

Abstract
Alterations in the function and expression of NMDA receptors are observed after in vivo and in vitro traumatic brain injury. We recently reported that mechanical stretch injury in cortical neurons transiently increases the contribution of NMDA receptors to network activity and results in an increase in calcium-permeable AMPA (CP-AMPA) receptor-mediated transmission 4 h postinjury (Goforth et al. 2011). Here, we evaluated changes in the function of synaptic vs. extrasynaptic GluN2B-containing NMDA receptors after injury. We also determined whether postinjury treatment with the GluN2B-selective antagonist Ro 25-6981 or memantine prevents injury-induced increases in CP-AMPA receptor activity. We found that injury increased extrasynaptic, GluN2B-containing NMDA receptor-mediated whole cell currents. In contrast, we found no differences in synaptic NMDA receptor-mediated transmission after injury. Furthermore, treatment with Ro 25-6981 or memantine after injury prevented injury-induced increases in CP-AMPA receptor-mediated activity. Together, our data suggest that increased NMDA receptor activity after injury is predominantly due to alterations in extrasynaptic, GluN2B-containing NMDA receptors and that activation of these receptors may contribute to the appearance of CP-AMPA receptors after injury.
Keywords: traumatic brain injury, NMDA receptors, GluN2B, cortical injury
traumatic brain injury (TBI) is a significant cause of disability and is accompanied by a wide range of emotional, cognitive, and motor disturbances that negatively impact quality of life. TBI produces a variety of neural alterations, including dysregulation of excitatory glutamatergic neurotransmission that can influence neuronal cell survival and impact learning and memory mechanisms such as LTP (e.g., Park et al. 2008; Cohen et al. 2007). Studies in both humans and rodents have found that injury-induced increases in neuronal Ca2+ influx can trigger a variety of intracellular signaling cascades that ultimately affect receptor-mediated neurotransmission and cell survival. Because N-methyl-d-aspartate (NMDA) receptors are one of the main sources of Ca2+ entry into cells and play important roles in synaptic plasticity and neuronal cell death, work in the TBI field has focused on understanding the role of these receptors in the production of TBI-induced neurological deficits. However, few studies have directly examined whether injury alters the function of NMDA receptors or differentially affects NMDA receptor subtypes.
NMDA receptors are heteromeric complexes composed of GluN1 and GluN2 subunits (GluN2A-D; see Collingridge et al. 2009 for NMDA receptor nomenclature). GluN2A- and GluN2B-containing receptors predominate in cortex (Groc et al. 2009; Luo et al. 1997), and a substantial amount of research has focused on understanding differences in the distribution and function of these NMDA receptor subtypes. While it is somewhat controversial (see Kohr 2006 for review), GluN2B-containing NMDA receptors appear to be enriched in extrasynaptic regions of the neuron (Prybylowski and Wendthold 2004; Rumbaugh and Vicini 1999), whereas GluN2A-containing receptors are enriched in synapses (Bartlett et al. 2007; Liu et al. 2004; Steigerwald et al. 2000). The precise role of extrasynaptic GluN2B-containing and synaptic GluN2A-containing NMDA receptors in learning-related mechanisms like LTP and LTD and neurotoxicity is not fully understood.
Current evidence suggests that extrasynaptic, GluN2B-containing receptors mediate LTD induction (Ge et al. 2010; Liu et al. 2004; Massey et al. 2004), whereas synaptic, GluN2A-containing receptors are crucial for LTP (Bartlett et al. 2007; Liu et al. 2004; Papouin et al. 2012). However, there are cases where blockade of GluN2B-containing receptors, which should predominantly affect extrasynaptic receptor populations, does not block LTD (Berberich et al. 2005; Morishita et al. 2007). The role of distinct populations of NMDA receptors in neurotoxicity is somewhat more complicated (e.g., Wroge et al. 2012; Martel et al. 2009), with examples of both extrasynaptic, GluN2B-containing (Hardingham et al. 2002; Hardingham and Bading 2003) and synaptic, GluN2A-containing (Papouin et al. 2012) receptors contributing to neuronal cell death.
We recently found that in vitro stretch injury of cultured cortical neurons produces an increase in NMDA receptor activity that appears immediately after injury and subsides after 4 h (Goforth et al. 2011). Evidence obtained from several different TBI models suggests that injury may differentially affect GluN2B-containing NMDA receptors (DeRidder et al. 2006; Osteen et al. 2004; Schumann et al. 2008; Spaethling et al. 2012). However, the specific role of these receptors in TBI-induced alterations has not been directly examined. For example, the activation of GluN2B-containing NMDA receptors has been implicated in the appearance of calcium-permeable AMPA (CP-AMPA) receptors after injury (Spaethling et al. 2012). CP-AMPA receptors provide an additional route of Ca2+ entry to the cell and contribute to delayed cell death following injury (Bell et al. 2009; Spaethling et al. 2008). The extrasynaptic enrichment of GluN2B-containing receptors and their role in the appearance of CP-AMPA receptors has led to the hypothesis that injury may selectively affect this population of NMDA receptors. However, no studies have directly evaluated the effect of TBI on the function of particular NMDAR subtypes or extrasynaptic vs. synaptic GluN2B-containing NMDA receptors.
To address this, we evaluated changes in NMDA receptor activity mediated by synaptic vs. extrasynaptic GluN2B-containing NMDA receptors. We also determined whether blockade of NMDA receptors with memantine or with the GluN2B-selective antagonist Ro 25-6981 after injury could prevent injury-induced increases in CP-AMPA receptor-mediated activity. We found that injury indeed selectively enhanced the contribution of extrasynaptic, GluN2B-containing receptors to NMDA-elicited activity without altering transmission mediated by synaptic NMDA receptors. In addition, treatment with Ro 25-6981 or memantine immediately after injury prevented the subsequent appearance of CP-AMPA receptors 4 h postinjury.
MATERIALS AND METHODS
Animals.
The procedures used were approved by the University of Michigan University Committee on the Use and Care of Animals in accordance with AAALAC and National Institutes of Health guidelines. Pregnant Sprague-Dawley rats (Harlan, Indianapolis, IN) obtained at 14–20 days of gestation were housed individually. One-day-old pups were used to prepare cortical neurons and astrocyte cultures.
Cortical cultures.
Primary neuronal cultures were prepared using standard methods (Huettner and Baughman 1986) as previously described (Goforth et al. 2011). Neocortices of postnatal day 1 rats were isolated, dissociated with papain (37°C, 1.5 h, 10 U/ml; Worthington Biochemical, Lakewood, NJ), and rinsed with trypsin inhibitors and conditioned MEM (5% calf serum, 25 mM glucose, 100 U/ml penicillin, 100 μg/l streptomycin, and 500 nM glutamine) that had been incubated overnight in flasks containing a confluent layer of astrocytes. Cells were plated at a density of 5 × 105 cells/25-mm well onto a confluent layer of astrocytes grown on deformable Silastic membranes (Flexcell International, Hillsborough, NC). Cultures were maintained at 37°C in 95% air-5% CO2, and one-half of the media was replaced twice weekly with fresh conditioned MEM. Cultures were used for experiments after 15–18 days in vitro.
Mechanical stretch injury.
Mechanical stretch injury was applied using a Cell Injury Controller II, as described previously (Tavalin et al. 1995). A 50-ms pulse of compressed air was used to deform the Silastic membranes of the six-well culture plate (2.5-cm diameter) by 6.5 mm, corresponding to 38% stretch of the membrane and attached cells (McKinney et al. 1996; Tavalin et al. 1995). Control cultures were treated identically, but did not receive mechanical injury.
General electrophysiological procedures.
All electrophysiology experiments were conducted 15 min postinjury and were carried out at room temperature. Recordings included in each data set were made from at least three different culture preparations. Reported Ns indicate number of individual neurons. Patch pipettes (6–7 MΩ) were pulled from 1.5-mm o.d. borosilicate glass capillaries (WPI, Sarasota, FL) using a horizontal puller (model P97; Sutter Instrument, Novato, CA), heat-polished, and then were filled with a solution containing (in mM): 135 CsAsp, 4 KCl, 2 NaCl, 10 EGTA, 0.2 CaCl2, 2 MgATP, 0.6 Na2GTP, and 10 HEPES (pH 7.2; 290–295 mOsm). Cells were continuously superfused with bath-applied external solution at a flow rate of ∼2 ml/min. Whole cell currents were measured from individual cortical pyramidal neurons using the tight-seal whole cell voltage clamp technique and an EPC 10 USB patch clamp amplifier (Heka Elektronik). Currents were filtered at 2 kHz and then digitized at 10 kHz using Patchmaster software (Heka Elektronik). Cortical pyramidal neurons were identified by their characteristic morphology. Neurons selected for analysis had stable series resistances (Rs) of <30 MΩ. Liquid junction potentials were not corrected in the recordings shown. All chemicals and drugs were purchased from Sigma Aldrich (St. Louis, MO) or Tocris Cookson (Bristol, UK).
Measurement of NMDA-elicited whole cell currents.
Standard external recording solution contained (in mM): 130 NaCl, 4 KCl, 3 CaCl2, 2 MgCl2, 10 HEPES, 11 glucose, 0.01 glycine, 0.0005 tetrodotoxin (TTX), and 30 μM bicuculline methiodide (BMI; pH 7.3, 300–305 mOsm). Current-voltage (I-V) relationships were determined by applying a voltage ramp (−100 mV to +40 mV, 16.7 mV/s) in the presence and absence of NMDA (100 μM) or NMDA plus the GluN2B-selective antagonist Ro 25-6981 (1 μM). Ro 25-6981 is an Ifenprodil derivative that is selective for GluN2B-containing NMDA receptors at the concentrations used here (Fischer et al. 1997; Kohr 2006). For I-V experiments, all drugs were bath applied and then voltage ramps were applied following the establishment of steady-state holding current (>2 min.)
To examine maximal NMDA-elicited currents, whole cell recordings were obtained using external solution containing reduced MgCl2 (0.05 mM without substitution of other ions) in the presence or absence of NMDA (100 μM), and with or without Ro 25-6981 (1 μM). Solutions containing NMDA and Ro 25-6981 were rapidly applied using an SF-77 Fast-Step rapid perfusion system (Warner Instrument, Hamden, CT), as previously described (Goforth et al. 1999). For both types of experiments, NMDA-elicited currents and the contribution of GluN2B-containing NMDA receptors to responses were measured in the same cell. Additional experiments confirmed that NMDA-elicited currents were blocked by coadministration of the nonselective NMDA receptor antagonist d-(-)-2-amino-5-phosphonopentanoic acid (APV; 20 μM) and that repeated application of NMDA did not result in appreciable current “run-down” (data not shown).
Measurement of extrasynaptic NMDA-elicited currents.
To isolate extrasynaptic NMDAR-mediated current, synaptically activated NMDA receptors were first blocked using the open channel blocker MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine-maleate] (Bengtson et al. 2008; Hardingham and Bading 2002; Harris and Pettit 2008; Huettner and Bean 1988; Ivanov et al. 2006). After achieving the whole cell configuration, standard extracellular recording solution containing MK-801 (50 μM) and BMI (30 μM), and without TTX, was bath applied for 6 min, followed by perfusion with extracellular recording solution containing TTX (0.5 μM for 3 min). These conditions promote spontaneous action potentials and glutamate release that allow MK-801 to block open NMDA receptors at synapses. Blockade of synaptic NMDA receptors was confirmed by examining the APV-sensitive component of miniature excitatory postsynaptic currents (mEPSCs) before and after MK-801 treatment in control and injured neurons. Following MK-801 treatment, NMDA-elicited currents were recorded by locally applying NMDA with and without Ro 25-6981 (1 μM) using standard external solution containing reduced MgCl2 (0.05 mM).
Measurement of NMDA receptor-mediated sEPSCs and mEPSCs.
Spontaneous excitatory postsynaptic currents (sEPSCs) were recorded using standard external solution containing BMI (30 μM) and MgCl2 (2 mM) with and without APV (20 μM). In addition, 5 mM QX314 was added to the intracellular solution to block Na2+-dependent action potentials in the cell from which sEPSC recordings were made. mEPSCs were recorded using standard external solution containing TTX (0.5 μM), BMI (30 μM), and minimal MgCl2 (0.05 mM) with and without APV (20 μM).
Treatment with Ro 25-6981 or memantine after injury and measurement of CP-AMPA receptor-mediated activity.
Cultures were injured as described above, and then vehicle, Ro 25-6981 (1 μM), or memantine (10 μM) was added to the well immediately after injury. Memantine is a noncompetitive antagonist of NMDA receptors believed to be an open-channel blocker (Wroge et al. 2012). While memantine does not show clear subunit selectivity, it can show selectivity for extrasynaptic NMDA receptors (e.g., Xia et al. 2010; Léveillé et al. 2008; Lipton et al. 2007) at low concentrations (1–10 μM). However, this selectivity is highly dependent on the open channel duration and is therefore affected by endogenous glutamate tone, magnesium levels, and possibly magnesium sensitivity of the NMDA receptor itself. The concentration used here is closer to therapeutic doses currently used to treat Alzheimer's and should not be assumed to solely affect extrasynaptic NMDA receptors. Cultures remained in the incubator in the presence of vehicle, Ro 25-6981, or memantine for 4 h. They were then thoroughly rinsed to wash out Ro 25-6981 and memantine prior to imaging. Control cultures were treated identically except that no injury was delivered in this case.
For [Ca2+]i imaging, cultures were incubated with fura 2-AM (5 μM; Invitrogen) in 0.01% pluronic acid (45 min, 37°C). Fura 2 fluorescence was imaged using an Olympus BX51W1 (Olympus, Tokyo, Japan) upright microscope equipped with a Hamamatsu Orca camera (Hamamatsu, Japan) and Ludl shutter/filter wheel (Ludl, Germany). Intracellular fura 2-AM was sequentially excited at 340 and 380 nm, the excitation wavelengths of Ca2+-bound and Ca2+-free fura 2-AM, respectively. Emitted fluorescence was collected at 510 nm every 2 s using IPLab 3.6.5 software (BioVision Technologies, Exton, PA). The ratio of fluorescence intensity generated in response to 340-nm and 380-nm excitation (F340/F380) was calculated and then plotted vs. time. Images were simultaneously collected from 8–15 neurons per well.
Spontaneous [Ca2+]i oscillations were monitored using standard external solution containing BMI (30 μM) and APV (20 μM), with and without the CP-AMPA receptor blocker 1-naphthylacetyl spermine trihydrochloride (NASPM; 50 μM) as previously described (Goforth et al. 2011). KCl (30 mM) was added at the end of each experimental run to confirm cell viability. Only those cells exhibiting an increase in [Ca2+]i in response to KCl were included in the analysis. Solutions were applied at a flow rate of ∼1 ml/min using a peristaltic pump. Changes in [Ca2+]i were determined at least 3 min after drug application to ensure complete solution exchange had occurred and to allow neuronal activity to reach a steady state. Recordings were made from control and injured neurons from the same culture preparation, and experiments were repeated using neurons from at least three different preparations. All experiments were conducted at room temperature.
Data analysis and statistics.
Data were analyzed using Igor Pro (WaveMetrics) and Mini-Analysis (Synaptosoft, Decatur, GA) software.
NMDA-elicited whole cell currents.
For each cell, current/voltage (I-V) curves were determined in the absence of drug, in the presence of NMDA, and in the presence of NMDA + Ro 25-6981. The NMDA-elicited current for each neuron was calculated as the difference between I-V curves measured in the presence and absence of NMDA. The GluN2B-sensitive component for each cell was calculated as the difference between I-V curves obtained in the presence of NMDA minus those obtained in the presence of NMDA + Ro 25-6981. Peak NMDA-elicited and Ro 25-6981-sensitive currents were then determined for each neuron. For experiments in reduced MgCl2 and after synaptic NMDA receptor blockade with MK-801, the maximal current elicited by NMDA and NMDA + Ro 25-6981 was determined for each cell, and the GluN2B-sensitive component was calculated as the difference between the maximal current observed in the presence of NMDA minus maximal current in the presence of NMDA + Ro 25-6981. Statistical analysis was performed using Prism 5 (GraphPad Software, San Diego, CA). For both sets of experiments, NMDA-elicited currents and the Ro 25-6981 sensitivity of these NMDA currents were measured within the same cell, but were graphed separately to simplify data presentation. Thus, post hoc analyses were conducted only after significant group × drug interaction using one-way ANOVA; P < 0.05.
In our cultures, sEPSCs occur in bursts that can exhibit complex waveforms (see Fig. 4A). To quantify the APV-sensitive component of sEPSCs, for each cell, sEPSC charge transfer was averaged from at least five bursts in the presence and absence of APV (20 μM) and compared between control and injured neurons. The NMDAR-mediated component was determined by subtracting the charge transfer in the absence and presence of APV. For studies of mEPSCs, individual mEPSC events were detected using an amplitude threshold of 5 pA (Synaptosoft, Decatur, GA) and were then confirmed visually. At least 85 events per drug treatment were used per neuron. The NMDA receptor-mediated and GluN2B-mediated components of the mEPSCs were calculated as differences in mEPSC amplitude or charge transfer (time integral of EPSC current) recorded in the presence or absence of APV or Ro 25-6981, respectively, in the same cell.
Fig. 4.

Injury does not alter synaptic NMDA receptor-mediated currents. A: mean (± SE) % block of sEPSC charge transfer by APV. An example trace is shown (right). B: mean (± SE) APV-sensitive (NMDA receptor-mediated) mEPSC peak amplitude and charge transfer. Average mEPSC wave form from an individual control neuron in the presence (gray) and absence (black) of APV (top, right) and NMDA receptor-mediated difference current, calculated as the average mEPSC in the absence of APV minus the average mEPSC in the presence of APV (right). C: mean (± SE) Ro 25-6981-sensitive (GluN2B-mediated) mEPSC peak amplitude and charge transfer. The average mEPSC wave form from an individual control neuron in the absence (black) and presence (gray) of Ro 25-6981 (top) and GluN2B receptor-mediated difference current, calculated as the average mEPSC in the absence of Ro 25-6981 minus the average mEPSC in the presence of Ro 25-6981 (right).
For calcium imaging data, [Ca2+]i levels were baseline subtracted and then analyzed using a custom program implemented in MatLab software (The MathWorks, Natick, MA). [Ca2+]i oscillation amplitude was calculated as the change in fura 2-AM ratio from baseline (ΔF340/F380). CP-AMPA receptor-mediated activity was calculated as the % inhibition of [Ca2+]i amplitude in the presence of NASPM.
RESULTS
Injury increases whole cell NMDA-elicited currents mediated by GluN2B-containing NMDA receptors.
To measure the NMDA-elicited whole cell currents of control and injured neurons, we used two complementary approaches. First, current-voltage relationships were measured in the presence and absence of NMDA (100 μM) in control and injured neurons bathed in solution containing 2 mM MgCl2. Mean peak NMDA-elicited current was significantly greater in injured vs. control neurons (Fig. 1A; control n = 12, injured n = 10: 2-tailed t-test: P < 0.05). In addition, the voltage at which the peak NMDA-elicited current occurred was hyperpolarized in injured vs. control neurons (Fig. 1B inset; 2-tailed t-test: P < 0.05). Importantly, NMDA-elicited current measured at the same voltage (−27 mV or −65 mV), rather than at peak current, was also significantly increased after injury (data not shown). Second, in separate experiments, NMDA was applied acutely to neurons voltage clamped to −65 mV and bathed in extracellular solution containing 0.05 mM MgCl2. These conditions minimize Mg2+ block of NMDA receptors to allow us to measure the maximal NMDA receptor conductance. Similar to results obtained from the I-V experiments, maximal NMDA-elicited current was significantly increased after injury (Fig. 1C; control n = 12, injured n = 5; 2-tailed t-test: P < 0.05). Representative I-V plots of the NMDA-elicited currents for control and injured neurons are shown in Fig. 1B, and representative NMDA-elicited currents in reduced MgCl2 are shown in Fig. 1D. Using either approach, NMDA-elicited current was increased in injured neurons.
Fig. 1.

Injury increases NMDA-elicited whole cell currents. A: mean (± SE) peak NMDA-elicited current recorded in 2 mM extracellular MgCl2. B: representative whole cell difference currents (presence of NMDA minus absence of NMDA) plotted against linear voltage ramps from −100 mV to +40 mV (6 s). Inset: average voltage at which the peak NMDA-elicited current occurred. C: mean (± SE) maximal NMDA-elicited current recorded from neurons voltage clamped to −65 mV in 0.05 mM MgCl2. D: representative whole cell NMDA-elicited currents. *P < 0.05.
To determine whether injury preferentially enhances the function of GluN2B-containing NMDA receptors, we tested the sensitivity of whole cell NMDA-elicited currents to the selective GluN2B antagonist Ro 25-6981. Representative whole cell difference currents (presence of NMDA minus NMDA + Ro 25-6981) plotted against linear voltage ramps from −100 mV to +40 mV (6 s) are shown in Fig. 2B. Analysis of I-V curves showed that the peak Ro 25-6981-sensitive NMDA elicited current was significantly greater in injured vs. control neurons (Fig. 2A; control n = 6, injured n = 6; 2-tailed t-test: P < 0.05). We also determined whole cell difference currents (presence of NMDA minus NMDA + Ro 25-6981) during local application of drug in reduced MgCl2. Similar to I-V results, the Ro 25-6981-sensitive NMDA-elicited current measured in reduced MgCl2 was also increased after injury, although this did not reach statistical significance [data not shown; control n = 12 (113.4 pA, ± 45.8), injured n = 5 (254.3 pA ± 117.3; P = 0.09)].
Fig. 2.

Injury increases GluN2B NMDA receptor-mediated whole cell currents. A: mean (± SE) peak Ro 25-6981-sensitive (GluN2B mediated) current recorded in 2 mM MgCl2. B: representative whole cell difference currents (presence of NMDA minus NMDA + Ro 25-6981) plotted against linear voltage ramps from −100 mV to +40 mV (6 s). *P < 0.05.
Injury increases extrasynaptic GluN2B receptor-mediated whole cell currents.
To test the hypothesis that injury-induced increases in NMDA receptor current were due to a change in extrasynaptic GluN2B-containing receptors, we first blocked synaptic NMDA receptors using a standard approach. Cultures were incubated with the NMDA receptor open channel blocker MK-801 for 6 min (see materials and methods for details). Successful blockade of synaptic NMDA receptors was confirmed by measurement of the D-APV-sensitive, NMDA receptor-mediated, component of the mEPSC after MK-801 treatment. As shown in Fig. 3, A and B, APV had no effect on mEPSC amplitude or charge transfer following MK-801 treatment (n = 5), confirming that MK-801 treatment selectively blocked synaptic NMDA receptors.
Fig. 3.
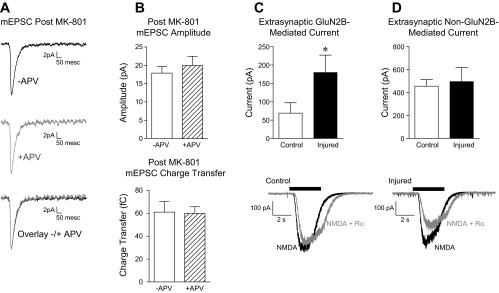
Injury increases extrasynaptic GluN2B-containing NMDA receptor-mediated currents. All recordings made in 0.05 mM extracellular MgCl2. A: treatment with MK-801 blocked synaptic NMDA receptors. The average mEPSC wave form recorded from an individual control neuron after MK-801 treatment in the absence (black) and presence (gray) of d-(-)-2-amino-5-phosphonopentanoic acid (APV). B: mean (± SE) mEPSC amplitude (top) and charge transfer (bottom) after MK-801 treatment. C: mean (± SE) maximal extrasynaptic Ro 25-6981-sensitive (GluN2B-mediated), NMDA-elicited current recorded after blockade of synaptic NMDA receptors. D: mean (± SE) maximal extrasynaptic Ro 25-6981-insensitive (non-GluN2B-mediated), NMDA-elicited current recorded after blockade of synaptic NMDA receptors. Representative extrasynaptic whole cell currents recorded in the presence of NMDA (black) or NMDA + Ro 25-6981 (gray) are shown at bottom of C and D. *P < 0.05.
We then measured Ro 25-6981-sensitive NMDA-elicited current in a separate set of control and injured neurons following MK-801 treatment. Consistent with the hypothesis that extrasynaptic GluN2B-containing receptors are potentiated by injury, the injury-induced increase in Ro 25-6981-sensitive whole cell current persisted even after synaptic NMDA receptors were blocked with MK-801. Thus, mean Ro 25-6981-sensitive NMDA-elicited current was significantly greater in injured vs. control neurons (Fig. 3C; control n = 8, injured n = 8; 1-tailed t-test, P < 0.05). In addition, no differences were observed in the residual Ro 25-6981-insensitive (i.e., non-GluN2B-mediated) extrasynaptic NMDA-elicited whole cell current of control vs. injured neurons (Fig. 3D). Together, these data strongly suggest that injury increases the activity of extrasynaptic, GluN2B-containing NMDA receptors. Representative traces from a control and injured neuron following MK-801 treatment are shown in the presence of NMDA and NMDA + Ro 25-6981 (Fig. 3, C and D).
Injury does not alter synaptic NMDA receptor-mediated currents.
Next, we examined the effect of injury on sEPSCs and mEPSCs mediated by NMDARs. As shown in Fig. 4A, in the presence of bicuculline, sEPSCs occur as bursts of multiple events. The NMDAR-mediated sEPSC charge transfer appeared increased in injured vs. control neurons, but this difference did not reach statistical significance (Fig. 4A; control n = 7, injured n = 9; 2-tailed t-test, P > 0.05). We observed no consistent effect of injury on the APV sensitivity of sEPSC bursts, which reflect varying degrees of presynaptic glutamate release and postsynaptic receptor activation at multiple synapses. Thus, to more directly evaluate injury-induced changes in postsynaptic NMDA receptors, we also recorded mEPSCs in minimal magnesium (0.05 mM). Consistent with our previous results (Goforth et al. 2011), injury did not alter the mean APV-sensitive component of mEPSC amplitude or charge transfer (Fig. 4B; control n = 6, injured n = 5). The average mEPSC waveform of an individual control neuron measured in the presence and absence of APV, and NMDA receptor-mediated difference currents, are shown in Fig. 4B. In addition, we found that mean Ro 25-6981-sensitive mEPSC amplitude and charge transfer were also unaffected by injury (Fig. 4C; control n = 11, injured n = 9). The average mEPSC waveforms for an individual control neuron measured in the presence or absence of Ro 25-6981 and resulting GluN2B receptor-mediated difference current are shown in Fig. 4C.
Blocking NMDA receptors following injury prevented increased CP-AMPA receptor activity.
Because NMDAR activation has been linked to the upregulation of CP-AMPA receptors following injury (Schumann et al. 2008; Spaethling et al. 2012), we determined whether blockade of NMDA receptors with memantine or GluN2B-containing NMDA receptors using Ro 25-6981 would in turn prevent injury-induced increases in CP-AMPA receptor-mediated activity (Bell et al. 2009; Goforth et al. 2011; Spaethling et al. 2008). We examined the contribution of CP-AMPAR activity to [Ca2+]i oscillations resulting from spontaneous network activity. Measurement of neuronal [Ca2+]i oscillations is a reliable measure of action-potential driven network activity and permits the examination of overall excitatory neurotransmission within a large network of neurons, rather than in single cells. In addition, we find good agreement between the amount of NASPM sensitivity of [Ca2+]i oscillations (∼20% increase after injury) and NASPM-sensitive synaptic currents (also ∼20% increased after injury) (Goforth et al. 2011).
Cultures were treated with vehicle, Ro 25-6981, or memantine immediately after injury, and the drug remained present for 4 h after injury. The contribution of CP-AMPA receptors to neuronal activity was then examined by determining the sensitivity of synaptically driven neuronal [Ca2+]i oscillations to the CP-AMPA receptor blocker NASPM as previously described (Goforth et al. 2011). The mean percent inhibition of [Ca2+]i by NASPM 4 h after injury is shown in Fig. 5. Consistent with previous reports, a 20% increase in NASPM sensitivity was observed 4 h after injury (Fig. 5, A and B; 1-way ANOVA, P < 0.05) (Bell et al. 2009; Goforth et al. 2011; Spaethling et al. 2008). Treatment with Ro 25-6981 (Fig. 5A) or memantine (Fig. 5B) immediately after injury prevented injury-induced increases in CP-AMPA receptor-mediated activity. Thus, blockade of GluN2B-containing or more general blockade of NMDA receptors were both able to prevent injury-induced increases in CP-AMPA receptor function even when the drugs were administered after injury. In addition, NAPSM sensitivity was significantly lower in injured vs. control neurons treated with Ro 25-6981 (2-tailed t-test, P = 0.008) but was similar for injured and control neurons treated with memantine.
Fig. 5.

Injury-induced increases in CP-AMPA receptor-mediated activity are blocked by treatment with Ro 25-6981 or memantine (Mem) after injury. A: mean (± SE) percent inhibition of [Ca2+]i oscillation amplitude by the CP-AMPA receptor blocker NASPM 4 h after injury with and without Ro 25-6981 treatment. Injury produced a significant increase in NASPM sensitivity (1-way ANOVA, P < 0.05; Con Veh vs. Inj Veh, *P < 0.05) that was blocked by treatment with Ro 25-6981 (Inj Veh vs. Inj Ro, #P < 0.05). B: mean (± SE) percent inhibition of [Ca2+]i oscillation amplitude by NASPM 4 h after injury with and without Mem treatment. Injury produced a significant increase in NASPM sensitivity (1-way ANOVA, P < 0.05; Con Veh vs. Inj Veh, *P < 0.05) that was blocked by treatment with Mem (Inj Veh vs. Inj Mem, #P < 0.05). C: representative traces of [Ca2+]i oscillations in control and injured cells treated with vehicle or Ro 25-6981. D: representative traces of [Ca2+]i oscillations in control and injured cells treated with vehicle or memantine.
DISCUSSION
Alterations in NMDA receptor expression and activity have been observed following in vivo and in vitro TBI (Beigon et al. 2004; Goforth et al. 2011; Kumar et al. 2002; Lea et al. 2003; Osteen et al. 2004; Zhang et al. 1996). We recently reported that mechanical stretch injury transiently increased NMDA receptor-mediated activity immediately after injury and increased CP-AMPA receptor-mediated transmission 4 h later (Goforth et al. 2011). Here, we directly evaluated injury-induced changes in the contribution of synaptic vs. extrasynaptic GluN2B-containing NMDA receptors to this enhancement. In addition, we determined the role of GluN2B-containing receptors in the upregulation of CP-AMPA receptor-mediated activity following injury.
Using complementary approaches, we found that injury-induced increases in NMDA receptor-mediated activity were predominantly due to an increase in the contribution of GluN2B-containing NMDA receptors located at extrasynaptic regions. No differences in synaptic NMDA receptor-mediated transmission were found. In addition, postinjury blockade of GluN2B receptors with Ro 25-6981 or NMDA receptors with memantine prevented injury-induced increases in CP-AMPA receptor-mediated activity.
Our finding that injury enhanced peak NMDA-elicited current (Fig. 1) is consistent with previous reports that mechanical injury increases NMDA receptor-mediated activity as measured by [Ca2+]i oscillations (Goforth et al. 2011) and single cell recordings (Lea et al. 2003; Zhang et al. 1996). Furthermore, the current results are consistent with reports of increased NMDA receptor expression (Schumann et al. 2008), binding (Beigon et al. 2004), and spontaneous activity (Goforth et al. 2011) after injury. Unlike our previous results (Zhang et al. 1996), the voltage dependence of NMDA-elicited currents was not strongly reduced by injury in the current study. However, there was a significant leftward shift in the peak voltage in injured neurons compared with controls (Fig. 1B, inset) that is consistent with a modest reduction in magnesium sensitivity predicted by modeling NMDA receptor properties (Li et al. 1996).
In our previous work (Zhang et al. 1996), neurons were not cultured on an astrocyte bed, as they were in the current study. However, it is not likely that this difference can account for the change in magnitude, as Lea et al. (2003) found that the presence or absence of astrocytes did not dramatically alter the ability of injury to reduce magnesium sensitivity. Another difference between our current and previous results is the number of days in vitro (15–18 DIV used here vs. 10–15 DIV previously) prior to injury and recording. Although both ranges of culture duration coincide with the presence of mature glutamatergic synapses, the balance of GluN2A/GluN2B-containing receptors in cultured cortical neurons does gradually increase from DIV 12–21 (Li et al. 1998), raising the intriguing possibility that this balance may influence the effect of injury on magnesium sensitivity. It is important to point out that the complement of GluN2A/GluN2B as well as the synaptic and extrasynaptic location of these receptors at the DIV used here closely corresponds to that found in postnatal rat brains in vivo (Monyer et al. 1994; Portera-Cailliau et al. 1996; Sheng et al. 1994). Indeed, expression of GluN2A at all developmental ages is lower than that of GluN2B and restricted to synapses (Flint et al. 1997; Li et al. 1998). This is consistent with our observation that the majority of the whole cell NMDA-elicited current was sensitive to blockade by Ro 25-6981 (Fig. 1A vs. Fig. 2A), while synaptic currents contained a Ro 25-6981-insensitive component (Fig. 4B vs. 4C).
While changes in overall NMDA receptor activity and subunit levels have been observed following TBI, this is the first report to our knowledge of subtype and subcellular localization-specific alterations in the function of NMDA receptors. Specifically, we observed an injury-induced potentiation of the function of GluN2B-containing NMDA receptors (Fig. 2). This result is consistent with previous reports of an increase in the ratio of GluN2B:GluN2A protein expression after in vivo injury (Osteen et al. 2004). Furthermore, the potentiation was selectively mediated by extrasynaptic receptors, as evidenced by the persistence of injury-induced increases in GluN2B receptor-mediated current despite the blockade of synaptic NMDA receptors with MK-801 (Fig. 3C). While we cannot rule out the possibility that MK-801 could block some extrasynaptic NMDA receptors activated by glutamate spillover from synapses, this is highly unlikely given that experiments were conducted 15 min after injury, when extracellular glutamate is no longer elevated (Li et al. 1996), and in a perfused bath.
In further support of a selective effect of injury on extrasynaptic NMDA receptors, injury did not alter the contribution of NMDARs to mEPSCs, nor could we detect a shift in the contribution of GluN2B-containing receptors to the mEPSCs (Fig. 4). Together, the data suggest that injury-induced increases in GluN2B-mediated whole cell currents are predominantly due to changes in the extrasynaptic NMDA receptor population. Future experiments will be needed to determine whether the enhanced NMDA-elicited currents we observed reflect changes in the properties and/or the expression of extrasynaptic NMDA receptors.
Extrasynaptic GluN2B-containing receptors have been linked to LTD induction (Massey et al. 2004) and altered signaling via CREB and ERK in neurons (Atkins et al. 2009; Hardingham et al. 2002; Ivanov et al. 2006). The balance between the activation of synaptic and extrasynaptic NMDA receptors can also influence cell survival (Hardingham et al. 2002; Hardingham 2009; Léveillé et al. 2008; Zhang et al. 2007). Thus, by enhancing the extrasynaptic, GluN2B-containing population of NMDA receptors, TBI may set the stage for altered neuroplasticity and help shape the response to subsequent neuronal insults. For example, a shift in the complement of extrasynaptic NMDA receptors and thus an alteration in the balance between synaptic and extrasynaptic receptors may contribute to complications associated with TBI, including posttraumatic epilepsy.
The activation of NMDA receptors has also been linked to the upregulation of CP-AMPA receptors after injury (Bell et al. 2009; Spaethling et al. 2008). CP-AMPA receptors are not usually abundant in mature cortical neurons and provide an additional route for calcium entry into the cell (Cull-Candy et al. 2006; Liu and Zukin 2007). Thus, their activity has been linked to injury-induced cell death (see Park et al. 2008 for review). In addition, although the role of CP-AMPA receptors in plasticity and cell signaling is not well defined (Man 2011), increases in CP-AMPA receptors have been associated with drug addiction (Conrad et al. 2008; Ferrario et al. 2011) and scaling after deprivation of synaptic activity (Turrigiano 2008). Thus the possibility that the appearance of CP-AMPA receptor after injury may represent a maladaptive “recovery” mechanism that warrants further investigation.
Spaethling et al. (2012) recently showed that blocking NMDA receptors at the time of injury attenuated AMPA-stimulated Ca2+ entry 4 h later. This suggests that NMDA receptor blockade may be able to prevent enhanced CP-AMPA receptor-dependent activity. We examined the effect of postinjury blockade of NMDA receptors with memantine or GluN2B-containing NMDA receptors with the selective antagonist Ro 25-6981 on CP-AMPA receptor-mediated [Ca2+]i oscillations 4 h after injury (compounds were present throughout the 4-h postinjury period, but not at the time of injury or at the time of imaging).
As expected, injury increased the CP-AMPA receptor-mediated component of synaptically driven [Ca2+]i oscillations, as indicated by an enhanced sensitivity of [Ca2+]i oscillation amplitude to the CP-AMPA receptor-selective blocker NASPM (Fig. 5). Blockade with either compound prevented this injury-induced increase. However, Ro 25-6981 treatment was more effective than memantine, suggesting that selectively targeting GluN2B-contatining receptors may be beneficial. In addition, both Ro 25-6981 and memantine produced the opposite effect in uninjured control neurons; that is, we observed a slight increase in CP-AMPA receptor-mediated [Ca2+]i oscillations in uninjured control neurons with either drug. This opposing effect on injured vs. uninjured neurons was more pronounced in memantine-treated cultures and is likely due to the differences between the mechanism of action of these two compounds. For example, memantine does not show clear subunit selectivity, but rather is affected by the duration of NMDA receptor channel opening, extracellular vs. synaptic glutamate levels, and magnesium concentration (for discussion see Wroge et al. 2012). Given that TBI in vivo affects some neuronal populations and not others, it will be important to determine, in future studies, whether the effects observed in healthy neurons negatively impact the benefits seen in the injured neurons. It is also interesting to consider these differences in light of our observed injury-induced shifts in NMDA receptor profile. Although speculative, it is possible that a shift towards a greater contribution of extrasynaptic GluN2B-containing receptors promotes CP-AMPA receptor activity. This could help explain the opposing effects in uninjured vs. injured neurons. It is also important to remember that while GluN2B-containing receptors are enriched in extrasynaptic regions, they are not absent from synapses, as can be seen by their small contribution to the mEPSC (Fig. 4C). Thus, the ability of Ro 25-6981 and memantine to prevent injury-induced increases in CP-AMPA receptor-mediated activity may not be limited to effects on extrasynaptic receptors.
In summary, the current study is the first to examine the effect of injury on the function of subpopulations of NMDA receptors (extrasynaptic and GluN2B containing). Injury-induced increases in NMDA receptor-mediated activity were predominantly due to a greater contribution of extrasynaptic, GluN2B-containing NMDA receptors, while no differences in synaptic NMDA receptor-mediated transmission were observed. In addition, blocking GluN2B-containing receptors for up to 4 h after injury prevented subsequent increases in CP-AMPA receptor-mediated activity. Thus, the data presented here support the idea that postinjury treatment using antagonists that target GluN2B-containing receptors can effectively block some of the deleterious effects of TBI.
GRANTS
Research was supported by National Institute of Neurological Disorders and Stroke Grants RO1-NS-49519 (to L. S. Satin) and NS-069629 (to C. R. Ferrario).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
C.R.F., L.S.S., and P.B.G. conception and design of research; C.R.F., B.O.N., and J.R. performed experiments; C.R.F., B.O.N., J.R., and P.B.G. analyzed data; C.R.F., L.S.S., and P.B.G. interpreted results of experiments; C.R.F. and P.B.G. drafted manuscript; C.R.F., B.O.N., L.S.S., and P.B.G. edited and revised manuscript; C.R.F., B.O.N., J.R., L.S.S., and P.B.G. approved final version of manuscript; P.B.G. prepared figures.
ACKNOWLEDGMENTS
We thank Benjamin Schwartz for Matlab programing.
REFERENCES
- Atkins CM, Falo MC, Alonso OF, Bramlett HM, Dietrich WD. Deficits in ERK and CREB activation in the hippocampus after traumatic brain injury. Neurosci Lett 459: 52–56, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett TE, Bannister NJ, Collett VJ, Dargan SL, Massey PV, Bortolotto ZA, Fitzjohn SM, Bashir ZI, Collingridge GL, Lodge D. Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of 2-week-old rat hippocampus. Neuropharmacol 52: 60–70, 2007 [DOI] [PubMed] [Google Scholar]
- Bell JD, Park E, Ai J, Baker AJ. PICK1-mediated GluR2 endocytosis contributes to cellular injury after neuronal trauma. Cell Death Diff 16: 1665–1680, 2009 [DOI] [PubMed] [Google Scholar]
- Bengtson CP, Dick O, Bading H. A quantitative method to assess extrasynaptic NMDA receptor function in the protective effect of synaptic activity against neurotoxicity. BMC Neurosci 9: 11, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berberich S, Punnakkal P, Jensen V, Pawlak V, Seeburg PH, Hvalby Ø, Köhr G. Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci 25: 6907–6910, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegon A, Fry PA, Paden CM, Alexandrovich A, Tsenter J, Shohami E. Dynamic changes in N-methyl-d-aspartate receptors after closed head injury in mice: implications for treatment of neurological and cognitive deficits. Proc Natl Acad Sci USA 101: 5117–5122, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AS, Pfister BJ, Schwarzbach E, Grady MS, Goforth PB, Satin LS. Injury-induced alterations in CNS electrophysiology. Prog Brain Res 161: 143–169, 2007 [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Olsen RW, Peters J, Spedding M. A nomenclature for ligand-gated ion channels. Neuropharmacol 56: 2–5, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454: 118–121, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S, Kelly L, Farrant M. Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr Opin Neurobiol 16: 288–297, 2006 [DOI] [PubMed] [Google Scholar]
- DeRidder MN, Simon MJ, Siman R, Auberson YP, Raghupathi R, Meaney DF. Traumatic mechanical injury to the hippocampus in vitro causes regional caspase-3 and calpain activation that is influenced by NMDA receptor subunit composition. Neurobiol Dis 22: 165–176, 2006 [DOI] [PubMed] [Google Scholar]
- Ferrario CR, Loweth JA, Milovanovic M, Ford KA, Galiñanes GL, Heng LJ, Tseng KY, Wolf ME. Alterations in AMPA receptor subunits and TARPs in the rat nucleus accumbens related to the formation of Ca2-permeable AMPA receptors during the incubation of cocaine craving. Neuropharmacol 61: 1141–1151, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer G, Mutel V, Trube G, Malherbe P, Kew JN, Mohacsi E, Heitz MP, Kemp JA. Ro 25-6981, a highly potent and selective blocker of N-methyl-d-aspartate receptors containing the NR2B subunit. Characterization in vitro. J Pharmacol Exp Ther 283: 1285–1292, 1997 [PubMed] [Google Scholar]
- Flint AC, Maisch US, Weishaupt JH, Kriegstein AR, Monyer H. NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J Neurosci 17: 2469–2476, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Dong Z, Bagot RC, Howland JG, Phillips AG, Wong TP, Wang YT. Hippocampal long-term depression is required for the consolidation of spatial memory. PNAS 107: 16697–16702, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goforth PB, Ellis EF, Satin LS. Enhancement of AMPA-mediated current after traumatic injury in cortical neurons. J Neurosci 19: 7367–7374, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goforth PB, Ren J, Schwartz BS, Satin LS. Excitatory synaptic transmission and network activity are depressed following mechanical injury in cortical neurons. J Neurophysiol 105: 2350–2363, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groc L, Bard L, Choquet D. Surface trafficking of N-methyl-d-aspartate receptors: physiological and pathological perspectives. Neuroscience 158: 4–18, 2009 [DOI] [PubMed] [Google Scholar]
- Hardingham GE. Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem Soc Trans 37: 1147–1160, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Coupling of extrasynaptic NMDA receptors to a CREB shut-off pathway is developmentally regulated. Biochim Biophys Acta 1600: 148–153, 2002 [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. The Yin and Yang of NMDA receptor signaling. Trends Neurosci 26: 81–89, 2003 [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5: 405–414, 2002 [DOI] [PubMed] [Google Scholar]
- Harris AZ, Pettit DL. Recruiting extrasynaptic NMDA receptors augments synaptic signaling. J Neurophysiol 99: 524–533, 2008 [DOI] [PubMed] [Google Scholar]
- Huettner JE, Baughman RW. Primary culture of identified neurons from the visual cortex of postnatal rats. J Neurosci 6: 3044–3060, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettner JE, Bean BP. Block of N-methyl-d-aspartate-activated current by the anticonvulsant MK-801: selective binding to open channels. Proc Natl Acad Sci USA 85: 1307–1311, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-Ari Y, Medina I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J Physiol 572: 789–798, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohr G. NMDA receptor function: subunit composition versus spatial distribution. Cell Tissue Res 326: 439–446, 2006 [DOI] [PubMed] [Google Scholar]
- Kumar A, Zou L, Yuan X, Long Y, Yang K. N-methyl-d-aspartate receptors: transient loss of NR1/NR2A/NR2B subunits after traumatic brain injury in a rodent model. J Neurosci Res 67: 781–786, 2002 [DOI] [PubMed] [Google Scholar]
- Lea PM, 4th, Custer SJ, Stoica BA, Faden AI. Modulation of stretch-induced enhancement of neuronal NMDA receptor current by mGluR1 depends upon presence of glia. J Neurotrauma 20: 1233–1249, 2003 [DOI] [PubMed] [Google Scholar]
- Léveillé F, El Gaamouch F, Gouix E, Lecocq M, Lobner D, Nicole O, Buisson A. Neuronal viability is controlled by a functional relation between synaptic and extrasynaptic NMDA receptors. FASEB J 22: 4258–4271, 2008 [DOI] [PubMed] [Google Scholar]
- Li YX, Bertram R, Rinzel J. Modeling N-methyl-d-aspartate-induced bursting in dopamine neurons. Neuroscience 71: 397–410, 1996 [DOI] [PubMed] [Google Scholar]
- Li JH, Wang YH, Wolfe BB, Krueger KE, Corsi L, Stocca G, Vicini S. Developmental changes in localization of NMDA receptor subunits in primary cultures of cortical neurons. Eur J Neurosci 10: 1704–1715, 1998 [DOI] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson Y, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 304: 1021–1024, 2004 [DOI] [PubMed] [Google Scholar]
- Liu SJ, Zukin RS. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci 30: 126–134, 2007 [DOI] [PubMed] [Google Scholar]
- Luo J, Wang Y, Yasuda RP, Dunah AW, Wolfe BB. The majority of N-methyl-d-aspartate receptor complexes in adult rat cerebral cortex contain at least three different subunits (NR1/NR2A/NR2B). Mol Pharmacol 51: 79–86, 1997 [DOI] [PubMed] [Google Scholar]
- Man HY. GluA2-lacking, calcium-permeable AMPA receptors–inducers of plasticity? Curr Opin Neurobiol 21: 291–298, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel MA, Wyllie DJ, Hardingham GE. In developing hippocampal neurons, NR2B-containing N-methyl-d-aspartate receptors (NMDARs) can mediate signaling to neuronal survival and synaptic potentiation, as well as neuronal death. J Neurosci 158: 334–343, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci 24: 7821–7828, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney JS, Willoughby KA, Liang S, Ellis EF. Stretch-induced injury of cultured neuronal, glial, and endothelial cells. Effect of polyethylene glycol-conjugated superoxide dismutase. Stroke 27: 934–940, 1996 [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev H, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of the four NMDA receptors. Neuron 12: 529–540, 1994 [DOI] [PubMed] [Google Scholar]
- Morishita W, Lu W, Smith GB, Nicoll RA, Bear MF, Malenka RC. Activation of NR2B-containing NMDA receptors is not required for NMDA receptor-dependent long-term depression. Neuropharm 52: 71–76, 2007 [DOI] [PubMed] [Google Scholar]
- Osteen CL, Giza CC, Hovda DA. Injury-induced alterations in N-methyl-d-aspartate receptor subunit composition contribute to prolonged calcium accumulation following lateral fluid percussion. Neuroscience 128: 305–322, 2004 [DOI] [PubMed] [Google Scholar]
- Papouin T, Ladépêche L, Ruel J, Sacchi S, Labasque M, Hanini M, Groc L, Pollegioni L, Mothet JP, Oliet SHR. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 150: 633–646, 2012 [DOI] [PubMed] [Google Scholar]
- Park E, Bell JD, Baker AJ. Traumatic brain injury: can the consequences be stopped? CMAJ 178: 1163–1170, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portera-Cailliau C, Price DL, Martin LJ. N-methyl-d-aspartate receptor proteins NR2A and NR2B are differentially distributed in the developing rat central nervous system as revealed by subunit-specific antibodies. J Neurochem 66: 692–700, 1996 [DOI] [PubMed] [Google Scholar]
- Prybylowski K, Wenthold RJ. N-methyl-d-aspartate receptors: subunit assembly and trafficking to the synapse. J Biol Chem 279: 9673–9676, 2004 [DOI] [PubMed] [Google Scholar]
- Rumbaugh G, Vicini S. Distinct synaptic and extrasynaptic NMDA receptors in developing cerebellar granule neurons. J Neurosci 19: 10603–10610, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann J, Alexandrovich GA, Biegon A, Yaka R. Inhibition of NR2B phosphorylation restores alterations in NMDA receptor expression and improves functional recovery following traumatic brain injury in mice. J Neurotrauma 25: 945–957, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Cumming J, Roldan LA, Jan YN, Jan LY. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368: 144–147, 1994 [DOI] [PubMed] [Google Scholar]
- Spaethling JM, Klein DM, Singh P, Meaney DF. Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J Neurotrauma 25: 1207–1216, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaethling JM, Le L, Meaney DF. NMDA receptor mediated phosphorylation of GluR1 subunits contributes to the appearance of calcium-permeable AMPA receptors after mechanical stretch injury. Neurobiol Dis 46: 646–654, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steigerwald F, Schulz TW, Schenker LT, Kennedy MB, Seeburg PH, Köhr G. C-terminal truncation of NR2A subunits impairs synaptic but not extrasynaptic localization of NMDA receptors. J Neurosci 20: 4573–4581, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavalin SJ, Ellis EF, Satin LS. Mechanical perturbation of cultured cortical neurons reveals a stretch-induced delayed depolarization. J Neurophysiol 74: 2767–2773, 1995 [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135: 422–435, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroge CM, Hogins J, Eisenman L, Mennerick S. Synaptic NMDA receptors mediate hypoxic excitotoxic death. J Neurosci 32: 6732–6742, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Chen HSV, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci 30: 11246–11250, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Rzigalinski BA, Ellis EF, Satin LS. Reduction of voltage-dependent Mg2+ blockade of NMDA current in mechanically injured neurons. Science 274: 1921–1923, 1996 [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Steijaert MN, Lau D, Schutz G, Delucinge-Vivier C, Descombes P, Bading H. Decoding NMDA receptor signaling: identification of genomic programs specifying neuronal survival and death. Neuron 53: 549–562, 2007 [DOI] [PubMed] [Google Scholar]