google Scholar]
- 57. Xu M, Platoshyn O, Makino A, Dillmann WH, Akassoglou K, Remillard CV, Yuan JX. Characterization of agonist-induced vasoconstriction in mouse pulmonary artery. Am J Physiol Heart Circ Physiol 294: H220–H228, 2008 [DOI] [PubMed] [Google Scholar]
- 58. Yamamoto Y, Koike K. α1-Adrenoceptor subtypes in the mouse mesenteric artery and abdominal aorta. Brit J Pharmacol 134: 1045–1054, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yamamura A, Guo Q, Yamamura H, Zimnicka AM, Pohl NM, Smith KA, Fernandez RA, Zeifman A, Makino A, Dong H, Yuan JX. Enhanced Ca2+-sensing receptor function in idiopathic pulmonary arterial hypertension. Circ Res 111: 469–481, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yoo H, Park S, Seo EY, Park K, Han JA, Kim K, Shin D, Earm Y, Zhang YH, Kim S. Role of thromboxane A2-activated nonselective cation channels in hypoxic pulmonary vasoconstriction of rat. Am J Physiol Cell Physiol 302: C307–C317, 2012 [DOI] [PubMed] [Google Scholar]
- 61. Yuan JX, Aldinger AM, Juhaszova M, Wang J, Conte JV, Jr, Gaine SP, Orens JB, Rubin LJ. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation 98: 1400–1406, 1998 [DOI] [PubMed] [Google Scholar]
- 62. Yuan XJ. Voltage-gated K+ currents regulate resting membrane potential and [Ca2+]i in pulmonary arterial myocytes. Circ Res 77: 370–378, 1995 [DOI] [PubMed] [Google Scholar]
- 63. Yuan XJ, Goldman WF, Tod ML, Rubin LJ, Blaustein MP. Hypoxia reduces potassium currents in cultured rat pulmonary but not mesenteric arterial myocytes. Am J Physiol Lung Cell Mol Physiol 264: L116–L123, 1993 [DOI] [PubMed] [Google Scholar]
- 64. Yuan XJ, Tod ML, Rubin LJ, Blaustein MP. Contrasting effects of hypoxia on tension in rat pulmonary and mesenteric arteries. Am J Physiol Heart Circ Physiol 259: H281–H289, 1990 [DOI] [PubMed] [Google Scholar]
- 65. Yuan XJ, Wang J, Juhaszova M, Gaine SP, Rubin LJ. Attenuated K+ channel gene transcription in primary pulmonary hypertension. Lancet 351: 726–727, 1998 [DOI] [PubMed] [Google Scholar]