

Abstract
Severe asthma is a chronic airway disease characterized by the Th2/Th17-polarized inflammation along with permanent airway remodeling. Despite past extensive studies, the exact role for Th2 and Th17 cytokines in asthmatic pathoetiology, particularly in the pathogenesis of bronchial epithelial-mesenchymal transition (EMT), is yet to be fully addressed. We herein conducted studies in 16-HBE cells and demonstrated that Th2-derived IL-4 and Th17-derived IL-17A provide a chronic inflammatory milieu that favors TGF-β1 to induce bronchial EMT. A synergic action was noted between TGF-β1, IL-4 and IL-17A in terms of induction of EMT. IL-4 and IL-17A synergized with TGF-β1 to induce epithelial cells re-entering cell cycle, and to promote epithelial to mesenchymal morphological transistion, and by which they enhanced the capacity of TGF-β1 to suppress E-cadherin expression, and to induce a-SMA expression in epithelial cells. Mechanistic studies revealed that this synergic action is coordinated by the regulation of ERK1/2 activity. Our results not only provide a novel insight into the understanding of the mechanisms underlying airway remodeling in asthmatic condition, but also have the potential for developing more effective therapeutic strategies against severe asthmatics in clinical settings.
Keywords: Severe asthma, airway remodeling, chronic inflammatory milieu, epithelial mesenchymal transition (EMT), ERK1/2
Introduction
Severe asthma is a chronic airway disease characterized by the Th2/Th17-polarized inflammation along with permanent structural changes termed airway remodeling [1-5]. The pathological changes of airway structural cells during airway remodeling include epithelial fragility and plasticity, increased number of activated fibroblasts/myofibroblasts, thickened basement membrane, airway smooth muscle hyperplasia/hypertrophy, and angiogenesis [6]. Previously, airway remodeling in asthma is considered to be an abnormal repair response secondary to persistent inflammation [7,8]. Recent investigations, however, have changed this concept from an outcome to a consequence of persistent inflammation, in which both inflammatory components and airway structural cells are involved equally and actively [9-11].
Given that the bronchial epithelial cells (BECs) undergo an abnormal proliferation in response to some noxious stimuli along with a phenotypic conversion from epithelial into mesenchymal morphology by a process termed epithelial-mesenchymal transition (EMT) [10,12-14], their role in airway remodeling receives much more attentions recently. Indeed, it was found that altered proliferation of bronchial epithelial cells is associated with a thickened epithelium and lamina reticularis in the airways of severe asthmatic subjects [12]. During the process of EMT, the bronchial epithelial cells are manifested by the removed cellular polarity and disrupted cell-cell contacts. Specifically, markers for polarized epithelial cells such as E-cadherin and cytokeratins are downregulated, while markers specific for mesenchymal cells such as a-SMA and vimentin are upregulated [15,16]. As a result, EMT is associated with increased cell motility and excessive deposition of extracellular matrix including collagen I, III, and fibronectin [17]. Once EMT occurs in the bronchial epithelium, the asthmatic subject is refractory to corticosteroids, the first line medication for treatment of asthma [18].
In subjects with severe asthma, the bronchial epithelium is exposed to a chronic and complex inflammatory environment enriched with Th2 and Th17 cytokines along with high levels of TGF-β1 [19-21], and particularly, TGF-β1 has been recognized as a key factor to induce EMT in multiple organs including the lung [22-26]. Indeed, studies in animals revealed that approximately 30% of the fibroblasts/myofibroblasts during the course of airway remodeling are actually derived from EMT [27]. However, there is also evidence indicating that the bronchial epithelial cells are much less sensitive to TGF-β1 in terms of EMT induction [23,27-29]. For example, the bronchial epithelial cells isolated from asthmatic subjects failed to acquire characteristic markers of mesenchymal cells such as a-SMA and vimentin following TGF-β1 stimulation [23]. Together, these observations support that additional factors other than TGF-β1 are necessary for the induction of EMT in asthma. Given the role of IL-4 and IL-17A played in disease severity and airway remodeling [4,30], we thus hypothesized that IL-4 and IL-17A provide a Th2/Th17-polarized inflammatory milieu that favors TGF-β1 to induce EMT during the process of airway remodeling in severe asthma. To test this hypothesis, we performed studies in 16HBE cells, an immortalized but nontransformed cell line derived from ciliated human bronchial epithelial cells that line the airway of the lung. Our data demonstrate that IL-4 and IL-17A synergize with TGF-β1 to induce 16HBE cell proliferation and morphological change along with the expression of mesenchymal markers. These results are not only important for better understanding the mechanisms underlying bronchial EMT, but also useful for developing novel therapeutic strategies for prevention and treatment of EMT in the setting of severe asthma.
Materials and methods
Cells and reagents
Human bronchial epithelial cell line 16HBE-14o (16-HBE) was kindly provided by Dr. Bing Li of Guangzhou Medical University. Minimal Essential Medium (MEM) and fetal bovine serum (FCS) were from Invitrogen (Carlsbad, CA, USA). Recombinant human TGF-β1, IL-4, and IL-17A were purchased from Peprotech (Rocky Hill, USA). The highly selective inhibitor for ERK1/2, U0126, was from Sigma (Sigma, St. Louis, MO). Antibodies against human E-cadherin, EGFR and phosphorylated EGFR (pEGFR) were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies for ERK1/2, phosphorylated ERK1/2 (pERK1/2), Smad3, and phosphorylated Smad3 (pSmad3) were obtained from Cell Signaling Technology (Danvers, MA). Antibodies against a-SMA and β-actin were purchased from Sigma (St. Louis, MO).
Cell culture
The culture and stimulation of 16HBE cells were performed as reported [27]. In brief, the cells were plated in MEM supplemented with 10% FCS in a humidified incubator at 37°C with a 5% CO2 atmosphere. Once the cells reached confluence, they were transferred into low serum medium (1% FCS) for 24 h for synchronization. The synchronized cells were subsequently treated with indicated cytokines for 24 h, or 48 h or 72 h, respectively. Treatment of cells with 0.1% DMSO was served as a control. For blockade of ERK1/2 activity, 20 μM of U0126 were added into the culture 1 h before cytokine stimulation.
Cell proliferation assay
Analysis of 16-HBE cell proliferation was carried out using a cell counting kit-8 (CCK-8) from Beyotime (Shanghai, China) as instructed. The Cells (1×104 cells/well) were seeded in 96-well plates and incubated overnight in the serum-free medium. TGF-β1 (10 ng/ml), or IL-4 (10 ng/ml), or IL-17A (10 ng/ml), or TGF-β1 (10 ng/ml) along with IL-4 (10 ng/ml) and IL-17A (10 ng/ml) (cocktail) were then added into the culture. The cells cultured with 10% FCS were served as a positive control, while cells cultured with 1% FCS were used as a negative control. After culturing the cells for another 24 h, 10 μl of cell counting kit-8 were added into each well. The cells were next subjected to fluorescence counting after 3 h of incubation. Cell proliferation was determined by measuring the absorbance of CCK-8 at 450 nm wavelength. Each assay included 3 replicates, and the experiments were repeated by 3 times.
RNA extraction and real-time RT-PCR
After 24 h of cytokine treatment, 16-HBE cells were harvested for RNA extraction using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. cDNA synthesis was then carried out using 2 μg of total RNA with a RevertAidTM First Strand cDNA Synthesis Kit (Shanghai, China). Equal quantities of cDNA (equivalent to 25 ng of total RNA) from each sample were used for real-time PCR analysis of E-cadherin and α-SMA. Real-time PCR analysis was carried out using a StepOne™ Real-Time PCR System (Applied Biosystems, Foster, CA) and a SYBR® Premix Ex Taq™ II kit (TakaRa, Shanghai) [31]. PCR amplifications were conducted in a 20 μl volume at 95°C for 10 min followed by 40 cycles of 95°C for 30 sec, 95°C for 5 sec, 60°C for 34 sec. The primers for each target gene are as follows: E-cadherin (forward, 5′-gag tgc caa ctg gac cat tca gta-3′; reverse, 5′-agt cac cca cct cta agg cca tc-3′), α-SMA (forward, 5′-gac aat ggc tct ggg ctc tgt aa-3′, reverse, 5′-ctg tgc ttc gtc acc cac gta-3′), and GAPDH (D379014, TakaRa, Shanghai). Relative expressions for each target were normalized by GAPDH and calculated as 2-ΔΔCt {2-ΔΔCt=2-[(Ct treated sample-Ct internal control)-(Ct untreated sample-Ct internal control)]}. PCR specificity was verified by melting curve analysis and agarose gel electrophoresis.
Western blot analysis
Whole cell lysates were prepared after 72 h of cytokine treatment from 16-HBE cells using RIPA lysis buffer with protease inhibitors (Beyotime, China). Total protein concentrations were determined by the BCA Protein Assay kit (Wellbiology, USA) as instructed. The loaded proteins (20 μg) were separated by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred onto PVDF membranes. After blocking with 5% milk, the membranes were probed with antibodies against β-actin, α-SMA, E-cadherin, EGFR, pEGFR, SMAD3, pSMAD3, ERK1/2 and pERK1/2, followed by incubation with a secondary antibody, respectively. The blots were developed using the ECL Plus reagents (Thermo pierce, USA) as previously reported [32]. The relative amount of each target protein was determined by the ratio with β-actin.
ELISA analysis of collagen I propeptides
Culture supernatants of 16HBE cells after 72 h of cytokine treatment were collected for ELISA analysis of amino-terminal propeptide of type I procollagen (PINP), a marker for collagen synthesis using a commercial kit (Uscn, Shanghai) using the established technique within the lab [33].
Statistical analysis
Results are expressed as mean ± SD and analyzed using SPSS 16.0 software. One-way ANOVA followed by the Student-Newman-Keuls multiple analysis was used to determine the significance of differences in multiple comparisons. For comparison of two groups, data were analyzed by the non-parametric Mann-Whitney U tests. For all analyses, p < 0.05 was considered with statistical significance.
Results
TGF-β1 induces 16-HBE cell proliferation in the presence of IL-4 and Il-17A
To assess the impact of TGF-β1, IL-4 and IL-17A stimulation on 16-HBE cell proliferation, the cells were first cultured in serum free medium overnight, and then stimulated with TGF-β1 (10 ng/ml), or IL-4 (10 ng/ml), or IL-17A (10 ng/ml), or TGF-β1 (10 ng/ml) combined with IL-4 (10 ng/ml) and IL-17A (10 ng/ml) in 1% FCS medium for 24 h. Cells cultured in 10% FCS medium were served as a positive control, while cells cultured in 1% FCS medium were used as a negative control. As shown in Figure 1, although TGF-β1, IL-4 and IL-17A alone showed certain feasibility to induce 16-HBE cell proliferation, but the results were not statistically significant. However, TGF-β1 displayed much higher potency to induce 16-HBE cell proliferation once it combined with IL-4 and IL-17A as compared with that of negative control cells (140 ± 16.7% vs. 100 ± 13.2%, p < 0.05). Together, our data suggest that the potency for TGF-β1 alone is relatively low in terms of induction of 16-HBE cells re-entering the cell cycle (removal of cellular polarity), but it becomes much more potent in the presence of IL-4 and IL-17A.
Figure 1.
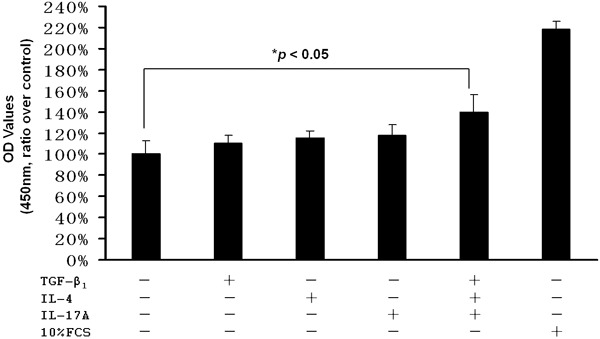
The impact of TGF-β1, IL-4, and IL-17A on 16-HBE proliferation. The cells were cultured overnight in serum-free medium for synchronization and then stimulated with TGF-β1 (10 ng/ml), or IL-4 (10 ng/ml), or IL-17A (10 ng/ml), or combined TGF-β1 (10 ng/ml), IL-4(10 ng/ml) and IL-17A (10 ng/ml) for 24 h. Cells cultured with 10% FCS were served as a positive control, while cells cultured in 1% FCS medium were used as a negative control.
The impact of TGF-β1 on the morphological changes of bronchial epithelial cells
Next, we intended to examine the impact of cytokine stimulation on the induction of morphological changes in bronchial epithelial cells. Under physiological condition, 16-HBE cells maintained a classic cobble-stone shape. Interestingly, unlike their impact on epithelial proliferation, IL-4 or IL-17A alone failed to induce a perceptible change for the 16-HBE cell morphology. In sharp contrast, after 72 h of 10 ng/ml TGF-β1 stimulation, a proportion of 16-HBE cells displayed a spindle-shape, fibroblast-like morphology with reduced cell-cell contact (Figure 2). Furthermore, much more cells showed a similar morphological change when they stimulated with combination of TGF-β1, IL-4 and IL-17A (Figure 2), suggesting that the presence of IL-4 and IL-17A enhanced the capacity of TGF-β1 to induce 16-HBE cells undergoing morphological change.
Figure 2.
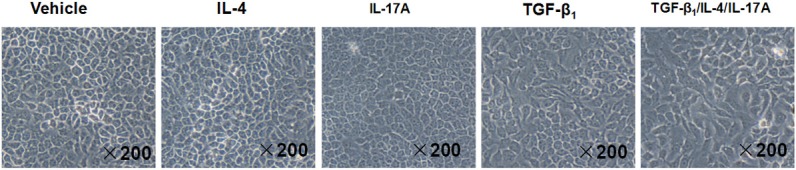
TGF-β1, IL-4 and IL-17A stimulation induces 16-HBE cells undergoing an epithelial to mesenchymal morphological transition. The 16-HBE cells were serum-starved overnight and then stimulated with TGF-β1 (10 ng/ml), or IL-4 (10 ng/ml), or IL-17A (10 ng/ml), or TGF-β1 (10 ng/ml) combined with IL-4 (10 ng/ml) and IL-17A (10 ng/ml) for 72 h, followed by morphological analysis under a light microscope.
IL-4 and IL-17A synergize with TGF-β1 to induce bronchial EMT
The above results prompted us to examine the impact of TGF-β1, IL-4 and IL-17A on the induction of EMT, an event commonly implicated in airway remodeling in severe asthma. We first performed real-time PCR analysis of EMT markers, E-cadherin and α-SMA, after 72 h of cytokine stimulation. It was noted that IL-4 stimulation did not show a perceptible impact on E-cadherin mRNA levels, but it slightly induced α-SMA expression in 16-HBE cells. On the contrary, IL-17A slightly decreased the levels of E-cadherin mRNA, but it did not induce a discenable change for α-SMA mRNA. In sharp contrast, TGF-β1 showed low potency to suppress E-cadherin mRNA levels and to induce α-SMA expression in 16-HBE cells (data not shown). Next, we treated 16-HBE cells with combined TGF-β1, IL-4 and IL-17A, and then analyzed E-cadherin and α-SMA mRNA after 24 h, 48 h and 72 h of stimulation. Remarkably, a time-dependent decrease of E-cadherin and increase of α-SMA expression were noted in 16-HBE cells following combined TGF-β1, IL-4 and IL-17A stimulation. Specifically, the expression of E-cadherin was suppressed by 4 fold 72 h after stimulation, while a 6.9 fold increase for α-SMA mRNA was noted at the same time point (Figure 3).
Figure 3.
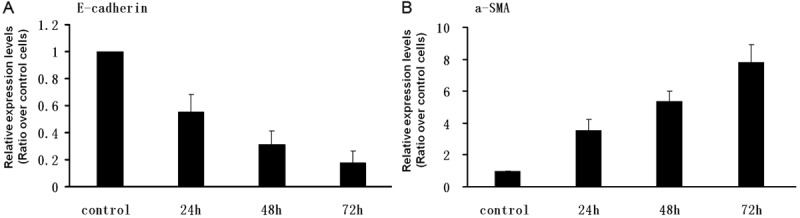
Real-time PCR analysis of E-cadherin and α-SMA expression following TGF-β1, IL-4 and IL-17A stimulation. The 16-HBE cells were serum-starved overnight and then stimulated with combined TGF-β1, IL-4 and IL-17A cocktail (10 ng/ml for each) for 24, 48 and 72 h, respectively. The cells were next harvested for analysis of mRNA levels for E-cadherin and α-SMA by Real-time RT-PCR as described. NAPDH was used for normalization. A. Relative expression levels for E-cadherin. B. Relative expression levels for α-SMA. The relative expression levels of target gene in the stimulated cells were presented as a ratio over control cells.
To confirm the above results, we next performed Western blot analysis of 16-HBE cell lysates 72 h after cytokine stimulation. As shown in Figure 4, IL-4 alone did not result in a significant change for the expressions of both E-cadherin and α-SMA. Similarly, a 30% decrease of E-cadherin was noted after IL-17A stimulation, but it did not show a perceptible impact on α-SMA protein levels. Of note, TGF-β1 combined with either IL-4 or IL-17A did not show a significant impact on E-cadherin expression as compared with that of TGF-β1 alone. However, a slight increase of α-SMA protein levels was noticed in cells treated with either TGF-β1/IL-17A or TGF-β1/IL-4. In shrap contrast, combined TGF-β1, IL-4 and IL-17A stimulation resulted in a 1.1 fold decrease of E-cadherin expression, while the expression of α-SMA increased by 2.4 fold. Taken all results together, our data suggest that IL-4 and IL-17A synergize with TGF-β1 to promote bronchia EMT.
Figure 4.
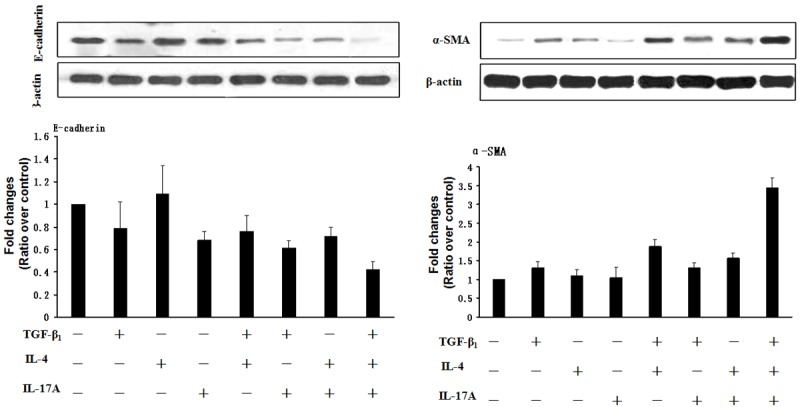
Western blot analysis of EMT markers E-cadherin and α-SMA after TGF-β1, IL-4 and IL-17A stimulation. The 16-HBE cells were serum-starved overnight and then stimulated with 10 ng/ml of TGF-β1, IL-4 and IL-17A alone or in combination for 72 h. The cells were next harvested for Western blot analysis of E-cadherin and α-SMA. β-actin was used for normalization, and the relative protein levels were presented as a ratio with control cells. The experiments were conducted with 3 replications.
Given that secretion of amino-terminal propeptide of type I procollagen (PINP) by activated epithelial cells is another characteristic feature of bronchia epithelial cells undergoing EMT, we thus examined PINP levels in the 16-HBE culture supernatants after 72 h of cytokine stimulation. As shown in Figure 5, TGF-β1 displayed high potency to induce 16-HBE cells secretion of PINP as manifested by a 3.2 fold increase of PINP levels following a 72 h TGF-β1 stimulation. Unexpectedly, we failed to observe a synergic effect for the combined TGF-β1, IL-4 and IL-17A stimulation on PINP secretion. Also, stimulation of 16-HBE cells with TGF-β1/IL-4 or TGF-β1/IL-17A or IL-4/IL-17A did not further enhance PINP secretion, rather, addition of IL-4 and/or IL-17A along with TGF-β1 significantly suppressed PINP secretion as compared with that of TGF-β1 alone.
Figure 5.
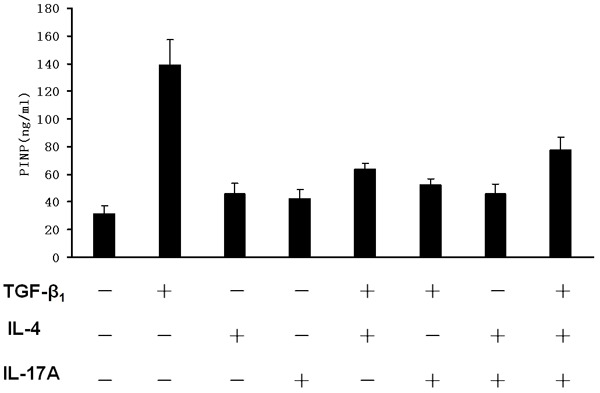
ELISA results for PINP secretion in 16-HBE cells. The 16-HBE cells were serum-starved overnight followed by stimulation with TGF-β1, IL-4 and IL-17A cocktail (10 ng/ml for each) for 72 h. Culture supernatants were next collected for ELISA analysis of PINP secretion as described.
IL-4 and IL-17A synergize with TGF-β1 to enhance ERK1/2 signaling
To dissect the mechanisms by which IL-4 and IL-17A synergize with TGF-β1 to promote 16-HBE cells undergoing EMT, we first exmined EGFR signaling, as EGFR signaling has been suggested to be a shared pathway for those cytokines [13,34-36]. For this purpose, 16-HBE cells were treated with indicated cytokines for 5 or 60 min and then harvested for Western blot analysis. Unexpectedly, TGF-β1, IL-4 and IL-17A alone or in combination failed to induce EGFR expression or activation as manifested by the similar levels for both EFGR and pEGFR after stimulation (Figure 6A).
Figure 6.
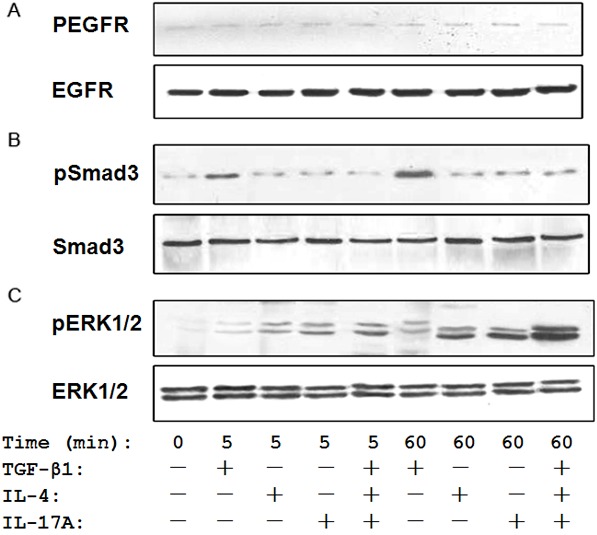
IL-4 and IL-17A synergize with TGF-β1 to promote ERK1/2 activation. The 16-HBE cells were first stimulated with indicated cytokines for 5 or 60 min, and then harvested for analysis of EGFR, Smad3 and ERK1/2 activation by Western blot analysis. A. TGF-β1, IL-4 and IL-17A failed to show a perceptible impact on EGFR activity. B. TGF-β1 was potent to stimulate Smad3 activation as manifested by the higher levels of pSmad3, while IL-4 and IL-17A failed to induce Smad3 activation. C. A synergic action was observed between TGF-β1, IL-4 and IL-17A on ERK1/2 activity, in which much higher levels of pERK1/2 were detected in cocktail stimulated cells as compared with that of cells stimulated with each cytokine alone.
Given the central role Smad3 played in TGF-β1-mediated EMT [37], we next examined the impact of TGF-β1, IL-4 and IL-17A stimulation on Smad3 activity. As expected, TGF-β1 did not enhance Smad3 expression, but a marked increase of pSmad3 was detected in 16-HBE cells following TGF-β1 stimulation. However, IL-4 and IL-17A alone or in combination failed to show a discernable effect on Smad3 expression or activation. Unexpectedly, the stimulatory effect for TGF-β1 on Smad3 activity disappeared once it combined with IL-4 and IL-17A (Figure 6B). This result prompted us to embark on the ERK1/2 signaling, a alternative pathway recently recognized in TGF-β1-mediated EMT [38]. Surprisingly, TGF-β1 alone did not dispaly a significant impact on ERK1/2 activity as manifested by the very mild increase of pERK1/2 after 5 or 60 min of TGF-β1 stimulation. In contrast, the potency was much higher for either IL-4 or IL-17A in terms of inducing ERK1/2 activation as compared with that of TGF-β1. It is worthy of note that a synergic action was noted when 16HBE cells stimulated with combined TGF-β1, IL-4 and IL-17A, in which 16HBE cells showed much higher ERK1/2 activity (pERK1/2) as compared with that of TGF-β1 or IL-4 or IL-17A alone (Figure 6C).
To further confirm the above results, we next treated 16HBE cells with U0126, an ERK1/2 specific inhibitor, before combined TGF-β1, IL-4 and IL-17A (cocktail) stimulation. We first demonstrated that U0126 indeed almost completely abolished cocktail induced ERK1/2 activation (Figure 7A). We then examined the impact of U0126 on cocktail induced expression changes for E-cadherin and a-SMA in 16-HBE cells. In line with our expectation, addition of U0126 almost completely abrogated cocktail induced desrease of E-cadherin expression (Figure 7B), and similarly, U0126 significantly attenuated cocktail induced a-SMA expression in 16-HBE cells (Figure 7C). All together, our data support that IL-4 and IL-17A synergize with TGF-β1 to enhance ERK1/2 activation, and by which they promote 16-HBE cells undergoing EMT.
Figure 7.
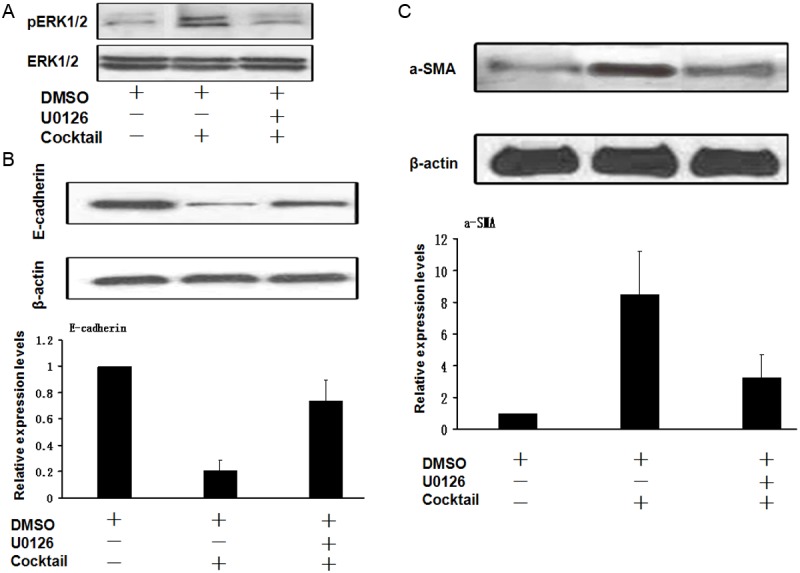
Blockade of ERK1/2 signaling almost completely abrogates the synergic action of TGF-β1, IL-4 and IL-17A on the induction of bronchial EMT. The 16-HBE cells were serum-starved overnight and then stimulated with indicated cytokine or cytokine cocktail in the presence or absence of U0126 for 60 min, followed by Western blot analysis of EMT markers. A. U0126 was potent to attenuate cytokine cocktail induced ERK1/2 activation. B. Pre-treatment of 16-HBE cells almost completely abrogated cytokine cocktail induced decrease of E-cadherin expression. C. U0126 potently attenuated cytokine cocktail induced α-SMA expression in 16-HBE cells.
Discussion
There is mounting evidence that the bronchial epithelium acts as a key player in coordinating airway remodeling. In normal individuals, the intact airway epithelium forms a physical, chemical and immunological barrier between the external environment and the internal milieu against environmental stimuli through cell-cell adhesion proteins such as E-cadherin, α-catenin, and β-catenin. In asthmatic subjects, the bronchial epithelium is exposed to the inhaled allergens, which would alter the properties for the epitehlial barrier. Interestingly, recent studies suggest that the bronchial epithelium displays heterogeneity in mild versus severe asthma. In mild asthma, the epithelium is featured by the disrupted barrier, while severe asthma is manifested by the thickened epithelium. It is believed that the inflammatory microenvironment in the airway is altered in severe asthma as compared with that in mild asthma [12,39]. Indeed, a Th2-polarized inflammatory response is assumed to be predominantly involved in mild asthma, whereas both Th2 and Th17-polarized responses are believed to be implicated in severe asthma [40].
Given that the bronchial epithelium in severe asthmatics is directly exposed to a complex and chronic inflammatory environment, Th2 and Th17 derived cytokines could synergize with TGF-β1 to promote airway remodeling. To address this assumption, we conducted studies in 16-HBE cells to assess the impact of IL-4 and IL-17A on TGF-β1 induced bronchial EMT. It was noted that TGF-β1 or IL-4 or IL-17A alone did not induce 16-HBE cells undergoing a significant proliferation, which is consistent with the studies conducted in primary bronchial epithelial cells [39,41]. However, combined TGF-β1, IL-4 and IL-17A showed potency to induce the removal of epithelial cellular polarity as manifested by the higher proliferation rate (Figure 1), demonstrating that IL-4 and IL-17 could provide a chronic inflammatory milieu that favors TGF-β1 to induce bronchia EMT. Indeed, Th2 and Th17 derived cytokines had been found to play a critical role in airway remodeling by stimulating the growth of bronchial epithelial cells [12,42]. In contrast to our findings, Semlali et al observed an inadequate proliferative response of epithelial cells isolated from subjects with asthma compared with that of normal subjects [39]. It is likely that this discrepancy is caused by the differences of disease states and severity.
Myofibroblasts are considered to be one of the important cellular elements in the ongoing airway remodeling process [43,44]. In line with this notion, the number of myofibroblasts in the layer of asthmatics has been found to be increased. However, the origin of these myofibroblasts in the airway layer is not entirely clear. Recently, a number of studies have demonstrated that the bronchial epithelial cells can contribute to myofibroblast pool by EMT, which provided an example of communication between bronchial epithelial cells and the underlying mesenchymal cells. During this process, downregulation of E-cadherin and acquisition of mesenchymal marker a-SMA are important components of EMT. In the present report, we demonstrated evidence indicating that 16-HBE cells undergo a typical morphological change from the classic cobble-stone shape to the spindle-shape, fibroblast-like morphology after 72 h of TGF-β1 stimulation (Figure 2). Interestingly, although IL-4 and IL-17A alone failed to stimulate a morphological change for 16-HBE cells, but they have significantly enhanced the capacity of TGF-β1 to induce such a morphological switch, which further supports the existence of a synergic action between TGF-β1, IL-4 and IL-17A in terms of induction of bronchial EMT. The most striking evidence is probably come from the following studies, in which both IL-4 and IL-17A alone or in combination failed to induce an obvious downregulation of E-cadherin or upregulation of α-SMA in 16-HBE cells. Remarkably, once IL-4 and IL-17A formed a cocktail with TGF-β1, this cytokine cocktail displayed high potency to suppress E-cadherin expression and to induce α-SMA expression in 16-HBE cells (Figures 3 and 4). Suppression of E-cadherin in epithelial cells would result in the reduction of cell-cell contact, the increase of cellular permeability to allergens, and the enhancement of susceptibility to injury. In contrast, induction of α-SMA expression in epithelial cells would provide a source of increased smooth muscle mass in the asthmatic airway [45]. These events are typical features of bronchial EMT, which would be in favor of airway remodeling associated with disease progression and severity. Given that TGF-β1 alone displayed much lower potency as compared with that of the above cytokine cocktail, our data further support that IL-4 and IL-17A may provide a chronic inflammatory milieu to favor TGF-β1 induction of bronchia EMT. Of note, the mRNA changes following stimulation were much more significantly than that of protein levels, this discrepancy is likely caused by the fact that EMT is a chronic and dynamic process, and a single time point may not reflect the entire disease process.
Interestingly, we failed to detect a synergic action between TGF-β1 and IL-4/IL-17A on PINP secretion as manifested by that TGF-β1 combined with either IL-4 or IL-17A or both did not further enhance 16-HBE cells secretion of PINP. In contrast, the presence of IL-4 and/or IL-17A significantly impaired TGF-β1 induction of PINP secretion. The reason for this unexpected observation is currently unknown. Given that subjects with severe asthma do not display an obvious fibrosis in the airway, our data suggest that procollagen deposition may not serve as a predominant risk factor prediposing to airway remodelng in severe asthma.
We also conducted studies to explore the mechanisms underlying the synergic action of TGF-β1, IL-4 and IL-17A cocktail in the induction of bronchial EMT. It has been noted that EGFR signaling stimulates breast cancer cells undergoing a transition from an epithelial to a spindle-like mesenchymal morphology, which is accompanied by the reduced expression of E-cadherin and increased expression of the mesenchymal proteins vimentin and TWIST [46]. Indeed, upon autocrine or paracrine ligand-mediated activation, EGFR induces EMT to promote cancer metastasis [47]. Unexpectedly, we failed to detect a discernable impact for TGF-β1, IL-4 and IL-17A alone or in combination on EGFR activity (Figure 6A). This discrepancy is probably due to the differences of disease etiology between cancer and asthma, in which cancer is manifested by the nonresolving inflammation. Given the role of Smad3 played in transducing signals for TGF-β1, we next assumed that IL-4 and IL-17A may coordinate with TGF-β1 to promote bronchial EMT. In contrast to our assumption, addition of IL-4 and/or IL-17 into the 16-HBE culture did not further enhance TGF-β1 induction of Smad3 activity (Figure 6B). We, therefore, next embarked on ERK1/2 as their activity has been considered to be a prerequisite for TGF-β1 induction of EMT [48]. Unlike EGFR and Smad3, enhanced ERK1/2 activity can be consistently detected in 16-HBE cells after IL-4 or IL-17A or TGF-β1 stimulation. As a result, the IL-4, IL-17A and TGF-β1 cocktail showed a synergic action in ERK1/2 activity (Figure 6C). Furthermore, treatment of 16-HBE cells with U0126, an ERK1/2 inhibitor, almost completely abolished IL-4, IL-17A and TGF-β1 cocktail induced epithelial to mesenchymal transition (Figure 7). Together, our data support that IL-4 and IL-17A synergize with TGF-β1 to enhance ERK1/2 activity, and by which they promote bronchia EMT during the course of severe asthma.
In summary, we provided the first evidence supporting that Th2 and Th17 derived cytokines, IL-4 and IL-17A, synergize with TGF-β1 to promote airway remodeling during the course of severe asthma, in which IL-4 and IL-17A provide a chronic inflammatory milieu that favors TGF-β1 to induce bronchial EMT. This synergic action is coordinated by the regulation of ERK1/2 activity. Our results not only provide novel information for better understanding of the mechanisms underlying airway remodeling in asthmatic condition, but also have the potential for developing more effective therapeutic strategies against severe asthmatics in clinical settings.
Acknowledgements
This work was supported by the 2011 Graduate Student Innovative Project of Hunan Province (CX2011B070), the Department of Science and Technology of Hunan Province (2010FJ2004), the National Natural Science Foundation of China (81130014), and the European Foundation for the Study of Diabetes (EFSD)/Chinese Diabetes Society (CDS)/Lilly Program for Collaborative Diabetes Research between China and Europe.
Disclosure of conflict of interest
The authors declare no competing financial interests.
References
- 1.Cho SH, Stanciu LA, Holgate ST, Johnston SL. Increased interleukin-4, interleukin-5, and interferon-gamma in airway CD4+ and CD8+ T cells in atopic asthma. Am J Respir Crit Care Med. 2005;171:224–230. doi: 10.1164/rccm.200310-1416OC. [DOI] [PubMed] [Google Scholar]
- 2.Doe C, Bafadhel M, Siddiqui S, Desai D, Mistry V, Rugman P, McCormick M, Woods J, May R, Sleeman MA, Anderson IK, Brightling CE. Expression of the T helper 17-associated cytokines IL-17A and IL-17F in asthma and COPD. Chest. 2010;138:1140–1147. doi: 10.1378/chest.09-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kiwamoto T, Ishii Y, Morishima Y, Yoh K, Maeda A, Ishizaki K, Iizuka T, Hegab AE, Matsuno Y, Homma S, Nomura A, Sakamoto T, Takahashi S, Sekizawa K. Transcription factors T-bet and GATA-3 regulate development of airway remodeling. Am J Respir Crit Care Med. 2006;174:142–151. doi: 10.1164/rccm.200601-079OC. [DOI] [PubMed] [Google Scholar]
- 4.Wang Q, Li H, Yao Y, Xia D, Zhou J. The overexpression of heparin-binding epidermal growth factor is responsible for Th17-induced airway remodeling in an experimental asthma model. J Immunol. 2010;185:834–841. doi: 10.4049/jimmunol.0901490. [DOI] [PubMed] [Google Scholar]
- 5.Zhao Y, Yang J, Gao YD, Guo W. Th17 immunity in patients with allergic asthma. Int Arch Allergy Immunol. 2010;151:297–307. doi: 10.1159/000250438. [DOI] [PubMed] [Google Scholar]
- 6.Bai TR. Evidence for airway remodeling in chronic asthma. Curr Opin Allergy Clin Immunol. 2010;10:82–86. doi: 10.1097/ACI.0b013e32833363b2. [DOI] [PubMed] [Google Scholar]
- 7.Homer RJ, Elias JA. Consequences of long-term inflammation. Airway remodeling. Clin Chest Med. 2000;21:331–343. ix. doi: 10.1016/s0272-5231(05)70270-7. [DOI] [PubMed] [Google Scholar]
- 8.Leigh R, Ellis R, Wattie JN, Hirota JA, Matthaei KI, Foster PS, O’Byrne PM, Inman MD. Type 2 cytokines in the pathogenesis of sustained airway dysfunction and airway remodeling in mice. Am J Respir Crit Care Med. 2004;169:860–867. doi: 10.1164/rccm.200305-706OC. [DOI] [PubMed] [Google Scholar]
- 9.Bossley CJ, Fleming L, Gupta A, Regamey N, Frith J, Oates T, Tsartsali L, Lloyd CM, Bush A, Saglani S. Pediatric severe asthma is characterized by eosinophilia and remodeling without T(H)2 cytokines. J Allergy Clin Immunol. 2012;129:974–982. e913. doi: 10.1016/j.jaci.2012.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopez-Guisa JM, Powers C, File D, Cochrane E, Jimenez N, Debley JS. Airway epithelial cells from asthmatic children differentially express proremodeling factors. J Allergy Clin Immunol. 2012;129:990–997. e996. doi: 10.1016/j.jaci.2011.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michalik M, Pierzchalska M, Wlodarczyk A, Wojcik KA, Czyz J, Sanak M, Madeja Z. Transition of asthmatic bronchial fibroblasts to myofibroblasts is inhibited by cell-cell contacts. Respir Med. 2011;105:1467–1475. doi: 10.1016/j.rmed.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 12.Cohen L, E X, Tarsi J, Ramkumar T, Horiuchi TK, Cochran R, DeMartino S, Schechtman KB, Hussain I, Holtzman MJ, Castro M NHLBI Severe Asthma Research Program (SARP) Epithelial cell proliferation contributes to airway remodeling in severe asthma. Am J Respir Crit Care Med. 2007;176:138–145. doi: 10.1164/rccm.200607-1062OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heijink IH, Postma DS, Noordhoek JA, Broekema M, Kapus A. House dust mite-promoted epithelial-to-mesenchymal transition in human bronchial epithelium. Am J Respir Cell Mol Biol. 2010;42:69–79. doi: 10.1165/rcmb.2008-0449OC. [DOI] [PubMed] [Google Scholar]
- 14.Trautmann A, Kruger K, Akdis M, Muller-Wening D, Akkaya A, Brocker EB, Blaser K, Akdis CA. Apoptosis and loss of adhesion of bronchial epithelial cells in asthma. Int Arch Allergy Immunol. 2005;138:142–150. doi: 10.1159/000088436. [DOI] [PubMed] [Google Scholar]
- 15.Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119:1438–1449. doi: 10.1172/JCI38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429–1437. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masszi A, Di Ciano C, Sirokmany G, Arthur WT, Rotstein OD, Wang J, McCulloch CA, Rosivall L, Mucsi I, Kapus A. Central role for Rho in TGF-beta1-induced alpha-smooth muscle actin expression during epithelial-mesenchymal transition. Am J Physiol Renal Physiol. 2003;284:F911–924. doi: 10.1152/ajprenal.00183.2002. [DOI] [PubMed] [Google Scholar]
- 18.Doerner AM, Zuraw BL. TGF-beta1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL-1beta but not abrogated by corticosteroids. Respir Res. 2009;10:100. doi: 10.1186/1465-9921-10-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Molet S, Hamid Q, Davoine F, Nutku E, Taha R, Page N, Olivenstein R, Elias J, Chakir J. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol. 2001;108:430–438. doi: 10.1067/mai.2001.117929. [DOI] [PubMed] [Google Scholar]
- 20.Redington AE, Madden J, Frew AJ, Djukanovic R, Roche WR, Holgate ST, Howarth PH. Transforming growth factor-beta 1 in asthma. Measurement in bronchoalveolar lavage fluid. Am J Respir Crit Care Med. 1997;156:642–647. doi: 10.1164/ajrccm.156.2.9605065. [DOI] [PubMed] [Google Scholar]
- 21.Walker C, Bauer W, Braun RK, Menz G, Braun P, Schwarz F, Hansel TT, Villiger B. Activated T cells and cytokines in bronchoalveolar lavages from patients with various lung diseases associated with eosinophilia. Am J Respir Crit Care Med. 1994;150:1038–1048. doi: 10.1164/ajrccm.150.4.7921434. [DOI] [PubMed] [Google Scholar]
- 22.Gal A, Sjoblom T, Fedorova L, Imreh S, Beug H, Moustakas A. Sustained TGF beta exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis. Oncogene. 2008;27:1218–1230. doi: 10.1038/sj.onc.1210741. [DOI] [PubMed] [Google Scholar]
- 23.Hackett TL, Warner SM, Stefanowicz D, Shaheen F, Pechkovsky DV, Murray LA, Argentieri R, Kicic A, Stick SM, Bai TR, Knight DA. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1. Am J Respir Crit Care Med. 2009;180:122–133. doi: 10.1164/rccm.200811-1730OC. [DOI] [PubMed] [Google Scholar]
- 24.Kojima T, Takano K, Yamamoto T, Murata M, Son S, Imamura M, Yamaguchi H, Osanai M, Chiba H, Himi T, Sawada N. Transforming growth factor-beta induces epithelial to mesenchymal transition by down-regulation of claudin-1 expression and the fence function in adult rat hepatocytes. Liver Int. 2008;28:534–545. doi: 10.1111/j.1478-3231.2007.01631.x. [DOI] [PubMed] [Google Scholar]
- 25.Nicolas FJ, Lehmann K, Warne PH, Hill CS, Downward J. Epithelial to mesenchymal transition in Madin-Darby canine kidney cells is accompanied by down-regulation of Smad3 expression, leading to resistance to transforming growth factor-beta-induced growth arrest. J Biol Chem. 2003;278:3251–3256. doi: 10.1074/jbc.M209019200. [DOI] [PubMed] [Google Scholar]
- 26.Sheppard D. Transforming growth factor beta: a central modulator of pulmonary and airway inflammation and fibrosis. Proc Am Thorac Soc. 2006;3:413–417. doi: 10.1513/pats.200601-008AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson JR, Roos A, Berg T, Nord M, Fuxe J. Chronic respiratory aeroallergen exposure in mice induces epithelial-mesenchymal transition in the large airways. PLoS One. 2011;6:e16175. doi: 10.1371/journal.pone.0016175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buckley ST, Medina C, Ehrhardt C. Differential susceptibility to epithelial-mesenchymal transition (EMT) of alveolar, bronchial and intestinal epithelial cells in vitro and the effect of angiotensin II receptor inhibition. Cell Tissue Res. 2010;342:39–51. doi: 10.1007/s00441-010-1029-x. [DOI] [PubMed] [Google Scholar]
- 29.Yang J, Liu Y. Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am J Pathol. 2001;159:1465–1475. doi: 10.1016/S0002-9440(10)62533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahn JH, Kim CH, Kim YH, Kim SJ, Lee SY, Kim YK, Kim KH, Moon HS, Song JS, Park SH, Kwon SS. Inflammatory and remodeling events in asthma with chronic exposure to house dust mites: a murine model. J Korean Med Sci. 2007;22:1026–1033. doi: 10.3346/jkms.2007.22.6.1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang S, Lv JW, Yang P, Yu Q, Pang J, Wang Z, Guo H, Liu S, Hu J, Li J, Leng J, Huang Y, Ye Z, Wang CY. Loss of dicer exacerbates cyclophosphamide-induced bladder overactivity by enhancing purinergic signaling. Am J Pathol. 2012;181:937–946. doi: 10.1016/j.ajpath.2012.05.035. [DOI] [PubMed] [Google Scholar]
- 32.Rao X, Zhong J, Zhang S, Zhang Y, Yu Q, Yang P, Wang MH, Fulton DJ, Shi H, Dong Z, Wang D, Wang CY. Loss of methyl-CpG-binding domain protein 2 enhances endothelial angiogenesis and protects mice against hind-limb ischemic injury. Circulation. 2011;123:2964–2974. doi: 10.1161/CIRCULATIONAHA.110.966408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhong J, Yang P, Muta K, Dong R, Marrero M, Gong F, Wang CY. Loss of Jak2 selectively suppresses DC-mediated innate immune response and protects mice from lethal dose of LPS-induced septic shock. PLoS One. 2010;5:e9593. doi: 10.1371/journal.pone.0009593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heijink IH, Marcel Kies P, van Oosterhout AJ, Postma DS, Kauffman HF, Vellenga E. Der p, IL-4, and TGF-beta cooperatively induce EGFR-dependent TARC expression in airway epithelium. Am J Respir Cell Mol Biol. 2007;36:351–359. doi: 10.1165/rcmb.2006-0160OC. [DOI] [PubMed] [Google Scholar]
- 35.Ip WK, Wong CK, Lam CW. Interleukin (IL)-4 and IL-13 up-regulate monocyte chemoattractant protein-1 expression in human bronchial epithelial cells: involvement of p38 mitogen-activated protein kinase, extracellular signal-regulated kinase 1/2 and Janus kinase-2 but not c-Jun NH2-terminal kinase 1/2 signalling pathways. Clin Exp Immunol. 2006;145:162–172. doi: 10.1111/j.1365-2249.2006.03085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zijlstra GJ, Ten Hacken NH, Hoffmann RF, van Oosterhout AJ, Heijink IH. Interleukin-17A induces glucocorticoid insensitivity in human bronchial epithelial cells. Eur Respir J. 2012;39:439–445. doi: 10.1183/09031936.00017911. [DOI] [PubMed] [Google Scholar]
- 37.Roberts AB, Tian F, Byfield SD, Stuelten C, Ooshima A, Saika S, Flanders KC. Smad3 is key to TGF-beta-mediated epithelial-to-mesenchymal transition, fibrosis, tumor suppression and metastasis. Cytokine Growth Factor Rev. 2006;17:19–27. doi: 10.1016/j.cytogfr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 38.Davies M, Robinson M, Smith E, Huntley S, Prime S, Paterson I. Induction of an epithelial to mesenchymal transition in human immortal and malignant keratinocytes by TGF-beta1 involves MAPK, Smad and AP-1 signalling pathways. J Cell Biochem. 2005;95:918–931. doi: 10.1002/jcb.20458. [DOI] [PubMed] [Google Scholar]
- 39.Semlali A, Jacques E, Plante S, Biardel S, Milot J, Laviolette M, Boulet LP, Chakir J. TGF-beta suppresses EGF-induced MAPK signaling and proliferation in asthmatic epithelial cells. Am J Respir Cell Mol Biol. 2008;38:202–208. doi: 10.1165/rcmb.2007-0031OC. [DOI] [PubMed] [Google Scholar]
- 40.Aujla SJ, Alcorn JF. T(H)17 cells in asthma and inflammation. Biochim Biophys Acta. 2011;1810:1066–1079. doi: 10.1016/j.bbagen.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 41.Inoue D, Numasaki M, Watanabe M, Kubo H, Sasaki T, Yasuda H, Yamaya M, Sasaki H. IL-17A promotes the growth of airway epithelial cells through ERK-dependent signaling pathway. Biochem Biophys Res Commun. 2006;347:852–858. doi: 10.1016/j.bbrc.2006.06.137. [DOI] [PubMed] [Google Scholar]
- 42.Ricciardolo FL, Di Stefano A, van Krieken JH, Sont JK, van Schadewijk A, Rabe KF, Donner CF, Hiemstra PS, Sterk PJ, Mauad T. Proliferation and inflammation in bronchial epithelium after allergen in atopic asthmatics. Clin Exp Allergy. 2003;33:905–911. doi: 10.1046/j.1365-2222.2003.01686.x. [DOI] [PubMed] [Google Scholar]
- 43.Cokugras H, Akcakaya N, Seckin, Camcioglu Y, Sarimurat N. Ultrastructural examination of bronchial biopsy specimens from children with moderate asthma. Thorax. 2001;56:25–29. doi: 10.1136/thorax.56.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Payne DN, Rogers AV, Adelroth E, Bandi V, Guntupalli KK, Bush A, Jeffery PK. Early thickening of the reticular basement membrane in children with difficult asthma. Am J Respir Crit Care Med. 2003;167:78–82. doi: 10.1164/rccm.200205-414OC. [DOI] [PubMed] [Google Scholar]
- 45.Zhang M, Zhang Z, Pan HY, Wang DX, Deng ZT, Ye XL. TGF-beta1 induces human bronchial epithelial cell-to-mesenchymal transition in vitro. Lung. 2009;187:187–194. doi: 10.1007/s00408-009-9139-5. [DOI] [PubMed] [Google Scholar]
- 46.Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei Y, Abbruzzese JL, Hortobagyi GN, Hung MC. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007;67:9066–9076. doi: 10.1158/0008-5472.CAN-07-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holz C, Niehr F, Boyko M, Hristozova T, Distel L, Budach V, Tinhofer I. Epithelial-mesenchymal-transition induced by EGFR activation interferes with cell migration and response to irradiation and cetuximab in head and neck cancer cells. Radiother Oncol. 2011;101:158–164. doi: 10.1016/j.radonc.2011.05.042. [DOI] [PubMed] [Google Scholar]
- 48.Hamilton LM, Puddicombe SM, Dearman RJ, Kimber I, Sandstrom T, Wallin A, Howarth PH, Holgate ST, Wilson SJ, Davies DE. Altered protein tyrosine phosphorylation in asthmatic bronchial epithelium. Eur Respir J. 2005;25:978–985. doi: 10.1183/09031936.05.00098604. [DOI] [PubMed] [Google Scholar]