

Abstract
It is becoming increasingly apparent that cAMP signals within the pulmonary endothelium are highly compartmentalized, and this compartmentalization is critical to maintaining endothelial barrier integrity. Studies demonstrate that the exogenous soluble bacterial toxin, ExoY, and heterologous expression of the forskolin-stimulated soluble mammalian adenylyl cyclase (AC) chimera, sACI/II, elevate cytosolic cAMP and disrupt the pulmonary microvascular endothelial barrier. The barrier-disruptive effects of cytosolic cAMP generated by exogenous soluble ACs are in contrast to the barrier-protective effects of subplasma membrane cAMP generated by transmembrane AC, which strengthens endothelial barrier integrity. Endogenous soluble AC isoform 10 (AC10 or commonly known as sAC) lacks transmembrane domains and localizes within the cytosolic compartment. AC10 is uniquely activated by bicarbonate to generate cytosolic cAMP, yet its role in regulation of endothelial barrier integrity has not been addressed. Here we demonstrate that, within the pulmonary circulation, AC10 is expressed in pulmonary microvascular endothelial cells (PMVECs) and pulmonary artery endothelial cells (PAECs), yet expression in PAECs is lower. Furthermore, pulmonary endothelial cells selectively express bicarbonate cotransporters. While extracellular bicarbonate generates a phosphodiesterase 4-sensitive cAMP pool in PMVECs, no such cAMP response is detected in PAECs. Finally, addition of extracellular bicarbonate decreases resistance across the PMVEC monolayer and increases the filtration coefficient in the isolated perfused lung above osmolality controls. Collectively, these findings suggest that PMVECs have a bicarbonate-sensitive cytosolic cAMP pool that disrupts endothelial barrier integrity. These studies could provide an alternative mechanism for the controversial effects of bicarbonate correction of acidosis of acute respiratory distress syndrome patients.
Keywords: soluble adenylyl cyclase, phosphodiesterase, edema, lung, compartmentalization
the pulmonary endothelial barrier regulates the flux of blood components from the vascular space into the underlying tissue and is critical to maintain efficient gas exchange in the lung (4, 16, 35, 36). Compartmentalized cAMP signals, localized to either the subplasma membrane compartment or the cytosolic compartment, modulate the paracellular permeability across the pulmonary endothelial barrier (45). It is well established that transmembrane adenylyl cyclases (ACs) generate cAMP within the subplasma membrane space, which strengthens the barrier through activation of plasma membrane targets, such as the actin-binding protein filamin (18, 29, 45, 47, 52). This subplasma membrane cAMP pool is confined to its compartment by diffusion barricades, such as phosphodiesterase (PDE) activity, which hydrolyzes cAMP before it escapes into the cytosolic compartment. Disruption of the PDE diffusion barricade permits cAMP to escape its compartment, activate cytosolic targets, such as the microtubule-associating protein endothelial tau, and disrupt the endothelial barrier (13). Indeed, emerging evidence reveals dual roles for cAMP in regulation of lung vascular permeability depending on whether the cAMP signal originates within the plasma membrane or cytosolic compartments (20, 44). While cAMP generated within the subplasma membrane compartment confers barrier protection, cAMP that gains access to or is generated within the cytosolic compartment confers barrier disruption.
The common lung pathogen Pseudomonas aeruginosa utilizes the syringe-like needle of the type three secretion system (TTSS) to deliver toxins into eukaryotic cells (62). One such toxin is the soluble AC, ExoY, which bypasses the subplasma membrane compartment and is inserted directly into the cytosolic compartment of pulmonary endothelial cells where it generates cAMP. ExoY induces endothelial cell gaps and increases the filtration coefficient (Kf) in the isolated lung (48). Furthermore, codon-optimized, conditionally expressed ExoY induces interendothelial gaps between pulmonary microvascular endothelial cells (PMVECs), demonstrating the barrier-disruptive effects of ExoY are independent of the bacteria (38). The role of compartmentalized cAMP signaling was further demonstrated using the exogenous AC chimera soluble mammalian adenylyl cyclase (sACI/II) (44, 46). This chimera lacks transmembrane domains and is exclusively expressed within the cytosolic compartment, yet retains forskolin-stimulated activity. Forskolin stimulation of PMVECs expressing sACI/II generates only a modest rise in cAMP within the cytosolic compartment, yet this small rise in cytosolic cAMP is sufficient to promote gaps between adjacent endothelial cells. Similar to the ExoY-cAMP pool, the sACI/II-cAMP pool is not regulated by PDE activity. Thus, collectively, these studies promote the idea that soluble ACs generate an endothelial barrier-disruptive cytosolic cAMP pool.
Recently, a novel mammalian soluble AC, AC isoform 10 (AC10) or commonly known as sAC, has been identified, which is an ortholog to cyanobacterial ACs (7, 11). Unlike the transmembrane isoforms AC1–9, AC10 lacks transmembrane domains and localizes within the cytosol as well as in association with microtubules, the centrosome, or within organelles such as the mitochondria and nucleus (10, 19, 22, 63). AC10 is molecularly distinct from isoforms AC1–9 and is insensitive to traditionally acknowledged activators of transmembrane ACs, Gsα and forskolin, but is uniquely stimulated by bicarbonate (but not CO2) in a pH-independent manner (7, 11, 25). Several kinetic studies also suggest AC10 is activated by calcium (24, 33). Thus, AC10 is poised to mediate intracellular cAMP functions and further substantiates a role for compartmentalized cAMP signaling. While expression of AC10 was initially thought to be restricted to sperm, its distribution in somatic tissues has now been clearly demonstrated (50, 63). Indeed, AC10 has been implicated in microtubule-mediated sperm motility (61) and ciliary beat frequency in airway epithelia (49), as well as pH homoeostasis in the kidney (41) and epididymis (42), regulation of oxidative phosphorylation (1), and apoptosis (6, 31, 59). Thus, AC10 is a bicarbonate sensor that localizes to the cytosolic compartment of the cell to generate cAMP.
The expression or function of AC10 in the pulmonary endothelium has not yet been described. In the present study, we identify AC10 in pulmonary conduit and capillary endothelium and examine the change in intracellular cAMP levels in response to increasing extracellular bicarbonate. Finally, we demonstrate bicarbonate regulation of pulmonary endothelial barrier integrity.
MATERIALS AND METHODS
Isolation and culture of rat PMVECs and pulmonary artery endothelial cells.
Isolation of rat PMVECs and pulmonary artery endothelial cells (PAECs) has been described in detail elsewhere (54) under the approval of the Animal Care and Use Committee of the University of South Alabama. Cell cultures where used from passage 8 through passage 15 and were maintained in EC media (high-glucose DMEM, 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin) at 5% CO2.
Isolation of RNA and RT-PCR.
Total RNA was extracted from confluent PMVECs and PAECs or freshly isolated lung or kidney tissue using TRIzol reagent (Life Technologies) following the manufacturer's protocol. Samples were stored at −20°C. RT-PCR was performed with an Eppendorf thermal cycler (Eppendorf, Hamburg, Germany) using a OneStep RT-PCR kit (1 μg template, 600 nM primers, 2 μl enzyme mix in a final volume of 50 μl and including Q solution; Qiagen) following the manufacturer's instructions. The reverse transcription step proceeded for 30 min at 50°C, PCR activation for 15 min at 95°C, followed by 40 denaturation, annealing, and extension cycles (94°C for 30 s, 50°C for 30 s, and 72°C for 1 min, respectively). A final extension was performed at 72°C for 10 min. PCR products were separated on a 1% (wt/vol) agarose gel and stained with ethidium bromide for visualization under ultraviolet light to confirm product size. Primers against rat sequences were as follows: AC10 (NM_021684) (17); primers for NCBe1 (NM_053424), sodium-bicarbonate cotransporter (NBC) e2 (NM_212512), NBCn1 (NM_178092), NCBE (NBCn2, NM_178092), sodium-driven chloride-bicarbonate cotransporters (NDCBE, NM_199497), and β-actin (NM_031144.2) have been described previously (56).
Isolation of total cellular proteins and Western analysis.
Confluent PMVECs or PAECs were rinsed in ice-cold PBS and then lysed on ice in ice-cold radioimmunoprecipitation assay buffer (10 mM sodium phosphate, pH 7.2, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS; Boston Bioproducts) with protease and phosphatase inhibitors (Halt; Thermo Scientific). For Western analysis of AC10, cells were lysed on ice in ice-cold extraction buffer (5 mM magnesium chloride, 1 mM EDTA, 40 mM Tris, pH 7.4, 130 mM sodium chloride, and 50 mM octylglucopyranoside) with protease and phosphatase inhibitors (Halt; Thermo Scientific). Following lysis, all samples were rotated at 4°C for 15 min and then centrifuged at 13,000 rpm, 4°C for 10 min (Micro-17R). The supernatant was collected and stored at −80°C for protein concentration assay. Samples were adjusted to equal protein concentration, Sample Buffer (62.4 mM Tris·HCl, pH 6.4, 10% glycerol, 1.8% β-mercaptoethanol, and 70 mM SDS) was added, and then samples were heated at 37°C for 30 min. Proteins were resolved by SDS-polyacrylamide gel electrophoresis (Nu-Page; Invitrogen) and transferred to nitrocellulose membranes (Bio-Rad). Nitrocellulose membranes were incubated in blocking buffer [Tris-buffered saline (TBS, 137 mM NaCl, 25 mM Tris, pH 7.4) supplemented with 0.5% Tween 20 (TTBS) and with 5% nonfat dried milk]. Following a TBS rinse, membranes were rocked overnight at 4°C with anti-AC10 antibody [R21 antibody was a gift from L. Levin and J. Buck (63), 1:500], anti-NBCe1A/B antibody [gift M. Bevensee (3), 1:1,000], anti-NBCn1 antibody [gift J. Praetorius, Aarhus, Denmark (14)], or anti-β-actin antibody (1:3,000; Santa Cruz) in TTBS containing 1% nonfat dried milk. The membranes were washed (3 × 10 min) in TTBS with rocking and then incubated with secondary antibody (IgG conjugated to horseradish peroxidase) in TTBS containing 1% nonfat dried milk for 1 h. Finally, membranes were rinsed (3 × 10 min) in TTBS, treated with either ECL reagent (Amersham Biosciences) or Femto (Pierce), and exposed to film.
Bicarbonate treatment of cells.
PMVECs or PAECs were grown to confluence in six-well dishes under normal culturing conditions (see isolation and culturing above). Cells were rinsed in PBS (37°C) and placed in HEPES (20 mM)-buffered low-bicarbonate (5 mM) DMEM (GIBCO) for 20 min at 0% CO2 and 37°C to maintain physiological pH. Bicarbonate concentration was adjusted in the presence or absence of rolipram (10 μM) with or without ionomycin (1 μM) for an additional 15 min at 37°C, and the CO2 tensions were adjusted to maintain physiological pH. For osmolality controls, 5 mM bicarbonate DMEM was adjusted to the same osmolality as 40 mM bicarbonate DMEM using either sodium chloride or sucrose. For sodium-free media (118 mM choline chloride, 1 mM MgCl2, 1.3 mM calcium gluconate, 5 mM glucose, 0.4 mM K2PO4, 1.6 K2HPO4, and 10 mM HEPES), choline chloride was used to replace sodium chloride, and choline bicarbonate was used to increase bicarbonate concentration of the media; sodium was added back to this media (118 mM NaCl and sodium bicarbonate) to demonstrate the feasibility of the buffer. An aliquot of the media was removed for determination of CO2, pH, and bicarbonate using the ABL 5 blood gas analyzer (Radiometer, Copenhagen, Denmark). After the media was removed, the cells were rinsed in PBS and lysed in 1 M HCl. The pH was neutralized by addition of 1 M NaOH, and 3-isobutyl-1-methylxanthine was added (100 μM). Samples were stored at −80°C before cAMP radioimmunoassay (RIA) and protein concentration.
Determination of whole cell cAMP.
Whole cell cAMP concentrations were determined using standard RIA following the manufacturer's instructions (Biomedical Technologies, Stoughton, MA). cAMP levels were normalized to protein content using the Micro Lowry with Peterson's modification method (Sigma, St. Louis, MO).
Calcium measurements.
Endothelial cells were seeded on 25-mm glass cover slips and grown to confluence. Intracellular calcium concentration was estimated with the fluorophore fura 2-acetoxymethyl ester (fura 2-AM; Molecular Probes) according to methods described previously (27, 55).
Transendothelial electrical resistance.
Endothelial cells were seeded on polycarbonate wells containing small-evaporated gold microelectrodes (10−3 cm2) in series with a large gold counterelectrode. Electrical cell-substrate impedance sensing (ECIS) was conducted using an Applied Biophysics Model 1600R instrument (Applied Biophysics, Troy, NY) to measure electrical resistance across the monolayer. Current was applied across the electrodes by a 4,000-Hz AC voltage source with amplitude of 1 V in series with a 1-M resistance to approximate a constant current source (∼1 μA). When cells had achieved confluency, transelectrical resistance (TER) was monitored at low bicarbonate (5 mM) to establish a baseline resistance (minimum 1 h) followed by real-time TER measurements after applying bicarbonate (maintained at 5 mM or adjusted to 40 mM). TER was recorded for an additional period, and data from each microelectrode were normalized to initial baseline resistance and plotted vs. time.
Lung isolation and perfusion.
The lungs and heart were removed en bloc from adult male Sprague-Dawley rats (250–300 g) as described previously (48). After the first Kf recording, bicarbonate, or a NaCl solution adjusted to the same osmolality as the sodium bicarbonate solution, was added, and the Kf was measured as described previously (48). A perfusate sample was withdrawn for blood gas analysis (ABL5; Radiometer) after each Kf measurement.
Graphing and statistical analysis.
Analytical data are reported as means ± SE. Comparisons between data groups were accomplished using one- or two-way ANOVA, in conjunction with a Bonferroni post hoc test as needed. In all cases, values of P < 0.05 were considered significant. Data were graphed using GraphPad.
RESULTS
AC10 transcripts and protein are detected in lung tissue and pulmonary endothelium.
Previous reports demonstrate AC10 transcripts are detected in human lung tissue (22). Utilizing RT-PCR and AC10-specific primers, we confirmed the presence of AC10 transcripts in rat whole lung tissue through amplification of PCR products at the expected amplicon size (Fig. 1A). To examine AC10 expression in pulmonary endothelial cells, we similarly performed RT-PCR using cultured rat PMVECs and PAECs and detected AC10 transcripts in both capillary and conduit endothelial cells, respectively. Furthermore, the presence of AC10 protein was confirmed by immunoblotting whole cell lysates from both PMVECs and PAECs using the R21 antibody, which recognizes a region of the protein at the end of the first catalytic domain (Fig. 1B). Two bands were detected at ∼50 and 48 kDa, consistent with previous findings in somatic and germ cells (17). The band density was greater in PMVECs compared with PAECs, suggesting greater expression in the endothelial cells derived from capillary vessels vs. from conduit vessels.
Fig. 1.

Pulmonary endothelial cells express the mammalian soluble adenylyl cyclase isoform 10 (AC10). A: using RT-PCR and specific primers, the AC10 transcript is detected in whole lung tissue and in pulmonary artery endothelial cells (PAECs) and pulmonary microvascular endothelial cells (PMVECs) B: AC10 protein is detected in both PAECs and PMVECs as 2 bands at ∼50 and 48 kDa using the R21 antibody, which recognizes an epitope around the second catalytic domain of AC10. There is greater expression of both AC10 isoforms in PMVECs compared with PAECs. Images are representative of at least three independent experiments.
Endothelial expression of sodium bicarbonate transporters.
Unlike transmembrane ACs, which are stimulated by Gsα signaling or pharmacologically with forskolin (except AC9), AC10 is insensitive to either of these agents and is stimulated by bicarbonate and calcium (24, 33). Bicarbonate is a hydrophilic molecule that does not readily cross the plasma membrane. Therefore, in order for extracellular bicarbonate to stimulate AC10, it must be transported into the cell through bicarbonate transporters. Thus, we wanted to determine whether bicarbonate transporters are expressed within the pulmonary endothelium.
The solute carrier 4 superfamily of bicarbonate transporters includes the sodium-independent exchangers or anion exchangers (AE1, AE2, and AE3) and the sodium-coupled bicarbonate cotransporters (NCBTs) (reviewed in Refs. 5, 34, and 39). The five NCBTs include the NBCs and the electroneutral NDCBE. NBCs transport bicarbonate in an electrogenic [NBCe1 (a.k.a. NBC1) and NBCe2 (a.k.a. NBC4)] or electroneutral [NBCn1 (a.k.a. NBC3) and NBCn2 (formerly known as NCBE)] manner (40). The activity of NBCs and NDCBE transporters normally leads to bicarbonate influx, with the exception of the 1Na+-3HCO3− mode of activity of NBCe1 in kidney proximal tubules (51). NBCe1 has been identified in rat lung tissue (14) while NBCn1 was initially cloned from rat aorta (12) and is also detected in the lung (43). We performed RT-PCR to examine the expression of NCBT transcripts in whole lung tissue and within the pulmonary endothelium using specific primers (Fig. 2A). While RT-PCR products were amplified with primers for NBCe1 and NBCn1 in both PMVEC and PAECs, the RT-PCR product for NBCn2 was only detected in PMVECs. No NBCe2 or NDCBE RT-PCR product was detected in either cell type. Western blot analysis revealed expression of NBCe1 and NBCn1 protein in both PMVECs and PAECs, thus confirming RT-PCR findings (Fig. 2B).
Fig. 2.

Pulmonary endothelial cells selectively express sodium-bicarbonate cotransporters. A: using RT-PCR and primers specific to the solute carrier 4A (SLC4A) family of sodium-bicarbonate cotransporters, the electroneutral sodium-driven chloride-bicarbonate cotransporter (NBC) e1 and NBCn1 transcripts are expressed in both PMVECs and PAECs, yet only the NBCn2 transcript is expressed in PMVECs. The NBCe2 and sodium-driven chloride-bicarbonate cotransporters (NDCBE) transcripts are not detected in either PMVECs or PAECs. B: PMVECs and PAECs express the NBCe1 isoforms A and/or B (band on top) and NBCn1 protein (band in middle). Images are representative of at least three independent experiments.
PMVECs have a bicarbonate- and PDE-sensitive cAMP pool that is dependent on sodium.
With mechanisms in place for bicarbonate to be transported across the plasma membrane of endothelial cells and provide a potential bicarbonate source for AC10 stimulation, we performed bicarbonate dose-response studies and measured whole cell cAMP. Indeed, an increase in the whole cell cAMP pool was detected in PMVECs in the presence of the PDE4 inhibitor rolipram (10 μM) over the dose range of 5 to 40 mM with a significant response occurring as bicarbonate increased from 25 to 30 mM; however, no increase in cAMP was detected in the absence of rolipram (Fig. 3A). Because normal plasma bicarbonate is 24 mM, this response corresponds to the range where plasma bicarbonate is considered elevated above normal values. PDE inhibitors are often required to reveal a highly compartmentalized cAMP signal that is diluted in whole cell analysis, yet these inhibitors may not be required to detect a physiological/pathophysiological response. This cAMP response was independent of any change in osmolality, since a 5 mM bicarbonate solution adjusted to the same osmolality as 40 mM with either NaCl or sucrose did not change the cAMP levels (Fig. 3B). NBCs require sodium to transport bicarbonate into the cell. Therefore, to demonstrate the functional role of sodium-coupled vs. sodium-independent bicarbonate transport in the bicarbonate-induced cAMP response in PMVECs, experiments were performed with media containing choline chloride to replace sodium chloride; choline bicarbonate was used to elevate bicarbonate concentrations in these experiments. In the absence of sodium, increasing bicarbonate did not lead to an increase in total cellular cAMP (Fig. 3C), yet, when sodium was added back, the bicarbonate response was restored. When N-methyl-d-glucamine was used to replace sodium chloride, identical results were obtained (data not shown). Thus, these data suggest that sodium-coupled bicarbonate transport is necessary to stimulate AC10 activity. In contrast to PMVECs, the baseline cAMP level was lower in PAECs, and cAMP did not increase in PAECs at any extracellular bicarbonate concentration tested (Fig. 3D). To determine the time course of the cAMP response, PMVECs and PAECs were treated with bicarbonate and rolipram over 30 min, and cAMP was measured. These studies demonstrate that the peak cAMP rise in PMVECs occurs at 15 min and is sustained at 30 min, yet at no time point was there a cAMP increase in PAECs (Fig. 3E).
Fig. 3.
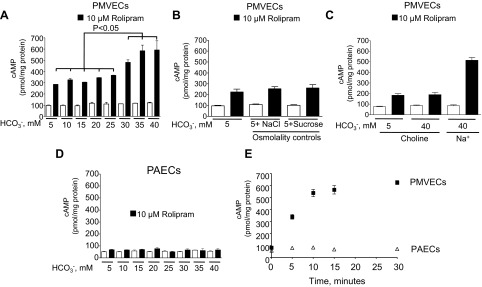
PMVECs but not PAECs have a bicarbonate-stimulated, rolipram-sensitive cAMP pool that is dependent upon sodium. A: in the presence of the phosphodiesterase (PDE) inhibitor rolipram (10 μM), and 15 min after the addition of increasing concentrations of bicarbonate to the extracellular media, there is an increase in whole cell cAMP that is not due to changes in osmolality (B). C: when choline chloride replaces NaCl and choline bicarbonate replaces sodium bicarbonate, the bicarbonate-induced cAMP response is lost, but, when sodium is replaced, the bicarbonate-induced cAMP response is restored. D: with increasing doses of extracellular bicarbonate and in the presence of rolipram (10 μM), there is no increase in intracellular cAMP in PAECs. E: following the addition of 40 mM bicarbonate and rolipram (10 μM), intracellular whole cell cAMP levels increase over time in PMVECs (■), but there is no such response in PAECs (△). Individual experiments are performed in triplicate with n = 3 for each condition. Values are presented as means ± SE.
AC10 is not stimulated by calcium in PMVECs.
Previous reports suggest that AC10 can be stimulated by calcium and bicarbonate (24, 33); thus, we used ionomycin (1 μM) to induce a global calcium response. To examine the intracellular calcium response to ionomycin, confluent PMVECs were loaded with the calcium indicator fura 2-AM, and the 340 nm-to-380 nm ratio was detected over time. Following baseline measurements over 200 s, 1 μM ionomycin was added to the cells, and a rapid increase in intracellular calcium was detected (Fig. 4A). Subsequently, we examined whether the ionomycin-induced rise in intracellular calcium was sufficient to activate AC10 in PMVECs. Under our experimental conditions, the ionomycin-induced calcium response did not increase either the 5 mM bicarbonate cAMP pool or the bicarbonate-induced rolipram-sensitive cAMP pool (Fig. 4B). Thus, it appears that the ionomycin-induced calcium response was not sufficient to increase AC10 activity in PMVECs.
Fig. 4.

In PMVECs, the AC10 cAMP pool is not potentiated by elevated intracellular calcium. A: PMVECs were loaded with the calcium indicator fura 2-AM in the presence of 2 mM extracellular calcium. Following the addition of 1 μM ionomycin at the 200-s time point, the ratios of the wavelengths of calcium-bound (340 nm) to -unbound (380 nm) for fura 2 fluorescence were recorded. EDTA (5 mM) was added to chelate calcium 20 min after the addition of ionomycin (n = 9). B: ionomycin did not potentiate the bicarbonate-induced, rolipram-sensitive cAMP pool even in the presence of rolipram at the 15-min time point. Individual experiments are performed in triplicate with at least n = 3 for each condition. Values are presented as means ± SE. NS, not significant.
Bicarbonate increases the Kf.
To address whether activation of AC10 and generation of an endogenous cAMP pool disrupt the endothelial barrier, we performed isolated perfused lung studies (Fig. 5A). Following an initial baseline Kf, the perfusate was either maintained at low bicarbonate concentration, maintained at low bicarbonate but with increased osmolality to mimic the elevated bicarbonate condition, or adjusted to 38.7 ± 5.8 mM bicarbonate. While there was no increase in the Kf in either low bicarbonate (data not shown) or low bicarbonate with elevated osmolality, there was a significant increase in the Kf when perfusate bicarbonate was increased to 38.7 ± 5.8 mM. We performed ECIS experiments to determine whether bicarbonate was sufficient to disrupt the PMVEC monolayer (Fig. 5B). Once PMVECs had achieved confluency, the media was changed to low bicarbonate (5 mM). Once a stable baseline resistance was achieved over a 60-min time period, the media was either maintained at 5 mM bicarbonate or increased to 40 mM bicarbonate. While there was no change in resistance under the 5 mM condition, increasing the bicarbonate concentration to 40 mM, with adjustment of CO2 tensions to maintain pH, there was a significant decrease in resistance. Similar studies were performed with PAECs (Fig. 5C), yet these cells are more sensitive to mechanical disruption alone such that when media of equal bicarbonate concentration is added to the cells there is a greater decrease in resistance compared with similar experiments performed with PMVECs. Thus, when adjusting for the effects of mechanical disruption, 40 mM bicarbonate induced a greater decrease in resistance in PMVECs compared with PAECs.
Fig. 5.
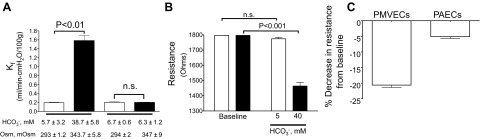
Bicarbonate increases the filtration coefficient (Kf) and PMVEC permeability. A: the filtration coefficient increases when perfusate bicarbonate concentration is increased from 5.7 ± 3.2 to 38.7 ± 5.8 mM, yet there was no response with controls where NaCl was used to increase perfusate osmolality to the same level as 38 mM bicarbonate (347 ± 9 vs. 343.7 ± 5.8 mosmol/kgH2O, respectively). Data represent n = 3 for each condition, and values are presented as means ± SE. B: resistance across the PMVEC monolayer decreases when extracellular bicarbonate is increased from 5 (baseline) to 40 mM while there is no change in resistance when bicarbonate is maintained at 5 mM. Data represent n = 3 for each with individual experiment performed as n = 4. Values are presented as means ± SE. C: PMVECs experience a greater decrease in resistance from baseline following the addition of 40 mM bicarbonate compared with PAECs. Data represent n = 3 for each with individual experiments performed as n = 4. Values are presented as means ± SE.
DISCUSSION
cAMP plays a dual role in regulation of lung vascular permeability. While cAMP synthesized at the plasma membrane strengthens the pulmonary endothelial barrier, cAMP generated in the cytosol by exogenous soluble ACs, such as ExoY or sACI/II, disrupt the endothelial barrier. Herein we provide further evidence that cytosolic cAMP disrupts the endothelial barrier, but, in contrast to studies that utilize exogenous expression of cytosolic ACs, the current studies reveal the role of the endogenous bicarbonate-responsive cytosolic AC, AC10. Indeed, because extracellular bicarbonate increased beyond the normal physiological range, stimulation of AC10 promoted pulmonary endothelial barrier failure and hyperpermeability.
These studies support a new cyclic nucleotide paradigm within pulmonary endothelial cells providing evidence for a second endogenous cAMP source not localized to the plasma membrane. Thus, two endogenous cAMP compartments exist: one localized to the plasma membrane and the second localized within the cytosol (47). Interestingly, while the cytosolic AC10-cAMP pool is regulated by PDE4, the cytosolic cAMP pool generated by the exogenous soluble ACs, ExoY and sACI/II, are not regulated by rolipram-sensitive PDE4 activity (46, 48). This is perhaps not surprising since endogenous ACs are thought to exist in tightly regulated signalosomes formed by A-kinase anchoring proteins which coordinate cAMP synthesis, degradation by PDEs, and downstream signaling (15). Meanwhile, lack of PDE regulation of ExoY or sACI/II could be because of lack of compartmentalization, since these ACs perhaps lack localization signals and are nondiscriminately localized within the cytosol and incapable of colocalizing with PDEs. Thus, regardless of the compartment where endogenous ACs synthesize cAMP, the plasma membrane, or the cytosol, the degradation of the signal is tightly controlled. There are over 20 distinct PDE4 isoforms encoded by four different genes (PDE4A-D) with multiple splice variants, which are all inhibited by rolipram. While all PDE4 isoforms have the same conserved catalytic domain, specific isoforms are differentially regulated and targeted within the cell (23). In PMVECs, the long PDE4D4 splice variant is found in the plasma membrane fraction (13); currently, it is not clear whether the same PDE4 isoform also regulates the AC10 cAMP pool. Regardless of the PDE4 isoform, it appears that the AC10-cAMP is similarly spatially and temporally regulated through hydrolysis to maintain compartmentalized cAMP signaling specificity and perhaps prevent the cAMP escaping beyond the functional compartment where it could possibly lead to off-target effects. Currently, the physiological significance of AC10 within the endothelium is unclear.
Intriguingly, elevations in extracellular bicarbonate led to increased cAMP in PMVECs but not PAECs despite protein expression in both cell types. The reason for this lack of bicarbonate responsiveness is unclear and could be due to either lower protein expression in PAECs or may be due to lack of bicarbonate availability. Indeed, our data reveal expression of NBCn2 in PMVECs but not PAECs. Heterogeneity between endothelial cells derived from different segments of the vascular bed has been well documented (53). Indeed, endothelial cells derived from the capillary bed vs. the conduit vessels display different structural and functional characteristics, such as permeability function and ion channel expression and function. Thus, AC10 expression and activity as well as NBCn2 expression also contribute to this endothelial heterogeneity.
In 1982, Garbers et al. reported that bicarbonate stimulated cAMP production in spermatozoa, and in 1999 Buck et al. cloned this bicarbonate sensor, the sAC otherwise known as AC10 (7, 21). While mammalian transmembrane ACs are class IIIa ACs, AC10 is a class IIIb AC, characterized by an aspartate substitution (D1018 of AC2C2 domain), responsible for ATP binding within the catalytic domain, most commonly to threonine or less commonly to serine (26). The threonine substitution, which renders these enzymes responsive to bicarbonate, is commonly found in prokaryotic ACs, such as cyaB of the common lung pathogen P. aeruginosa, as well as the mammalian AC10 (8, 9, 58). CyaB is a bicarbonate-sensitive AC restricted to the bacterial cytoplasm that generates cAMP to activate the cAMP-binding protein Vfr, which upregulates the TTSS (60). Thus, during P. aeruginosa infection, bicarbonate could act as a signal not only to activate the TTSS and the pulmonary endothelial barrier-disruptive soluble AC toxin ExoY but also further stimulate endogenous cytosolic cAMP production in PMVECs via AC10, and further potentiate endothelial barrier disruption (48). Interestingly, some prokaryotic ACs and eukaryotic AC10 have two distinct but homologous catalytic domains, C1 and C2, in contrast to eukaryotic class IIIa transmembrane ACs, which have a single catalytic center and a pseudocatalytic domain (8). Furthermore, phylogenetic analysis demonstrates that the amino acid sequence of AC10 catalytic domains is more closely related to bacteria. How AC10 evolved or became part of the eukaryotic genome has yet to be determined.
Previous studies demonstrate that AC10 is regulated by calcium (24, 33). In contrast, our data reveal that ionomycin-induced elevated intracellular calcium is not sufficient to increase either the basal or the bicarbonate-stimulated cAMP pool in PMVECs despite the presence of the PDE inhibitor rolipram over the time course of the experiment. The reason for this discrepancy is unclear; however, in vitro studies revealed calcium chloride activation of the truncated AC10 isoform with an EC50 ≈750 μM, which is not physiologically relevant in PMVECs. A second study examined the calcium sensitivity of the full-length AC10 and revealed an EC50 of 393 ± 140 nM, whereas a truncated form of the enzyme, which more closely mimics the isoform detected in pulmonary endothelium, has a higher EC50 (≈700 nM). Indeed, there appears to be some ambiguities within the literature as to the calcium concentrations necessary to activate AC10 and isoform variability. In the future, in vitro studies or alternative mechanisms to increase intracellular calcium might reveal calcium regulation of AC10 in PMVECs.
Interestingly, our studies reveal that AC10 activity increases as extracellular bicarbonate rises beyond the normal physiological serum range. Indeed, patients with elevated CO2 reaching levels above 65–70 mmHg will experience a corresponding increase in bicarbonate (30 mM or higher). Furthermore, as a consequence of prolonged periods of mechanical ventilation using the low tidal volume protective strategy, patients generally acquire elevated arterial CO2 tensions, known as permissive hypercapnia. Concomitant with permissive hypercapnia is a decrease in plasma pH culminating in hypercapnic acidosis, which is often compensated through renal mechanisms or, as recommended by the acute respiratory distress syndrome net, by sodium bicarbonate infusion (2, 30, 57). These mechanisms lead to elevated plasma bicarbonate, and, although buffering hypercapnia is a common practice, it is controversial (28). It was proposed that bicarbonate would lead to elevated CO2, which would diffuse into the cell causing a paradoxical acidosis. In the pulmonary circulation, animal studies support the detrimental use of elevated plasma bicarbonate such as an ischemia-reperfusion rabbit lung injury model in which permeability increased following buffering hypercapnic acidosis with an associated elevated perfusate bicarbonate (32). Furthermore, in a rat model of lipopolysaccharide or Escherichia coli-induced lung injury, renal buffering of hypercapnia promoted lung damage compared with nonbuffered normocapnic controls (37). In conjunction with our data, we suggest that this elevated bicarbonate could also act directly to activate AC10, increase endothelial permeability, and provide a mechanism to promote further lung injury.
In summary, our data reveal that there are two unique mechanisms for generating cAMP in pulmonary endothelial cells: the well-established transmembrane AC6 that resides in lipid raft domains and the newly described bicarbonate-stimulated AC10. We reveal AC10 expression and activity in PMVECs, which is tightly regulated by PDE4 activity. While AC10 is detected in PAECs, it appears to lack activity, either because of low expression levels or insufficient availability of cytosolic bicarbonate. While the function of AC10 in endothelium has not yet been established, elevated bicarbonate increases permeability not only across the endothelial monolayer but also increases permeability in the isolated perfused lung.
GRANTS
This work was supported by American Heart Association Grant AHA11GRNT7430039 (S. L. Sayner), the Parker B. Francis Fellowship Foundation Award (S. L. Sayner), and National Heart, Lung, and Blood Institute Grant HL-076125 (R. Kunstadt).
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: B.O. and S.L.S. conception and design of research; B.O., W.C., N.X., R.K., B.R., J.N., and S.L.S. performed experiments; B.O., W.C., N.X., and S.L.S. analyzed data; B.O., N.X., and S.L.S. interpreted results of experiments; W.C., N.X., R.K., J.N., and S.L.S. prepared figures; W.C. and S.L.S. edited and revised manuscript; W.C., N.X., R.K., and S.L.S. approved final version of manuscript; S.L.S. drafted manuscript.
ACKNOWLEDGMENTS
We thank Dr. Troy Stevens for careful review of this manuscript, Linn Ayers and Anna Buford of the Center for Lung Biology Tissue Culture Core for contributions with cell seeding, and Jonathan Daigle for technical contributions.
REFERENCES
- 1. Acin-Perez R, Salazar E, Kamenetsky M, Buck J, Levin LR, Manfredi G. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab 9: 265–276, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. ARDSNet. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 342: 1301–1308, 2000 [DOI] [PubMed] [Google Scholar]
- 3. Bevensee MO, Schmitt BM, Choi I, Romero MF, Boron WF. An electrogenic Na+-HCO3− cotransporter (NBC) with a novel COOH-terminus, cloned from rat brain. Am J Physiol Cell Physiol 278: C1200–C1211, 2000 [DOI] [PubMed] [Google Scholar]
- 4. Bogatcheva NV, Verin AD. The role of cytoskeleton in the regulation of vascular endothelial barrier function. Microvasc Res 76: 202–207, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boron WF, Chen L, Parker MD. Modular structure of sodium-coupled bicarbonate transporters. J Exp Biol 212: 1697–1706, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buck J, Levin LR. Physiological sensing of carbon dioxide/bicarbonate/pH via cyclic nucleotide signaling. Sensors (Basel) 11: 2112–2128, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buck J, Sinclair ML, Schapal L, Cann MJ, Levin LR. Cytosolic adenylyl cyclase defines a unique signaling molecule in mammals. Proc Natl Acad Sci USA 96: 79–84, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cann M. Bicarbonate stimulated adenylyl cyclases. IUBMB Life 56: 529–534, 2004 [DOI] [PubMed] [Google Scholar]
- 9. Cann MJ, Hammer A, Zhou J, Kanacher T. A defined subset of adenylyl cyclases is regulated by bicarbonate ion. J Biol Chem 278: 35033–35038, 2003 [DOI] [PubMed] [Google Scholar]
- 10. Chen J, Levin LR, Buck J. Role of soluble adenylyl cyclase in the heart. Am J Physiol Heart Circ Physiol 302: H538–H543, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen Y, Cann MJ, Litvin TN, Iourgenko V, Sinclair ML, Levin LR, Buck J. Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science 289: 625–628, 2000 [DOI] [PubMed] [Google Scholar]
- 12. Choi I, Aalkjaer C, Boulpaep EL, Boron WF. An electroneutral sodium/bicarbonate cotransporter NBCn1 and associated sodium channel. Nature 405: 571–575, 2000 [DOI] [PubMed] [Google Scholar]
- 13. Creighton J, Zhu B, Alexeyev M, Stevens T. Spectrin-anchored phosphodiesterase 4D4 restricts cAMP from disrupting microtubules and inducing endothelial cell gap formation. J Cell Sci 121: 110–119, 2008 [DOI] [PubMed] [Google Scholar]
- 14. Damkier HH, Nielsen S, Praetorius J. An anti-NH2-terminal antibody localizes NBCn1 to heart endothelia and skeletal and vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 290: H172–H180, 2006 [DOI] [PubMed] [Google Scholar]
- 15. Dessauer CW. Adenylyl cyclase–A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol Pharmacol 76: 935–941, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol 91: 1487–1500, 2001 [DOI] [PubMed] [Google Scholar]
- 17. Farrell J, Ramos L, Tresguerres M, Kamenetsky M, Levin LR, Buck J. Somatic ‘soluble’ adenylyl cyclase isoforms are unaffected in Sacy tm1Lex/Sacy tm1Lex ‘knockout’ mice. PLoS One 3: e3251, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Farrukh IS, Gurtner GH, Michael JR. Pharmacological modification of pulmonary vascular injury: possible role of cAMP. J Appl Physiol 62: 47–54, 1987 [DOI] [PubMed] [Google Scholar]
- 19. Feng Q, Zhang Y, Li Y, Liu Z, Zuo J, Fang F. Two domains are critical for the nuclear localization of soluble adenylyl cyclase. Biochimie 88: 319–328, 2006 [DOI] [PubMed] [Google Scholar]
- 20. Fischmeister R. Is cAMP good or bad? Depends on where it's made. Circ Res 98: 582–584, 2006 [DOI] [PubMed] [Google Scholar]
- 21. Garbers DL, Tubb DJ, Hyne RV. A requirement of bicarbonate for Ca2+-induced elevations of cyclic AMP in guinea pig spermatozoa. J Biol Chem 257: 8980–8984, 1982 [PubMed] [Google Scholar]
- 22. Geng W, Wang Z, Zhang J, Reed BY, Pak CY, Moe OW. Cloning and characterization of the human soluble adenylyl cyclase. Am J Physiol Cell Physiol 288: C1305–C1316, 2005 [DOI] [PubMed] [Google Scholar]
- 23. Houslay MD. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci 35: 91–100, 2009 [DOI] [PubMed] [Google Scholar]
- 24. Jaiswal BS, Conti M. Calcium regulation of the soluble adenylyl cyclase expressed in mammalian spermatozoa. Proc Natl Acad Sci US A 100: 10676–10681, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kamenetsky M, Middelhaufe S, Bank EM, Levin LR, Buck J, Steegborn C. Molecular details of cAMP generation in mammalian cells: a tale of two systems. J Mol Biol 362: 623–639, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kanacher T, Schultz A, Linder JU, Schultz JE. A GAF-domain-regulated adenylyl cyclase from Anabaena is a self-activating cAMP switch. EMBO J 21: 3672–3680, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kelly JJ, Moore TM, Babal P, Diwan AH, Stevens T, Thompson WJ. Pulmonary microvascular and macrovascular endothelial cells: differential regulation of Ca2+ and permeability. Am J Physiol Lung Cell Mol Physiol 274: L810–L819, 1998 [DOI] [PubMed] [Google Scholar]
- 28. Kindig NB, Filley GF. Intravenous bicarbonate may cause transient intracellular acidosis (Abstract). Chest 83: 712, 1983 [DOI] [PubMed] [Google Scholar]
- 29. Kobayashi H, Kobayashi T, Fukushima M. Effects of dibutyryl cAMP on pulmonary air embolism-induced lung injury in awake sheep. J Appl Physiol 63: 2201–2207, 1987 [DOI] [PubMed] [Google Scholar]
- 30. Kollef MH, Schuster DP. The acute respiratory distress syndrome. N Engl J Med 332: 27–37, 1995 [DOI] [PubMed] [Google Scholar]
- 31. Kumar S, Kostin S, Flacke JP, Reusch HP, Ladilov Y. Soluble adenylyl cyclase controls mitochondria-dependent apoptosis in coronary endothelial cells. J Biol Chem 284: 14760–14768, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Laffey JG, Engelberts D, Kavanagh BP. Buffering hypercapnic acidosis worsens acute lung injury. Am J Respir Crit Care Med 161: 141–146, 2000 [DOI] [PubMed] [Google Scholar]
- 33. Litvin TN, Kamenetsky M, Zarifyan A, Buck J, Levin LR. Kinetic properties of “soluble” adenylyl cyclase. Synergism between calcium and bicarbonate J Biol Chem 278: 15922–15926, 2003 [DOI] [PubMed] [Google Scholar]
- 34. Majumdar D, Bevensee MO. Na-coupled bicarbonate transporters of the solute carrier 4 family in the nervous system: function, localization, and relevance to neurologic function. Neuroscience 171: 951–972, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev 86: 279–367, 2006 [DOI] [PubMed] [Google Scholar]
- 36. Michel CC, Curry FE. Microvascular permeability. Physiol Rev 79: 703–761, 1999 [DOI] [PubMed] [Google Scholar]
- 37. Nichol AD, O'Cronin DF, Howell K, Naughton F, O'Brien S, Boylan J, O'Connor C, O'Toole D, Laffey JG, McLoughlin P. Infection-induced lung injury is worsened after renal buffering of hypercapnic acidosis. Crit Care Med 37: 2953–2961, 2009 [DOI] [PubMed] [Google Scholar]
- 38. Ochoa CD, Alexeyev M, Pastukh V, Balczon R, Stevens T. Pseudomonas aeruginosa exotoxin Y is a promiscuous cyclase that increases endothelial tau phosphorylation and permeability. J Biol Chem 287: 25407–25418, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Parker MD, Boron WF. The divergence, actions, roles, and relatives of sodium-coupled bicarbonate transporters. Physiol Rev 93: 803–959, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parker MD, Musa-Aziz R, Rojas JD, Choi I, Daly CM, Boron WF. Characterization of human SLC4A10 as an electroneutral Na/HCO3 cotransporter (NBCn2) with Cl- self-exchange activity. J Biol Chem 283: 12777–12788, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pastor-Soler N, Beaulieu V, Litvin TN, Da Silva N, Chen Y, Brown D, Buck J, Levin LR, Breton S. Bicarbonate-regulated adenylyl cyclase (sAC) is a sensor that regulates pH-dependent V-ATPase recycling. J Biol Chem 278: 49523–49529, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pastor-Soler NM, Hallows KR, Smolak C, Gong F, Brown D, Breton S. Alkaline pH- and cAMP-induced V-ATPase membrane accumulation is mediated by protein kinase A in epididymal clear cells. Am J Physiol Cell Physiol 294: C488–C494, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Romero MF, Fong P, Berger UV, Hediger MA, Boron WF. Cloning and functional expression of rNBC, an electrogenic Na+-HCO3− cotransporter from rat kidney. Am J Physiol Renal Physiol 274: F425–F432, 1998 [DOI] [PubMed] [Google Scholar]
- 44. Sayner S, Stevens T. Soluble adenylate cyclase reveals the significance of compartmentalized cAMP on endothelial cell barrier function. Biochem Soc Trans 34: 492–494, 2006 [DOI] [PubMed] [Google Scholar]
- 45. Sayner SL. Emerging themes of cAMP regulation of the pulmonary endothelial barrier. Am J Physiol Lung Cell Mol Physiol 300: L667–L678, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sayner SL, Alexeyev M, Dessauer CW, Stevens T. Soluble adenylyl cyclase reveals the significance of cAMP compartmentation on pulmonary microvascular endothelial cell barrier. Circ Res 98: 675–681, 2006 [DOI] [PubMed] [Google Scholar]
- 47. Sayner SL, Balczon R, Frank DW, Cooper DM, Stevens T. Filamin A is a phosphorylation target of membrane but not cytosolic adenylyl cyclase activity. Am J Physiol Lung Cell Mol Physiol 301: L117–L124, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sayner SL, Frank DW, King J, Chen H, VandeWaa J, Stevens T. Paradoxical cAMP-induced lung endothelial hyperpermeability revealed by Pseudomonas aeruginosa ExoY. Circ Res 95: 196–203, 2004 [DOI] [PubMed] [Google Scholar]
- 49. Schmid A, Sutto Z, Nlend MC, Horvath G, Schmid N, Buck J, Levin LR, Conner GE, Fregien N, Salathe M. Soluble adenylyl cyclase is localized to cilia and contributes to ciliary beat frequency regulation via production of cAMP. J Gen Physiol 130: 99–109, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sinclair ML, Wang XY, Mattia M, Conti M, Buck J, Wolgemuth DJ, Levin LR. Specific expression of soluble adenylyl cyclase in male germ cells. Mol Reprod Dev 56: 6–11, 2000 [DOI] [PubMed] [Google Scholar]
- 51. Soleimani M, Grassi SM, Aronson PS. Stoichiometry of Na+-HCO-3 cotransport in basolateral membrane vesicles isolated from rabbit renal cortex. J Clin Invest 79: 1276–1280, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stelzner TJ, Weil JV, O'Brien RF. Role of cyclic adenosine monophosphate in the induction of endothelial barrier properties. J Cell Physiol 139: 157–166, 1989 [DOI] [PubMed] [Google Scholar]
- 53. Stevens T. Functional and molecular heterogeneity of pulmonary endothelial cells. Proc Am Thorac Soc 8: 453–457, 2011 [DOI] [PubMed] [Google Scholar]
- 54. Stevens T, Creighton J, Thompson WJ. Control of cAMP in lung endothelial cell phenotypes. Implications for control of barrier function. Am J Physiol Lung Cell Mol Physiol 277: L119–L126, 1999 [DOI] [PubMed] [Google Scholar]
- 55. Stevens T, Fouty B, Cornfield D, Rodman DM. Reduced PO2 alters the behavior of Fura-2 and Indo-1 in bovine pulmonary artery endothelial cells. Cell Calcium 16: 404–412, 1994 [DOI] [PubMed] [Google Scholar]
- 56. Taylor CJ, Nicola PA, Wang S, Barrand MA, Hladky SB. Transporters involved in regulation of intracellular pH in primary cultured rat brain endothelial cells. J Physiol 576: 769–785, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tobin MJ. Mechanical ventilation. N Engl J Med 330: 1056–1061, 1994 [DOI] [PubMed] [Google Scholar]
- 58. Topal H, Fulcher NB, Bitterman J, Salazar E, Buck J, Levin LR, Cann MJ, Wolfgang MC, Steegborn C. Crystal structure and regulation mechanisms of the CyaB adenylyl cyclase from the human pathogen Pseudomonas aeruginosa. J Mol Biol 416: 271–286, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tresguerres M, Buck J, Levin LR. Physiological carbon dioxide, bicarbonate, and pH sensing. Pflugers Arch 460: 953–964, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wolfgang MC, Lee VT, Gilmore ME, Lory S. Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev Cell 4: 253–263, 2003 [DOI] [PubMed] [Google Scholar]
- 61. Xie F, Garcia MA, Carlson AE, Schuh SM, Babcock DF, Jaiswal BS, Gossen JA, Esposito G, van Duin M, Conti M. Soluble adenylyl cyclase (sAC) is indispensable for sperm function and fertilization. Dev Biol 296: 353–362, 2006 [DOI] [PubMed] [Google Scholar]
- 62. Yahr TL, Vallis AJ, Hancock MK, Barbieri JT, Frank DW. ExoY, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. Proc Natl Acad Sci USA 95: 13899–13904, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zippin JH, Chen Y, Nahirney P, Kamenetsky M, Wuttke MS, Fischman DA, Levin LR, Buck J. Compartmentalization of bicarbonate-sensitive adenylyl cyclase in distinct signaling microdomains. FASEB J 17: 82–84, 2003 [DOI] [PubMed] [Google Scholar]