

Abstract
This study addresses how depletion of human cardiac left ventricle (LV) mitochondrial DNA (mtDNA) and epigenetic nuclear DNA methylation promote cardiac dysfunction in human dilated cardiomyopathy (DCM) through regulation of pyrimidine nucleotide kinases. Samples of DCM LV and right ventricle (n = 18) were obtained fresh at heart transplant surgery. Parallel samples from nonfailing (NF) controls (n = 12) were from donor hearts found unsuitable for clinical use. We analyzed abundance of mtDNA and nuclear DNA (nDNA) using qPCR. LV mtDNA was depleted in DCM (50%, P < 0.05 each) compared with NF. No detectable change in RV mtDNA abundance occurred. DNA methylation and gene expression were determined using microarray analysis (GEO accession number: GSE43435). Fifty-seven gene promoters exhibited DNA hypermethylation or hypomethylation in DCM LVs. Among those, cytosolic thymidine kinase 1 (TK1) was hypermethylated. Expression arrays revealed decreased abundance of the TK1 mRNA transcript with no change in transcripts for other relevant thymidine metabolism enzymes. Quantitative immunoblots confirmed decreased TK1 polypeptide steady state abundance. TK1 activity remained unchanged in DCM samples while mitochondrial thymidine kinase (TK2) activity was significantly reduced. Compensatory TK activity was found in cardiac myocytes in the DCM LV. Diminished TK2 activity is mechanistically important to reduced mtDNA abundance and identified in DCM LV samples here. Epigenetic and genetic changes result in changes in mtDNA and in nucleotide substrates for mtDNA replication and underpin energy starvation in DCM.
Keywords: dilated cardiomyopathy, mitochondria, mitochondrial DNA, thymidine kinase
cardiomyopathy (cm), a prominent cause of heart failure, may be defined as a primary weakness of the myocardium, impaired myocardial perfusion, or an infiltrative process. CM is functionally classified as ischemic (I)CM (poor tissue perfusion from myocardial infarction) and nonischemic (D)CM (toxic, metabolic, infectious, or idiopathic etiologies) (28). Both ICM and DCM lead to heart failure with a poor prognosis (14). Compared with the normal heart, CM is likened to an “energy-starved engine” where the mitochondrially derived “fuel” of high energy phosphate is depleted (1, 13, 30, 41).
Mitochondria are critical to myocardial energetics (16, 27). Mitochondrial structural and functional abnormalities occur in CM, and reciprocally, genetic diseases of mitochondria include CM as a clinical component (2, 5, 32). Oxidative phosphorylation (OXPHOS) provides ATP for the myocardium by transferring electrons through the electron transport chain (ETC). Nuclear DNA encodes 80% of ETC polypeptides, while only 13 are encoded by mtDNA, making those encoded by mtDNA critical and potentially limiting in energy production. Therefore, preservation of mtDNA abundance and mtDNA genetic integrity are critical in providing the enzymatic machinery necessary for normal cardiac energetics.
Deoxynucleosides and their phosphorylated products serve as the building blocks of mtDNA. They are phosphorylated to deoxynucleoside monophosphates by deoxynucleoside kinases (reviewed in Refs. 3, 42). Deoxythymidine (dThd) phosphorylation is accomplished cytoplasmically by thymidine kinase 1 (TK1) in the cytosol of dividing cells. Another TK isoform, TK2 (mitochondrial), exhibits high kinase activity in heart, whereas cardiac TK1 activity is reported to be low (15, 29).
Genetic evidence serves as proof of concept for the “energy starvation hypothesis” since mtDNA depletion is found in genetic diseases of mtDNA in which energy depletion is an important part (8, 36–39). The mitochondrial dNTP pool is pathogenetically important in genetic mtDNA depletion syndrome (MIM 251880, a heterogeneous group of inherited diseases sharing depletion of mtDNA and organ failure). Alterations in nucleotide pools, specifically dThd nucleotides, can elicit mtDNA depletion and altered contractility. Two separate mutations in human TK2 (Hisl21Asn, Ile212Asn) with different clinical severity were individually associated with infantile myopathy and mtDNA depletion in muscle (40).
We embrace the hypothesis that energy starvation is a pathophysiological underpinning of CM in the left ventricle (LV) and promotes the CM phenotype via depletion of mtDNA. While data exist to support some events in energy starvation in DCM, the hypothesis is difficult to test rigorously since nonfailing (NF) control human heart samples are rare. As a unique feature, the present study provides such controls in the form of samples from NF hearts deemed unsuitable as donor organs for heart transplant recipients. This study uses molecular and biochemical studies to compare DCM and NF anatomically matched samples.
Decreased mtDNA abundance was found in the DCM LV. In contrast, right ventricle (RV) mtDNA abundance was unchanged. Gene promoter microarrays coupled with mRNA microarrays identified unique gene signatures of DCM. The promoter of the cytosolic TK1 was hypermethylated in DCM, with a corresponding decrease in its mRNA transcript. Enzyme assays reveal reduced TK2 activity in DCM. These results provide novel insights into the role of epigenetic control via DNA methylation of TK gene promoters, effects on their mRNA transcription, and TK total and isoform activity in DCM.
MATERIALS AND METHODS
Human heart samples.
Adult human heart samples from DCM LVs (n = 18) were obtained fresh from surgically removed native hearts at Emory University in accordance with Institutional Review Board protocols. Samples from 12 adult human NF controls were obtained from Loyola University Health System's Cardiovascular Institute Tissue Repository and from the Gift of Hope Organ and Tissue Donor Network. The investigation conformed to the principles outlined in the Declaration of Helsinki. Other details of the sample procedures are included in the accompanying paper.
mtDNA abundance.
Methods utilized are similar to those described previously (26). DNA sequences for primers and probes used for quantitation of mitochondrial and nuclear DNA analyzed the COX I gene of the mtDNA (forward primer, 5′-TTC GCC GAC CGT TGA CTA TT-3′; reverse primer, 5′-AAG ATT ATT ACA AAT GCA TGG GC-3′) and the POLG2 gene of the nuclear DNA (forward primer, 5′-GAG CTG TTG ACG GAA AGG AG-3′; reverse primer, 5′-CAG AAG AGA ATC CCG GCT AA-3′). Amplification was performed using the Lightcycler 480 system (Roche, Indianapolis, IN).
DNA methylation.
DNA was extracted as previously described with a MagNAPure DNA Extraction System (Roche) (10). Total cellular DNA from 10 NF and 10 DCM samples was quantitated and diluted in 10 mM Tris·HCl, pH 8.5 at a final concentration of 30 ng/μl. The DNA was sonicated to obtain an average fragment size of 200–500 bp. A sample of DNA was set aside for later normalization (denoted “input”), and then a portion of the sonicated DNA was enriched using the MethylCollector Ultra kit (Active Motif, Carlsbad, CA) following the manufacturer's directions. Enriched DNA was subsequently cleaned, concentrated, and denoted as “methylated.” Both the methylated and input DNA were amplified by whole genome amplification (Sigma-Aldrich, St. Louis, MO). The amplified DNA was cleaned and verified for enrichment of methylated DNA with the provided PCR primers (Xist and GAPDH) in the MethylCollector Ultra Kit.
For DNA methylation analysis, Roche Nimblegen 2.1M Deluxe Promoter Arrays were utilized (Roche). Following the manufacturer's instructions, the DNA was fluorescently labeled and hybridized to arrays overnight at 42°C. Arrays were washed and then scanned on a Roche Nimblegen MS200 scanner. Images were analyzed by Nimblescan software as directed by the manufacturer (which included normalizing to the input DNA), resulting in a both log2 ratio values of methylated DNA compared with input for each probe and a final analysis utilizing a nonparametric, one-sided Kolmogorov-Smirnov (KS) test to determine a −log10 peak P value of the detected methylated DNA peaks (GEO accession number: GSE43435). Results were annotated to the gene locations.
Identification of differentially methylated genes.
The processed data files from Nimblegen with ratio of the methylated DNA sample to the input (total DNA) sample for each DNA set relative to the peaks within promoter regions were used for analysis. A total of 19,156 unique genes were represented by at least one peak in any of the samples, and these genes were used to generate an m*n matrix, where m = 19,156 genes and n = 20 samples, 10 from each group. A score of 0 was assigned if a gene was not found enriched in a sample. An average relative score was used for genes represented by more than one peak.
The data were transformed by using a log10(x + 1) transformation, where x is the matrix representing number of peaks uniquely mapping to a gene promoter. A two-stage gene selection process was used next to identify differentially methylated genes. The Bioconductor software for R was used for statistical analyses. In stage 1, differentially expressed genes were identified using two-sample Welch test, two-sample Wilcoxon test, and limma at false discovery rate (FDR) adjusted significance level of 0.05. Operationally, a gene was classified as being differentially methylated if it was selected by any one of the three methods. This approach enabled us to identify 131 differentially methylated gene promoters. In stage 2, a binary particle swarm optimization (PSO) algorithm combined with DAMIP was used to identify genes that displayed changes in promoter methylation (11, 12, 24, 34). We reported the results with 100% 10-fold cross validation accuracy for both DCM and NF groups. Stage 2 analysis identified 57 differentially methylated gene promoters.
mRNA expression arrays.
RNA was extracted from 10 NF and 10 DCM human tissues with the Qiagen Fibrous Tissue RNeasy kit (Qiagen, Valencia, CA). RNA was analyzed on a gel to ensure high-quality RNA extraction and subsequently was quantitated. Up to 10 μg of total RNA were used to synthesize double-stranded cDNA with the SuperScript Double-Stranded cDNA Synthesis Kit (Invitrogen/Life Technologies, Grand Island, NY). cDNA was then labeled with Cy3 and hybridized to a 12 × 135 kb human expression array (Roche) overnight at 42°C. Expression arrays were washed and subsequently scanned on the Roche Nimblegen MS200 scanner. Images were analyzed by Nimblescan software as directed by the manufacturer, including RMA normalization and generation of expression data (GEO accession number: GSE43435). All NF and DCM samples were averaged together in their respective sets, and outliers were determined by a Grubbs test. Only those expression results relating to thymidine de novo or salvage pathways were analyzed. Means were generated for each cDNA analyzed, and a P value was determined between the NF and DCM samples for each cDNA BY a Student's t-test. Significance was determined as those cDNAs exhibiting a greater than twofold change compared with NF and a P < 0.05.
Immunoblots.
Approximately 50 mg of heart tissue was placed in 1 ml RIPA buffer (Sigma Aldrich) with protease inhibitor cocktail (Sigma Aldrich). A steel ball bearing was placed in the tube, and the samples were lysed for 2 min at 30 Hz using a TissueLyser (Qiagen). Lysates were centrifuged at 1,000 g for 5 min, and the supernatant was transferred to new tubes and saved as the tissue lysate.
Lysates were analyzed with SDS-PAGE precast Bio-Rad AnyKD gels (Bio-Rad, Hercules, CA). Proteins were transferred to PVDF membranes and processed according to standard procedures. Protein detection utilized chemiluminescent substrate and film. TK1 band intensity was normalized to GAPDH band intensity. TK1 and GAPDH antibodies were obtained from Novus Biologicals (Littleton, CO). A total of seven NF and seven DCM samples were utilized for immunoblot analysis.
TK activity assays.
TK activity was determined as previously described (7, 9). Briefly, heart samples were lysed to preserve enzymatic activity. Assays utilized [3H]methyl-thymidine (Perkin Elmer, Waltham, MA) as the substrate and proceeded for 1 h at 37°C. Total TK activity was determined for NF and DCM samples. TK2 activity was inhibited with the presence of 10 mM deoxycytidine, allowing quantitation of TK1 activity alone. TK2 activity was determined as the difference between total TK activity and TK1 activity. Activity was determined by liquid scintillation. For the activity assay, seven NF and 10 DCM samples were utilized.
Statistical methods.
Statistical methods (not including the DNA methylation array analysis) utilized GraphPad Prism software. A P < 0.05 was determined to be significant in all cases unless otherwise noted. We used a Student's t-test when comparing NF to DCM, and a Grubbs test was used to determine outliers. The sample size is identified for each experimental set.
RESULTS
Human heart samples.
Adult human DCM samples (n = 18) were obtained fresh from surgically removed native hearts at the time of heart transplantation. Samples from 12 adult human NF controls came from donor specimens deemed unsuitable for clinical use. A total of 20 samples were selected for additional analysis, 10 from each group. The mean age (± SE) of NF donors was 53.0 ± 4.3 yr; mean age of DCM donors was 46.1 ± 2.6 years (Table 1, P = not significant). Both sample sets included males and females. Anatomically matched LV and RV tissue cubes (1 cm3) were obtained from each heart, with all samples being treated identically upon collection.
Table 1.
Human patient data
| Heart Sample | Age, yr | Sex | Cause of Death |
|---|---|---|---|
| NF-2 | 70 | M | ICH/Stroke |
| NF-3 | 44 | M | SDH |
| NF-4 | 20 | F | Drug overdose |
| NF-5 | 60 | M | ICH/Stroke |
| NF-6 | 56 | F | SAH |
| NF-7 | 59 | M | CVA |
| NF-9 | 50 | F | ICH/Stroke |
| NF-10 | 56 | M | Drug intoxication, methyl alcohol |
| NF-11 | 63 | M | ICH/Stroke |
| NF-12 | 52 | M | ICH/Stroke |
| Mean Age | 53.0 ± 4.3 | ||
| DCM-1 | 43 | M | |
| DCM-2 | 47 | M | |
| DCM-3 | 60 | M | |
| DCM-4 | 59 | F | |
| DCM-5 | 44 | M | |
| DCM-6 | 42 | M | |
| DCM-7 | 49 | M | |
| DCM-8 | 35 | M | |
| DCM-9 | 44 | F | |
| DCM-10 | 38 | M | |
| Mean Age | 46.1 ± 2.6 |
NF, nonfailing; DCM, dilated cardiomyopathy; M, male; F, female; CVA, cerebrovascular accident; ICH, intracerebral hemorrhage; SAH, subarachnoid hemorrhage; SDH, subdural hemorrhage.
mtDNA abundance.
Reduction of cardiomyocytic energy production leads to cardiac dysfunction and heart failure in mouse models, and its reduction in human DCM required further exploration (22, 25, 26). Clinical data support the role of decreased OXPHOS and ATP production in heart failure (35, 43). We determined mtDNA abundance with quantitative PCR. In human DCM explants, LV mtDNA abundance was decreased by 50% compared with NF (Fig. 1A). This observed decrease in mtDNA abundance was absent in RV samples (Fig. 1B). Together, these results underscore the importance of maintenance of mtDNA abundance in cardiac function and implicate mtDNA depletion in the LV as a potential mechanism for decreased OXPHOS and for LV dysfunction.
Fig. 1.
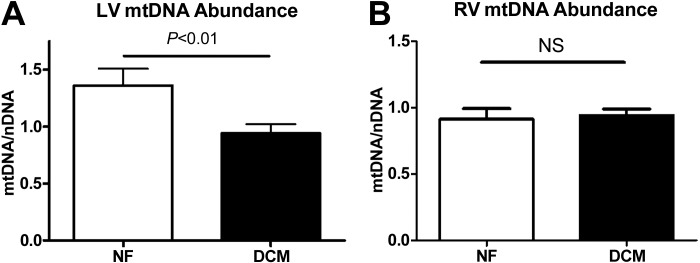
Mitochondrial DNA abundance in dilated cardiomyopathy (DCM). Quantitative PCR was utilized to determine the abundance of mtDNA in nonfailing (NF) and DCM samples. Left ventricle (LV) samples (A) from DCM samples displayed a nearly 50% reduction in mtDNA compared with NF samples. Right ventricle (RV) samples (B) demonstrated no change in mtDNA abundance between NF and DCM samples. NS, not significant.
DNA methylation and predictive analysis.
We examined epigenetic regulation of nuclear gene transcription as a factor contributing to mtDNA depletion in the failing human heart. We utilized a two-stage selection process to determine differentially methylated peaks. In stage 1, all detectable promoter peaks were combined for a list of 19,156 gene promoters as a common reference. Gene promoter selection from statistical and clustering methods revealed 121 hypermethylated and 10 hypomethylated gene promoters in the DCM compared with NF. In stage 2, an in-house feature selection algorithm was utilized, with 10,000 iterations and 10-fold cross-validation. This second stage produced a stringent analysis of the DNA methylation patterns of gene promoters. It has been applied successfully to other disease databases in predictive analyses (34).
Using this two-stage process, we identified 57 gene promoters with changes in DNA methylation (Table 2); 51 displayed hypermethylation in DCM samples, and six displayed hypomethylation. Some of the genes were previously defined as critical for cardiac function (e.g., thromboxane A2 receptor). Others have no known association with cardiac function. Among the latter, TK1 has not been previously implicated in human DCM, and TK1 is expressed at low levels in nonreplicating cells like cardiac myocytes (33). We explored activity of both TK isoforms in DCM LV.
Table 2.
| Gene ID | Name | Gene ID | Name |
|---|---|---|---|
| Hypermethylated Gene Promoters | |||
| 1104 | regulator of chromosome condensation 1; SNHG3-RCC1 | 57576 | kinesin family member 17 |
| 2524 | fucosyltransferase 2 (secretor status included) | 57758 | signal peptide, CUB domain, EGF-like 2 |
| 4837 | nicotinamide N-methyltransferase | 64150 | DIO3OS DIO3 opposite strand/antisense RNA (nonprotein coding) |
| 5020 | oxytocin, prepropeptide | 81027 | tubulin, beta 1 |
| 5879 | ras-related C3 botulinum toxin substrate 1 (Rac1) | 84856 | LOC84856 |
| 6122 | ribosomal protein L3; similar to 60S ribosomal protein L3 (L4) | 92340 | chromosome 17 open reading frame 72 |
| 6614 | sialic acid binding Ig-like lectin 1, sialoadhesin | 93408 | myosin, light chain 10, regulatory |
| 6621 | small nuclear RNA activating complex, polypeptide 4, 190 kDa | 114881 | oxysterol binding protein-like 7 |
| 6663 | SRY (sex determining region Y)-box 10 | 123099 | degenerative spermatocyte homolog 2, lipid desaturase (Drosophila) |
| 6794 | serine/threonine kinase 11 | 124220 | zymogen granule protein 16 homolog B |
| 6915 | thromboxane A2 receptor | 140873 | chromosome 20 open reading frame 173 |
| 7083 | thymidine kinase 1, soluble | 146713 | hexaribonucleotide binding protein 3 |
| 7122 | claudin 5 | 153579 | butyrophilin-like 9 |
| 7409 | vav 1 guanine nucleotide exchange factor | 164656 | transmembrane protease, serine 6 |
| 8740 | tumor necrosis factor (ligand) superfamily, member 14 | 201243 | chromosome 17 open reading frame 74 |
| 8869 | ST3 beta-galactoside alpha-2,3-sialyltransferase 5 | 219537 | smoothelin-like 1 |
| 9212 | aurora kinase B | 246778 | interleukin 27 |
| 9289 | G protein-coupled receptor 56 | 283284 | immunoglobulin superfamily, member 22 |
| 10250 | serine/arginine repetitive matrix 1 | 338773 | transmembrane protein 119 |
| 23541 | SEC14-like 2 (S. cerevisiae) | 339240 | keratin pseudogene |
| 23646 | phospholipase D family, member 3 | 386672 | keratin associated protein 10-4 |
| 23704 | potassium voltage-gated channel, Isk-related family, member 4 | 388468 | POTE ankyrin domain family, member D; POTE ankyrin domain family, member C; POTE ankyrin domain family, member B |
| 26071 | family with sequence similarity 127, member B | 641515 | hypothetical protein LOC641515 |
| 29990 | paired immunoglobin-like type 2 receptor beta | 650662 | C14orf184 |
| 51126 | N-acetyltransferase 5 (GCN5-related, putative) | 730971 | FLJ36777 |
| 57215 | THAP domain containing 11 | ||
| Hypomethylated Gene Promoters | |||
| 1469 | cystatin SN | 54948 | mitochondrial ribosomal protein L16 |
| 9536 | prostaglandin E synthase | 84440 | RAB11 family interacting protein 4 |
| 51699 | vacuolar protein sorting 29 homolog | 283869 | neuropeptide W |
TK1.
Of the 57 differentially methylated gene promoters, mathematical modeling revealed that pyrimidine biosynthesis through TK1 serves as a common thread to changes in nucleotide substrates for mtDNA replication. Previous studies showed alterations in the mitochondrial isoform (TK2) produce cardiac dysfunction and decreased mtDNA abundance (10, 22). Other studies suggest reduced TK1 activity worsens pre-existing deficiencies in TK2 and promote a mitochondrial dysfunction phenotype (7).
DNA methylation changes were found in regions of the TK1 promoter and gene. Log2 ratios of methylated DNA to input DNA were plotted against the probed regions of chromosome 17 where the TK1 gene is located (Fig. 2A). In DCM samples, regions in both the TK1 promoter and the TK1 gene exhibit hypermethylation compared with those regions in NF samples. Certain areas of the TK1 gene and promoter exhibit higher levels of DNA methylation in DCM than NF, including ∼4 kb upstream of the TK1 gene transcription start site and 2 kb following the transcription start site. We analyzed the ratio data for significant peaks in DNA methylation using a sliding-window KS test (Fig. 2B). When we used a peak P value >2, the most significant change was seen 4 kb upstream of the TK1 transcriptional start site, where a significant peak in DCM was seen and no peak was present in NF. The remaining regions also show higher peak P values in DCM compared with NF. These data demonstrate increased DNA methylation in the TK1 promoter in DCM.
Fig. 2.
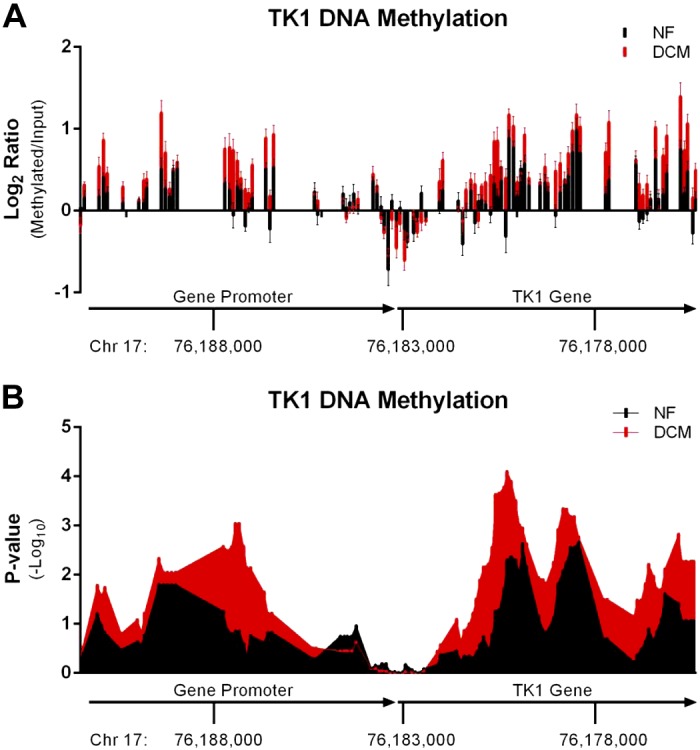
Methylation of the thymidine kinase (TK) 1 gene and promoter. Human LV samples from NF and DCM samples (n = 10) were analyzed for DNA methylation changes in the gene promoter of TK1. NF peak scores are shown in black, while DCM peak scores are shown in red. A: log2 ratio files were determined for each probe in the TK1 promoter and gene. Ratios are plotted along chromosome 17 and show significant increases in DNA methylation 4 kb upstream and 2 kb downstream of the TK1 transcriptional start site. B: array analysis software calculated the peak score at each oligonucleotide probe based on a sliding-window Kolmogorov-Smirnov test. Each peak score was plotted against the position of the oligonucleotide along the TK1 gene and promoter region. Results show a higher level of DNA methylation in DCM samples compared with NF, with the greatest difference seen around 4 kb upstream of the TK1 start site.
Using expression arrays for all 20 samples, we determined changes in expression of TK1 mRNA and compared those to changes in other thymidine-related genes. A 55% reduction in TK1 expression was found in DCM (Table 3). In contrast, expression of other genes related to either de novo dThd synthesis or to dTMP salvage was unchanged. This suggests that modulation of TK1 gene expression does not alter the expression of other dTMP-related genes. TK1 abundance was determined by quantitative immunoblotting. TK1 immunoblots revealed a 32% decrease in TK1 polypeptide abundance that mirrored its RNA expression (Fig. 3). Together, these data indicate that TK1 mRNA and protein modulation follows nuclear DNA methylation of the TK1 gene promoter in DCM LVs.
Table 3.
Fig. 3.

TK1 protein abundance. Immunoblots were analyzed for TK1 protein abundance, using 7 NF and 7 DCM LV samples. TK1 band intensity was normalized to GAPDH band intensity, and relative TK1 protein abundance was determined. A 32% decrease in TK1 protein abundance was observed in the DCM LV compared with NF LV. *P < 0.01, Student's t-test.
TK activity.
To determine if changes in TK1 expression and protein abundance altered thymidine phosphorylation, we assayed TK activity in the cardiac tissue samples. Total TK activity was determined as was the relative isoform contributions of TK1 and TK2. First we normalized TK1 and TK2 activity to total TK. TK1 accounted for 16.5% of the TK activity in NF samples and TK2 accounted for the remainder (Table 4). This ratio of TK isoform activity in DCM changed. TK1 accounted for 24.1% of the total, representing nearly 50% increase of TK1 in DCM LV compared with NF LV controls (P = 0.005). Using a similar comparison for TK2, we found a 9% decrease in the relative activity of TK2 in DCM LV compared with NF (P = 0.005).
Table 4.
Thymidine kinase activity
| Enzyme Activity, pmol·min−1·mg protein−1 |
Percent Total TK Activity |
||||
|---|---|---|---|---|---|
| TK1 | TK2 | TK1/TK | TK2/TK | Ratio of TK2/TK1 | |
| NF | 0.116 ± 0.02 | 0.584 ± 0.07 | 16.5 ± 2.3 | 83.5 ± 2.3 | 5.88 ± 1.1 |
| DCM | 0.126 ± 0.07 | 0.407 ± 0.05 | 24.1 ± 1.5 | 75.9 ± 1.5 | 3.25 ± 0.3 |
TK, thymidine kinase.
Because total TK activity decreased ∼30% in DCM, TK2 activity was compared with TK1 activity directly since TK1 activity was more consistent between DCM and NF samples. On the basis of this approach, we observed a 45% reduction in TK2 activity in DCM LVs compared with corresponding NF controls (P = 0.02, Table 4). These data demonstrate a significant reduction in the TK2/TK1 ratio in DCM. They further suggest that TK1 DNA methylation and its reduced expression may serve as a compensatory mechanism to promote mitochondrial TK activity.
DISCUSSION
It has been suggested for some time that the failing human heart is a transition from normal, to compensated left ventricular hypertrophy, to decompensated failure with LV dilation (4, 6). We combined this fundamental concept with that of energy starvation and hypothesized that human DCM is a cardiac phenotype of defective LV mtDNA replication and ETC with OXPHOS dysfunction. There is an intrinsic limitation to the study because of the relatively few patients that undergo transplantation and fewer donors deemed unacceptable for the procedure. Despite a relatively small sample size here, the homogeneity of the results within the cohorts and significance of differences between the cohorts speak to the robust nature of the findings, which are underpinned by extensive and predictive mathematical studies. The power of these results is further bolstered by the presence of authentic controls in the NF group. This unique resource allowed meaningful comparisons to be made and conclusions to be drawn.
This work utilizes results obtained from our associated study (18). In that work, we describe in more detail the two individual computational analyses utilized to identify differentially methylated gene promoters in DCM. We first employed subtractive analysis to identify DNA methylation peaks detected in eight out of 10 DCM samples with no peaks detected in NF samples. We combined this analysis with gene expression microarray results to identify 158 genes exhibiting differential DNA methylation in the promoters and altered gene expression. In a second analysis, we used a two-stage approach combined a PSO feature selection algorithm and a discriminant analysis via mixed integer programming (DAMIP) classifier to identify differentially methylated gene promoters (23). The first stage used standard statistical approaches that define differentially methylated gene promoters with FDR used as a criterion. The second stage “predicts” important differentially methylated gene promoters by training the support vector machine (DAMIP) to accurately classify NF and DCM. For these experiments, seven NF and seven DCM samples were used to “train” a computer to identify NF and failing hearts. Using the final three NF and three DCM samples, we can determine whether a candidate gene promoter can be used to blindly predict if a sample is NF or DCM. Using this approach, we could use 57 of these genes to accurately classify a human heart sample as NF or DCM. Since the subtractive analysis and DAMIP analysis were independent methods, cross-analysis of the results with the gene expression results identified four candidate genes warranting further studies. TK1 (along with AURKB, CLDN5, and BTNL9) was identified by both methods. This article serves as a molecular study to identify the cause of TK1 promoter methylation.
Findings underscore the importance of LV mtDNA depletion in the DCM heart and document epigenetic regulation of expression of a mitochondrially relevant gene that is intimately involved in mtDNA replication and energy homeostasis. Together, data here support the energy starvation hypothesis in DCM based on depleted mtDNA in the LV, document changes in pyrimidine substrates for mtDNA (i.e., dTTP) necessary for mtDNA replication by pol γ and link them to decompensated heart failure in DCM. Furthermore, they bolster mitochondrial dysfunction as a core feature of decompensated human heart failure in DCM. In our prior studies with transgenic mice, we engineered changes in nucleotide pools in the heart or fostered dysfunction of mtDNA replication at the level of the mitochondrial DNA replicase (pol γ) (10, 19, 21, 22). Those transgenic manipulations resulted in LV mtDNA depletion, oxidative stress, and cardiac dysfunction (10, 20, 21, 23). In the translational human studies here, mtDNA also was found to be depleted in the LV of DCM compared with LV of NF controls. No mtDNA depletion occurred in the corresponding RV. These data from LV complement those from Karamanlidis and colleagues (17), who documented significant mtDNA depletion in the failing LV at similar levels. In that published work, no comparable RV data were included. Together, results underscore mtDNA depletion in DCM exclusively in the LV per se and absence of such depletion in the NF LV or either DCM or NF RV.
In combination with mouse studies by others, findings suggest that the human heart may respond to depletion of mitochondrial TK2 activity by reducing cytosolic TK1 activity. This is accomplished in part by epigenetic modulation through DNA methylation of the promoter of the nuclear encoded TK1 gene. Our current hypothesis is that decreased TK2 activity drives TK1 promoter methylation and expression decline, possibly providing an epigenetic marker of a mitochondrial enzymatic deficiency that leads to downstream energy starvation. In studies performed in mutant TK2 mice, Hirano's group (7) found that heterozygous mutant TK2 +/− mice showed a modest though not significant decrease in TK1 activity. For this heterozygous mouse, TK2 activity decreased by ∼50%. However, homozygous mutant TK2 −/− mice (which have a >80% reduction in TK2 activity) did show a significant decrease in TK1 activity. This work suggests that the amount of TK2 activity decline (and possibly duration of TK2 functional deficit) impacts TK1 activity. For our studies, we observed a 30% decline in TK2 activity in heart samples. Their work and ours suggest interdependence between TK1 and TK2 activity, though how it is controlled is unknown. We hypothesize that DNA methylation is one mechanism, though it is fair to say there might be other mechanisms. The precise mechanism of TK1 gene promoter methylation in heart failure remains to be further clarified, but TK1 expression was the only thymidine-related gene affected in this way. Other pathways of thymidine biosynthesis were unchanged in DCM. On the basis of analyses here, it appears that expression of de novo and thymidine salvage pathway enzymes was not altered (Table 3).
DCM LV tissue responds to a reduction of mitochondrial dThd by altering TK1 expression. It follows that alterations in steady-state thymidine nucleotide pools may result in LV cells in DCM. If lower TK2 activity were the main force operating, reduced dTMP concentration intramitochondrially would result in decreased dTTP. Decreased mtDNA abundance could result from decreased availability of pyrimidine nucleotide substrate dTTP for replication of mtDNA by the pol γ replicase, or increased mutation in nascent mtDNA may result. Data supporting this latter scenario are available from both human genetic diseases and from the genetically engineered mouse studies. Reduced mitochondrial TK2 activity reduces mtDNA abundance in selected tissues (10, 39, 44, 45). It may be possible that mtDNA mutation rates are increased in parallel. Reduction in TK2 enzymatic activity could be a feedback response to excess dTTP and result in reduced mtDNA replication fidelity and further decrease native mtDNA abundance (31, 46). It may be possible that a previously uncharacterized mechanism may account for decreased intramitochondrial dTMP and result in decreased mtDNA replication.
In summary, this study documents the regulatory effects of epigenetic DNA methylation on nuclear gene promoters, resultant changes in TK gene mRNA expression, and downstream alterations in steady-state TK polypeptide abundance in human DCM. These together impact mtDNA abundance by regulating intramitochondrial dTTP as a substrate for pol γ, the mtDNA replicase. Rigorous mathematical analyses of nuclear DNA methylation used here indicate that a core of gene promoters is a common target of enhanced nuclear DNA methylation in the LV of human DCM and these promoters may impact mtDNA replication at the level of substrate for the replicase. Methylation of the TK1 promoter coupled with decreased steady-state abundance of TK1 mRNA and TK1 protein argue for TK1/TK2 events as newly recognized compensatory responses to mtDNA depletion in DCM. These new findings underscore the importance of maintenance of mtDNA replication homeostasis, the crucial nature of mtDNA depletion in the organellar dysfunction, and resultant energy starvation phenotype observed in adult human DCM. They emphasize nuclear epigenetic regulation of mitochondrial defects in energy starvation found in DCM.
GRANTS
Supported by National Institute on Drug Abuse Grant DA-030996 to W. Lewis.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: C.A.K., A.M.S., and W.L. conception and design of research; C.A.K., E.J.F., A.B., S.H., N.P., and A.M.S. performed experiments; C.A.K., E.J.F., A.B., S.H., and E.K.L. analyzed data; C.A.K., R.A.T., E.J.F., E.K.L., and W.L. interpreted results of experiments; C.A.K., R.A.T., A.B., and E.K.L. prepared figures; C.A.K. and W.L. drafted manuscript; C.A.K., R.A.T., and W.L. edited and revised manuscript; C.A.K., R.A.T., and W.L. approved final version of manuscript.
REFERENCES
- 1. Ahmad F, Seidman JG, Seidman CE. The genetic basis for cardiac remodeling. Ann Rev Genom Hum Genet 6: 185–216, 2005 [DOI] [PubMed] [Google Scholar]
- 2. Arbustini E, Diegoli M, Fasani R, Grasso M, Morbini P, Banchieri N, Bellini O, Dal Bello B, Pilotto A, Magrini G, Campana C, Fortina P, Gavazzi A, Narula J, Vigano M. Mitochondrial DNA mutations and mitochondrial abnormalities in dilated cardiomyopathy. Am J Pathol 153: 1501–1510, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arner ES, Eriksson S. Mammalian deoxyribonucleoside kinases. Pharmacol Ther 67: 155–186, 1995 [DOI] [PubMed] [Google Scholar]
- 4. Baandrup U, Olsen EG. Critical analysis of endomyocardial biopsies from patients suspected of having cardiomyopathy. I. Morphological and morphometric aspects. Br Heart J 45: 475–486, 1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bohlega S, Tanji K, Santorelli FM, Hirano M, al-Jishi A, DiMauro S. Multiple mitochondrial DNA deletions associated with autosomal recessive ophthalmoplegia and severe cardiomyopathy. Neurology 46: 1329–1334, 1996 [DOI] [PubMed] [Google Scholar]
- 6. Chien KR, Olson EN. Converging pathways and principles in heart development and disease: CV@CSH. Cell 110: 153–162, 2002 [DOI] [PubMed] [Google Scholar]
- 7. Dorado B, Area E, Akman HO, Hirano M. Onset and organ specificity of Tk2 deficiency depends on Tk1 down-regulation and transcriptional compensation. Hum Mol Genet 20: 155–164, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Elpeleg O. Inherited mitochondrial DNA depletion. Pediatr Res 54: 153–159, 2003 [DOI] [PubMed] [Google Scholar]
- 9. Franzolin E, Rampazzo C, Perez-Perez MJ, Hernandez AI, Balzarini J, Bianchi V. Bromovinyl-deoxyuridine: a selective substrate for mitochondrial thymidine kinase in cell extracts. Biochem Biophys Res Commun 344: 30–36, 2006 [DOI] [PubMed] [Google Scholar]
- 10. Hosseini SH, Kohler JJ, Haase CP, Tioleco N, Stuart T, Keebaugh E, Ludaway T, Russ R, Green E, Long R, Wang L, Eriksson S, Lewis W. Targeted transgenic overexpression of mitochondrial thymidine kinase (TK2) alters mitochondrial DNA (mtDNA) and mitochondrial polypeptide abundance: transgenic TK2, mtDNA, and antiretrovirals. Am J Pathol 170: 865–874, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucl Acid Res 37: 1–13, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protocol 4: 44–57, 2009 [DOI] [PubMed] [Google Scholar]
- 13. Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res 95: 135–145, 2004 [DOI] [PubMed] [Google Scholar]
- 14. Jessup M, Brozena S. Heart failure. N Engl J Med 348: 2007–2018, 2003 [DOI] [PubMed] [Google Scholar]
- 15. Johansson M, Karlsson A. Cloning of the cDNA and chromosome localization of the gene for human thymidine kinase 2. J Biol Chem 272: 8454–8458, 1997 [DOI] [PubMed] [Google Scholar]
- 16. Jullig M, Hickey AJ, Chai CC, Skea GL, Middleditch MJ, Costa S, Choong SY, Philips AR, Cooper GJ. Is the failing heart out of fuel or a worn engine running rich? A study of mitochondria in old spontaneously hypertensive rats. Proteomics 8: 2556–2572, 2008 [DOI] [PubMed] [Google Scholar]
- 17. Karamanlidis G, Nascimben L, Couper GS, Shekar PS, del Monte F, Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res 106: 1541–1548, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koczor CA, Lee EK, Torres RA, Boyd A, Vega JD, Uppal U, Yuan F, Fields EJ, Samarel AM, Lewis W. Detection of differentially methylated gene promoters in failing and nonfailing human left ventricle myocardium using computation analysis. Physiol Genomics 10.1152/physiolgenomics.00013.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kohl NE, Emini EA, Schleif WA, Davis LJ, Heimbach JC, Dixon RA, Scolnick EM, Sigal IS. Active human immunodeficiency virus protease is required for viral infectivity. Proc Natl Acad Sci USA 85: 4686–4690, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kohler JJ, Cucoranu I, Fields E, Green E, He S, Hoying A, Russ R, Abuin A, Johnson D, Hosseini SH, Raper CM, Lewis W. Transgenic mitochondrial superoxide dismutase and mitochondrially targeted catalase prevent antiretroviral-induced oxidative stress and cardiomyopathy. Lab Invest 89: 782–790, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kohler JJ, Hosseini SH, Cucoranu I, Hoying-Brandt A, Green E, Johnson D, Wittich B, Srivastava J, Ivey K, Fields E, Russ R, Raper CM, Santoianni R, Lewis W. Murine cardiac mtDNA: effects of transgenic manipulation of nucleoside phosphorylation. Lab Invest 89: 122–130, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kohler JJ, Hosseini SH, Green E, Hoying-Brandt A, Cucoranu I, Haase CP, Russ R, Srivastava J, Ivey K, Ludaway T, Kapoor V, Abuin A, Shapoval A, Santoianni R, Saada A, Elpeleg O, Lewis W. Cardiac-targeted transgenic mutant mitochondrial enzymes: mtDNA defects, antiretroviral toxicity and cardiomyopathy. Cardiovasc Toxicol 8: 57–69, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kohler JJ, Hosseini SH, Lewis W. Mitochondrial DNA impairment in nucleoside reverse transcriptase inhibitor-associated cardiomyopathy. Chem Res Toxicol 21: 990–996, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee EK. Large-scale optimization-based classification models in medicine and biology. Ann Biomed Eng 35: 1095–1109, 2007 [DOI] [PubMed] [Google Scholar]
- 25. Lemieux H, Semsroth S, Antretter H, Hofer D, Gnaiger E. Mitochondrial respiratory control and early defects of oxidative phosphorylation in the failing human heart. Int J Biochem Cell Biol 43: 1729–1738, 2011 [DOI] [PubMed] [Google Scholar]
- 26. Lewis W, Day BJ, Kohler JJ, Hosseini SH, Chan SS, Green EC, Haase CP, Keebaugh ES, Long R, Ludaway T, Russ R, Steltzer J, Tioleco N, Santoianni R, Copeland WC. Decreased mtDNA, oxidative stress, cardiomyopathy, and death from transgenic cardiac targeted human mutant polymerase gamma. Lab Invest 87: 326–335, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marin-Garcia J, Goldenthal MJ. Mitochondrial centrality in heart failure. Heart Fail Rev 13: 137–150, 2008 [DOI] [PubMed] [Google Scholar]
- 28. Mestroni L, Gilbert EM, Lowes BD, Bristow MR. Dilated cardiomyopathy. In: Hurst's The Heart, edited by Fuster V, O'Rourke RA, Poole-Wilson P, Walsh RA. New York: McGraw Hill, 2008, p. 803–821 [Google Scholar]
- 29. Munch-Petersen B, Cloos L, Tyrsted G, Eriksson S. Diverging substrate specificity of pure human thymidine kinases 1 and 2 against antiviral dideoxynucleosides. J Biol Chem 266: 9032–9038, 1991 [PubMed] [Google Scholar]
- 30. Neubauer S. The failing heart–an engine out of fuel. N Engl J Med 356: 1140–1151, 2007 [DOI] [PubMed] [Google Scholar]
- 31. Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 283: 689–692, 1999 [DOI] [PubMed] [Google Scholar]
- 32. Palmieri L, Alberio S, Pisano I, Lodi T, Meznaric-Petrusa M, Zidar J, Santoro A, Scarcia P, Fontanesi F, Lamantea E, Ferrero I, Zeviani M. Complete loss-of-function of the heart/muscle-specific adenine nucleotide translocator is associated with mitochondrial myopathy and cardiomyopathy. Hum Mol Genet 14: 3079–3088, 2005 [DOI] [PubMed] [Google Scholar]
- 33. Perez-Perez MJ, Hernandez AI, Priego EM, Rodriguez-Barrios F, Gago F, Camarasa MJ, Balzarini J. Mitochondrial thymidine kinase inhibitors. Curr Topic Med Chem 5: 1205–1219, 2005 [DOI] [PubMed] [Google Scholar]
- 34. Querec TD, Akondy RS, Lee EK, Cao W, Nakaya HI, Teuwen D, Pirani A, Gernert K, Deng J, Marzolf B, Kennedy K, Wu H, Bennouna S, Oluoch H, Miller J, Vencio RZ, Mulligan M, Aderem A, Ahmed R, Pulendran B. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat Immunol 10: 116–125, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rosca MG, Hoppel CL. New aspects of impaired mitochondrial function in heart failure. J Bioenerg Biomembr 41: 107–112, 2009 [DOI] [PubMed] [Google Scholar]
- 36. Saada-Reisch A. Deoxyribonucleoside kinases in mitochondrial DNA depletion. Nucleoside Nucleotide Nucl Acid 23: 1205–1215, 2004 [DOI] [PubMed] [Google Scholar]
- 37. Saada A. Deoxyribonucleotides and disorders of mitochondrial DNA integrity. DNA Cell Biol 23: 797–806, 2004 [DOI] [PubMed] [Google Scholar]
- 38. Saada A, Ben-Shalom E, Zyslin R, Miller C, Mandel H, Elpeleg O. Mitochondrial deoxyribonucleoside triphosphate pools in thymidine kinase 2 deficiency. Biochem Biophys Res Commun 310: 963–966, 2003 [DOI] [PubMed] [Google Scholar]
- 39. Saada A, Shaag A, Elpeleg O. mtDNA depletion myopathy: elucidation of the tissue specificity in the mitochondrial thymidine kinase (TK2) deficiency. Mol Genet Metab 79: 1–5, 2003 [DOI] [PubMed] [Google Scholar]
- 40. Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet 29: 342–344, 2001 [DOI] [PubMed] [Google Scholar]
- 41. Samarel A, Walsh RW. Molecular and cellular biology of the normal, hypertrophied and failing heart. In: Hurst's The Heart, edited by Fuster V, O'Rourke RA, Poole-Wilson P, Walsh RA. New York: McGraw Hill, 2008, p. 123–134 [Google Scholar]
- 42. Van Rompay AR, Johansson M, Karlsson A. Substrate specificity and phosphorylation of antiviral and anticancer nucleoside analogues by human deoxyribonucleoside kinases and ribonucleoside kinases. Pharmacol Ther 100: 119–139, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ventura-Clapier R, Garnier A, Veksler V, Joubert F. Bioenergetics of the failing heart. Biochim Biophys Acta 1813: 1360–1372, 2011 [DOI] [PubMed] [Google Scholar]
- 44. Villarroya J, Dorado B, Vila MR, Garcia-Arumi E, Domingo P, Giralt M, Hirano M, Villarroya F. Thymidine kinase 2 deficiency-induced mitochondrial DNA depletion causes abnormal development of adipose tissues and adipokine levels in mice. PLoS One 6: e29691, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang L, Eriksson S. Tissue specific distribution of pyrimidine deoxynucleoside salvage enzymes shed light on the mechanism of mitochondrial DNA depletion. Nucleoside Nucleotide Nucl Acid 29: 400–403, 2010 [DOI] [PubMed] [Google Scholar]
- 46. Wang L, Sun R, Eriksson S. The kinetic effects on thymidine kinase 2 by enzyme-bound dTTP may explain the mitochondrial side effects of antiviral thymidine analogs. Antimicrob Agent Chemother 55: 2552–2558, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]