

Abstract
The relative impermeability of the blood-brain barrier (BBB) results from tight junctions and efflux transport systems limits drug delivery to the central nervous system (CNS), and thus severely restricts the therapy of many central nervous system diseases. In order to enhance the brain-specific drug delivery, we employed a 12-mer phage display peptide library to isolate peptides that could target the drug delivery system to the brain. A 12-amino-acid-peptide (denoted as Pep TGN) which was displayed by bacteriophage Clone 12-2 was finally selected by rounds of in vivo screening. Pep TGN was covalently conjugated onto the surface of poly (ethyleneglycol)-poly (lactic-co-glycolic acid) (PEG-PLGA) based nanoparticles (NPs). The cellular uptake of Pep TGN decorated nanoparticles was significantly higher than that of unmodified nanoparticles when incubated with bEnd.3 cells. Enhanced brain accumulation efficiency together with lower accumulation in liver and spleen was observed in the nude mice intravenously injected with Pep TGN conjugated nanoparticles compared with those injected with plain nanoparticles, showing powerful brain selectivity of Pep TGN. Coumarin 6 was used as a fluorescent probe for the evaluation of brain delivery properties. The brain Drug Targeting Index (DTI) of coumarin 6 incorporated in targeted nanoparticles was significantly higher than that of coumarin 6 incorporated in plain nanoparticles. In conclusion, the Pep TGN is a motif never been reported before and Pep TGN modified nanoparticles showed great potential in targeted drug delivery across the blood brain barrier.
Keywords: Phage display, blood-brain barrier (BBB), brain delivery, biodegradable nanoparticle
1. Introduction
Brain diseases such as brain tumors, Alzheimer’s disease and Parkinson’s disease have caused heavy burden on the affected individual and the society in terms of disability, loss of productivity, premature mortality and health costs. Due to the blood-brain barrier (BBB) which performs as a formidable obstacle, 98% of small-molecule drugs and 100% of large-molecule drugs, including peptides, recombinant proteins, monoclonal antibodies, genes and short interfering RNAs cannot cross the blood-brain barrier.
BBB is formed by endothelial tight junctions and plays an effective way to protect the brain from harmful and toxic substances while hampering the systemic delivery of therapeutically important drugs from the blood into the brain [1, 2]. The preferable characteristics of drugs to traverse the BBB are: <400Da and lipophilic, non-substrate of the efflux system such as P-glycoprotein. However, very few drugs could meet these demands simultaneously to cross the BBB and reach their action targets within the brain parenchyma [3, 4]. Proteins and gene drugs are restricted to enter the CNS from systemic circulation due to their hydrophilicities, protein bound properties and large molecular weights. Therefore, to reach the brain, most substances must cross the BBB through interaction with specific transporters and/or receptors expressed at the luminal (blood) side of the endothelial cells [5].
To solve the problem of drug delivery across the BBB, quite a few CNS delivery strategies have been developed [6, 7], among which the most promising approach is the receptor-mediated transport (RMT) [8]. By coupling drug-loaded vehicles with ligands which specifically recognize receptors on the BBB, the RMT strategy combines the advantages of brain targeting, high incorporation capacity, reduction of side effects, and circumvention of the multidrug efflux system [9]. Some receptors are highly expressed on the endothelial cells forming the BBB, such as the insulin receptor, transferrin receptor, low-density lipoprotein receptor (LDLR) and its related protein, and others [10]. There have been many reports about RMT strategy employing transferrin, lactoferrin, OX26 or angiopeps as the specific ligands to enable nanocarriers crossing the BBB [11-14]. Research is still on-going to identify new receptors and their respective ligands.
Therefore, identification of brain-specific markers is critical to the improved therapy of brain diseases. Finding such brain-specific targets may help drugs get into brain more specifically through targeted delivery, thus providing higher therapeutic efficiency while simultaneously decreasing systemic toxicity. We chose a powerful tool called phage display technique for brain specific ligand identification in our study. Phage display is a well-developed technique used to obtain peptide sequences that interact with a particular molecule [15]. It has been used for the selection of peptides which can bind to defined proteins, cultured cells and even inorganic materials [16-18].
Since the in vivo phage display was first introduced by Pasqualini in 1996 [19], this technique has been expanded and has provided tissue-specific peptides as targeting moieties for tumors and organs. The three-amino-acid sequence Arg-Gly-Asp (RGD) is one of the most successful targeting ligands for tumor vascular endothelial cells screened by phage display technique [20]. The in vivo screening method was also conducted to search for tissue-homing peptides [19, 21, 22]. Wan XM applied a C7C phage display library intranasally to rats and recovered phage from the brain tissue and finally gained a peptide sequence (ACTTPHAWLCG) that can bypass the BBB through the nasal-to-brain passage [23]. Rooy et al selected two 15 amino acid-peptides (GLA and GYR) that can bind to the murine brain in an in situ brain perfusion model [24].
The aim of our study is to identify peptides that could traverse the BBB from the system circulation and to construct a drug delivery system that can target the brain using the identified peptide. In our study, a random 12-mer peptide library displayed on the surface of filamentous phage M13 was screened for peptides that could lead nanocarriers to traverse BBB into the CNS. A longer circulation time was arranged according to the brain/blood ratios of phage particles. Poly (ethyleneglycol) - poly (lactide-co-glycolic acid) nanoparticles (PEG-PLGA NP) were prepared by the emulsion/evaporation method, and then the ligands screened from phage display were attached covalently to the surface of PEG-PLGA NP. The physicochemical characteristics of the nanoparticles were investigated. The brain-targeting efficiency of this system was evaluated in vitro and in vivo using coumarin 6 as a probe. The in vitro cytotoxicity of the nanoparticle system was investigated by CCK-8 assay.
2. Materials and Methods
2.1. Materials and animals
Ph.D.-12™ phage display library Kit was purchased from New England Biolabs (Beverly, MA, USA). E. coli ER2738 (an F+ strain) was used for M13 phage propagation and was cultured on Luria-Bertani agar or broth at 37°C. Mouse anti-M13 monoclonal antibody was obtained from GE Healthcare(Piscataway, NJ, USA); Cy3 labeled goat anti-mouse IgG, 5-bromo-4-chloro-3-indolyl-beta-d-galactopyranoside (Xgal), coumarin 6 and coumarin 7 were all purchased from Sigma-Aldrich (St. Louis, MO, USA); 4,6-diamidino-2-phenylindole (DAPI) was purchased from Molecular Probes (Eugene, OR, USA); Isopropyl-beta-d-thiogalastoside (IPTG) was purchased from Merck (Germany); Optimal Cutting Temperature-compound ‘Tissue-Tek’ (O.C.T. compound) was from Miles Laboraories Inc. (USA). Methoxy-polyethyleneglycol Poly (lactic-co-glycolic acid) (MePEG3000-PLGA40000 (25:75)) copolymer, Maleimide-polyethyleneglycol Poly (lactic-co-glycolic acid) (Mal-PEG3400-PLGA40000 (25:75)) copolymer were prepared by University of Electronic Science and Technology of China. Plastic cell culture dishes, plates and flasks were obtained from Corning Incorporation (Lowell, MA); Dulbecco’s Modified Eagle Medium (DMEM) (high glucose) cell culture medium and fetal bovine serum (FBS) were from Gibco (Carlsbad, CA); Cell counting kit-8 (CCK-8) was obtained from Dojindo Laboratories (Japan). Deionized water (Millipore, Bedford, MA) was used through the entire study.
Adult male nude mice (16-20g) and ICR mice (18-22g) were obtained from the Sino-British Sippr/BK Lab. Animals were maintained at 22±2°C on a 12h light-dark cycle with access to food and water ad libitum. The animal experiments were carried out in accordance with the protocols evaluated and approved by the ethical committee of Fudan University.
2.2. Screening of phage libraries in vivo
To obtain brain-homing phage-displayed peptide, the appropriate time to recover phages from brain is determined according to the brain/blood ratio. The brain/blood ratio was calculated by dividing the phage titer (in TU/g tissue, TU is transducing unit) in the brain at a given time by the phage titer in blood (in TU/ml) at that time. Mice were injected in the tail vein with 1011 plaque forming units (pfu) of Ph.D.-12 ™ Phage Display Library in 100μL TBS. The phages were allowed to circulate in the mice for 15, 30 min and 1, 2, 4, 8, 12, 16, 20, 24, 30 and 36 h. The mice were then sacrificed by cervical dislocation. Blood was collected and the brain was withdrawn and weighed.
A total of 4 rounds of screening were performed with adult male ICR mice (18-22g). For the first round, ICR mice (n=3) were injected intravenously (i.v.) with 1011 pfu of Ph.D.-12 ™ Phage Display Library in 100μL TBS. Phages were allowed to circulate for a period of time prior to recovery from brain. Then, the mice were perfused through the heart with 500mL of sterile normal saline (containing 1% heparin). The brain tissue was withdrawn, weighed and homogenized under a bacteria-free environment. The homogenate was mixed together with rapidly growing Escherichia coli (ER2738 host strain, New England Biolabs Inc.). Phages in the homogenate were amplified by Escherichia coli ER2738 infection and subsequently pooled for the next round of biopanning (Txt S1). From round2 to round3, BSA was added in to TBS (round 2: 0.5%; round3: 1% BSA; round4: without BSA). Other procedures were repeated as described in round 1.
20 bacteriophage clones from the last round were randomly picked up and were subjected to DNA sequencing (ABI3730). The peptide-encoding nucleotide sequences were determined with -96g III primer (5’-HOCCC TCA TAG TTA GCG TAA CG-3’) included in Ph.D.-12™ phage display library.
2.3. Immunohistochemistry
Clone 12-2 (displayed the Pep TGN) and native M13 phage were given to two groups of ICR mice (n=3) via tail vein respectively (1011pfu in 100μL TBS). Mice were sacrificed 1h later. Cerebrum samples were fixed in 4% paraformaldehyde, followed by dehydration in 15% sucrose for 24h and 30% sucrose till deposition. Samples were frozen quickly in O.C.T. compound. Immunofluorescent detection of phages was performed by using Anti-M13 monoclonal antibody and a secondary Cy3 conjugated Anti-mouse IgG (Txt S1). Distribution of Clone 12-2 and native M13 phage in cerebrum was observed.
2.4. Peptide synthesis
All peptides were synthesized by Shanghai Sangon Biological Engineering Technology & Services Co., Ltd using standard solid-phase FMOC method and purified to >95% by high-performance liquid chromatography (HPLC). All peptides were verified using a mass spectrometer (lcms-2010a, Shimadzu, Japan).
2.5. Peptide competition
The competitive inhibitions of Clone 12-2 homing to brain were detected by the addition of synthetic Pep TGN. Clone 12-2 phages were mixed with increasing concentrations of synthetic Pep TGN respectively and then injected into mouse tail vein. The competitive inhibitory effect by added Pep TGN was quantified by evaluating phage titers.
2.6. Preparation of NP and TGN-NP
PEG-PLGA nanoparticles were prepared using the emulsion/solvent evaporation method. The ratio of MePEG-PLGA and Maleimide-PEG-PLGA is 9:1(weight). Coumarin-6-loaded nanoparticles were prepared with the same procedure except that 0.1% of coumarin 6 was added to the dichloromethane solution before emulsification and the obtained nanoparticles were subjected to a 1.5×20 cm sepharose CL-4B column (Pharmacia Biotech, Inc., Sweden) eluted with 0.05 M HEPES buffer (containing 0.15 M NaCl, pH 7.0) to remove the unentrapped coumarin 6.
Nanoparticles modified with Pep TGN (TGN-NP) were prepared by maleimide-thiol coupling reaction at room temperature for 8h. The thiol of Pep TGN was reacted with Maleimide of the nanoparticle at 1:3 or 1:1 molar ratio. The resulted TGN-modified nanoparticles were named TGN-NP (1:3) and TGN-NP(1:1), respectively. The products were then eluted with 0.01 M phosphate buffered saline buffer(PBS, pH7.4) through the 1.5×20 cm sepharose CL-4B column to remove the unconjugated proteins.
2.7. Characterization of NP and TGN-NP
The morphological examination of nanoparticles was carried out by transmission electron microscope (TEM) (H-600, Hitachi, Japan) with 2% phosphotungstic acid staining. The mean diameter and ζ-potential of the nanoparticles were determined by dynamic light scattering (DLS) using a Zeta Potential/Particle Sizer NICOMP™ 380 ZLS (PSS.NICOMP PARTICLE SIZE SYSTEM, Santa Barbara, CA). The coumarin 6 loading capacity (DLC) of NP and TGN-NP were determined by HPLC analysis after dissolving the nanoparticles in 3-time volume of acetonitrile and calculated as described previously [25]. NP and TGN-NP were lyophilized using ALPHA 2-4 Freeze Dryer (0.070 Mbar Vacuum, -80°C, Martin Christ, Germany). The surface composition of C, O and N was determined with XPS. This analysis was performed on a PHI 5000 C ESCA system (USA).
2.8. In vitro cytotoxicity of NP and TGN-NP on bEnd.3 cells
bEnd.3 cells (the immortalized mouse brain endothelial cell line) were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% FBS, 100U/mL penicillin and 100μg/m L streptomycin. 100μL of bEnd.3 Cells were seeded into 96-well plates at the density of 5×104 cells/mL and incubated at 37°C with 5%CO2 for 24h to allow cell attachment. NP, TGN-NP (1:3) and TGN-NP (1:1) were diluted with corresponding culture media to different concentrations. Cells without exposure to samples were used as control. Cell viability was evaluated by CCK-8 [26] method by calculating the percentage of absorbance for sample groups in comparison with that for the control.
2.9. In vitro uptake of NP and TGN-NP in bEnd.3 cells
bEnd.3 cells were cultured over night on the polylysine-coated glass cover slip (seeded at a density of 104 cells/cm2). After equilibrated with HBSS for 15 min, the cells were incubated with freshly prepared coumarin-6-loaded NP and TGN-NP suspensions (50μg/mL in HBSS, pH 7.4) for 0.5, 1 and 2 h at 37 °C, respectively. The uptake experiment was terminated by aspirating the test samples and washing the cell monolayer with ice-cold 0.01M PBS three times. Each cell monolayer was fixed with 4% paraformaldehyde for 20 min. Afterwards, they were washed three times with 0.01M PBS, mounted in Dako fluorescent mounting medium and observed under fluorescent microscope (Olympus, IX71, Japan).
2.10. In vivo brain distribution of NP and TGN-NP
2.10.1. In vivo imaging analysis
The near infrared dye DiR was employed as a probe. 0.1% DiR was added to the dichloromethane solution before emulsification. The DiR loaded NP or TGN-NP was prepared using the emulsion/solvent evaporation method. Free DiR was separated by Sepharose CL-4B column. NP formulations were injected into the tail vein of nude mice (0.5mgDiR/kg) which were under anesthesia. 1h after injection, images were taken by the Maestro in vivo imaging system (CRI, MA). Brains and other main organs were harvested 2h after injection for relative accumulation comparison.
2.10.2. In vivo quantitative distribution of coumarin-6-loaded NP and TGN-NP
Three groups of mice (n=4) received i.v tail-vein administration of coumarin-6-loaded TGN-NP (1:3), coumarin-6-loaded TGN-NP (1:1) and coumarin-6-loaded NP (30μg/kg) respectively. Mice were sacrificed at 0.083h, 0.25h, 0.5h, 1h, 2h, 4h, 8h, 12h, and 24h after injection. Brains were collected immediately, washed twice with normal saline solution. Coumarin 6 concentration was measured by Agilent 1100 High Performance Liquid Chromatography system (HPLC) with a C-18 column (4.6×200mm) (Txt S1).
2.11. Statistical analysis
The statistical analysis of the samples was undertaken using a Student’s t-test when one group was compared with the control group. P-values <0.05 were considered statistically significant. All statistical analyses were performed using Stata version 8.0. All data reported are means±standard deviations, unless otherwise noted.
3. Results and Discussion
3.1. In vivo phage display
Though using culture cells for biopanning is more convenient, it is well known that the in vitro situation can be quite different from the in vivo situation. In vivo phage display has proven to be very effective in selecting phages with high organ specificity upon systemic injection [15, 19]. Therefore, we decided to select in vivo for brain targeting peptides. Previous phage display biopanning for brain homing peptide have been performed during a very short circulation time in vivo, usually 5-15 minutes being allowed before phages recovery from brain tissues [19, 22]. However the screening results can be affected by high backgrounds of phages resulted from vein administration of phages. Phages recovered from the brain could be easily “polluted” by phages from the circulation especially the first few hours after intravenous injection (Fig S1.). Jun Zou et al reported that the accumulation of intact infectious phage particles in tissues (calculated by dividing the titer (in TU/g tissue) in a given organ at a given time by the titer in blood (in TU/ml) at that time) differed [27]. We found that the brain/blood ratios of phage particles from library we employed in this article peaked at 24h (Fig 1B). Thus, we chose 24h as the optimal time point to recover phages from the brain after tail-vein administration.
Fig 1. (A) Four rounds of in vivo screening.
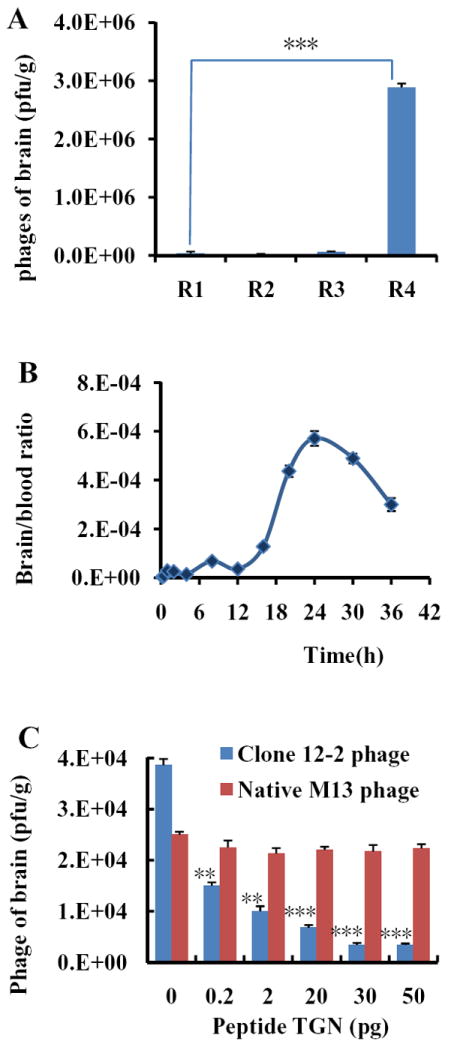
Phages were injected i.v. into four groups of ICR mice. Rescued phages from the fourth round had a great augment (mean±S.D., n=3, *** p<0.005). (B) The time curve of brain/blood ratio of M13 phages titer. The accumulation of intact infectious phage particles in brains was analyzed by the ratio of phage particles in brain and in blood. The accumulation of phages in brain peaked at 24h. (mean±S.D., n=3). (C) Pep TGN competition with Clone 12-2. At the amount of 20μg, Pep TGN efficiently prevent the Clone 12-2 entering into the brain. (mean±S.D.), n=3 **p< 0.01, ***p<0.005).
Four rounds of consecutive biopanning were conducted to select peptide-ligands that could improve transport efficiency of macromolecules into the brain. The efficiency of phage to reach the brain increased after each round of panning and the amount of recovered phage increased about 250 times compared to the first round (Fig 1A). After the final round of biopanning, total 20 peptides were sequenced from randomly selected individual phage clones. The sequences of the displayed peptides were shown in Tab 1.
Tab 1.
Sequences of individual phage clones
| phage clone | Sequence | Frequency |
|---|---|---|
| 1 | AHSLKSITNHGL | 1 |
| 2 | TGNYKALHPHNG | 12 |
| 3 | GPSYLHRLVPAF | 12 |
| 4 | TGNYKALHPHNG | 12 |
| 5 | LPDPHATNILFR | 1 |
| 6 | TGNYKALHPHNG | 12 |
| 7 | YLPDQQLTWFPS | 12 |
| 8 | TGNYKALHPHNG | 12 |
| 9 | SISTSFHAYKLK | 1 |
| 10 | TGNYKALHPHNG | 1 |
| 11 | TPQLPIDVNADR | 1 |
| 12 | TGNYKALHPHNG | 1 |
| 13 | TGNYKALHPHNG | 12 |
| 14 | TGNYKALHPHNG | 12 |
| 15 | SEAVRHLAGPPR | 12 |
| 16 | TGNYKALHPHNG | 12 |
| 17 | TGNYKALHPHNG | 1 |
| 18 | GLNLPQNKVSFS | 1 |
| 19 | TGNYKALHPHNG | 12 |
| 20 | TGNYKALHPHNG | 12 |
A multiple sequence-alignment analysis using Lasergene (version 7.1.0) program revealed that 60% of the peptides shared a consensus sequence. Consensus amino acid motifs appearing with high frequencies were commonly observed in phage display experiments and were considered as an important evidence of the phage display selection with the progressive enrichment in phage titers along with the succeeding rounds of biopanning. The consensus motifs between phages selected via biopanning on living cells or upon injection in vivo, often appear to be three or four amino acid sequences [19, 28, 29]. Smith et al identified a brain-homing motif (AC-SYTSSTM-CGGGS) only at a frequency of 25% [30]. It is rare that in our study, 60% of the peptides share the same sequence. It indicated that the peptide sequences we identified in this study would have a higher potential to transport into the brain. We named this sequence Pep TGN for short and the phage clone encoding Pep TGN was named as Clone 12-2. The sequence of Pep TGN was analyzed by BLAST (www.ncbi.nlm.nih.gov) search database. There were no putative conserved domains detected.
The competitive inhibitions of Clone 12-2 homing to brain by synthetic Pep TGN were detected by the addition of synthetic Pep TGN. This assay was performed to determine whether the synthetic Pep TGN and Clone 12-2 competed for the same binding site. The Clone 12-2 phage homing to the brain was competitively inhibited by Pep TGN in a dose-dependent manner while there wasn’t significant competitive effect of Pep TGN on native M13 phage (Fig 1C). For Clone 12-2, the presence of Pep TGN in the concentration from 0.2pg to 50pg resulted in an inhibition of the phage homing from 61% to 91%. The competitive-inhibition effect might be due to the binding sites on the BBB which Clone 12-2 interacted with were saturated by the free Pep TGN. Less binding sites remained for Clone 12-2 will lead to decrease of Clone 12-2 transport into the brain.
3.2. Immunohistochemistry
Fluorescence microscopy examination revealed that the phage Clone 12-2 encoding Pep TGN showed a significant superiority on transport efficiency into the brain compared with native M13 phage (Fig 2). Especially, they distributed extensively in the third ventricle, the lateral ventricle, the periventricular region of the third ventricle and cortex (Fig 2 A, B, C and D respectively). In contrast, there were very weak signal in those regions mentioned above for native M13 phages under the same background level (Fig 2 E, F, G, and H respectively). Clone 12-2 was hardly observed in lung, spleen, heart, liver and kidney while native M13 phage had certain distribution in spleen, liver and kidney (Fig S2). These results revealed significant superiority of Clone 12-2 to native M13 phages on transport efficiency into the brain and therefore demonstrated the specific affinity of Pep TGN to the brain. Consequently, the Pep TGN could be available as a leading ligand to facilitate efficient transport of drugs into the brain.
Fig 2. Brain distribution of Bacteriophage in brain.
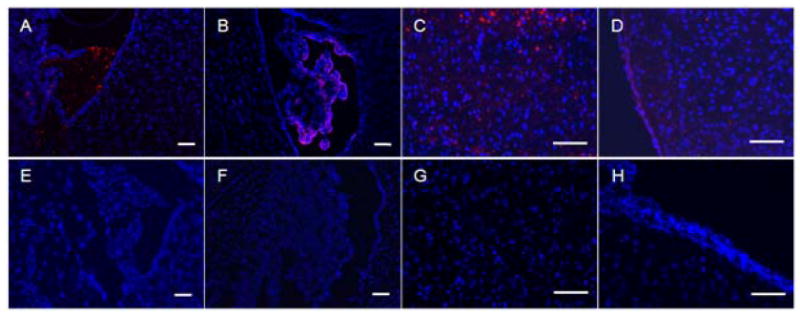
Distribution of Clone 12-2 (A-D) and native M13 phage (E-H) in third ventricle (A,E), lateral ventricle (B,F), periventricular region of the third ventricle (C,G) and cortex (D,H) visualized 1h after i.v. administration in mouse caudal vein. Red: a secondary Cy3 conjugated Anti-mouse IgG; Blue: cell nuclei stained with 1μg/mL DAPI for 10min. The magnification bar represents 100μm.
3.3. Preparation and characterization of NP and TGN-NP
Nanoparticle formulations were prepared by the emulsion/solvent evaporation method. Representative transmission electron micrographs (Fig 3) illustrated that NPs had characteristic round shape. Quantitative analysis of particle size, zeta potential and coumarin 6 encapsulation efficiency were shown in Tab 2. After conjugation with Pep TGN, the average diameter increased slightly. The polydispersity of all the formulations also showed quite narrow size distribution. The zeta potential of the NP formulations was negative (from -18mV to -24mV). In terms of drug encapsulation efficiency (EE), the slightly lower EE for the TGN-NP was most likely due to drug loss during the incubation. The elemental composition percentages of carbon, oxygen and nitrogen on the surface of unconjugated NPs were 64%, 35.9% and undetectable, respectively; while those on the surface of TGN-NP (1:1) were 63.6%, 36.1% and 0.3%, respectively (Tab S1). Nitrogen signal not detected on the surface of unconjugated NPs was observed on the surfaces of TGN-NPs instead, thus confirming Pep TGN conjugation to the NP surface (Table S1).
Fig 3. Transmission electron micrographs of NP and TGN-NP.
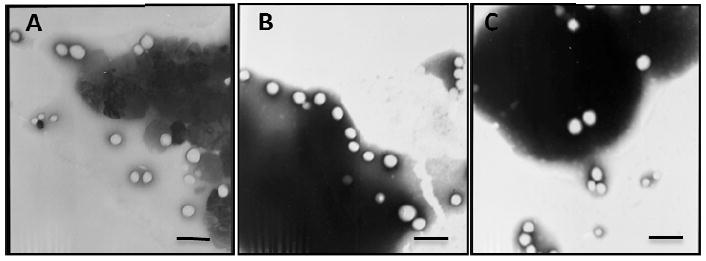
Different nanoparticles were negatively stained with phosphotungstic acid solution and observed under transmission electron micrographs: (A) NP; (B) TGN-NP (1:3); (C) TGN-NP (1:1). The bar is 200nm.
Tab 2.
Physical characterization of different nanoparticles (Data represent mean±SD, n=3)
| Formulation* | Size (nm) | PDI† | Zeta potential (mV) | EE(%)‡ |
|---|---|---|---|---|
| NP | 104.17±3.45 | 0.123±0.023 | -24.43±0.22 | – |
| TGN-NP(1:3) | 109.67±5.46 | 0.118±0.013 | -21.90±0.84 | – |
| TGN-NP(1:1) | 119.67±1.01 | 0.113±0.014 | -20.24±0.11 | – |
| coumarin-6-loaded NP | 109.27±0.55 | 0.134±0.007 | -23.44±0.52 | 64.38±0.35 |
| coumarin-6-loaded TGN-NP(1:3) | 115.23±1.04 | 0.128±0.009 | -19.78±1.05 | 59.32±1.94 |
| coumarin-6-loaded TGN-NP(1:1) | 121.46±0.76 | 0.137±0.001 | -18.25±0.88 | 58.53±2.50 |
NP: unmodified nanoparticles
TGN-NP (1:3): nanoparticles conjugated with Pep TGN at a molar ratio of 1:3 (Pep TGN: Maleimide);
TGN-NP (1:1): nanoparticles conjugated with Pep TGN at a molar ratio of 1:1 (Pep TGN: Maleimide);
NP/coumarin 6: coumari-6-loaded nanoparticles;
Refers to polydispersity index.
Refers to encapsulation efficiency. EE(%)= (drug loaded in nanoparticles/total amount of drug fed) × 100%
3.4. Cytotoxicity assay
In vitro cytotoxicity of plain NP, TGN-NP (1:3) and TGN-NP (1:1) were investigated with bEnd.3 cells after 4h, 24h incubation at 37°C by the CCK-8 assay (Fig 4). There was no significant difference in the toxicity of different formulations at any of the given concentrations (P >0.05). Cell viability was slightly reduced at higher concentrations. The PLGA polymers are generally accepted as being of low cytotoxicity with good biocompatibility and biodegradability. Thus, PLGA NP was regarded as the safety control group. No notable difference was observed between NP and TGN-NP and this confirmed the relative safety of TGN-NP compared with NP. Accordingly, TGN-NP is a promising drug carrier without any significant cytotoxic effects on cells tested.
Fig 4. In vitro cytotoxicity.
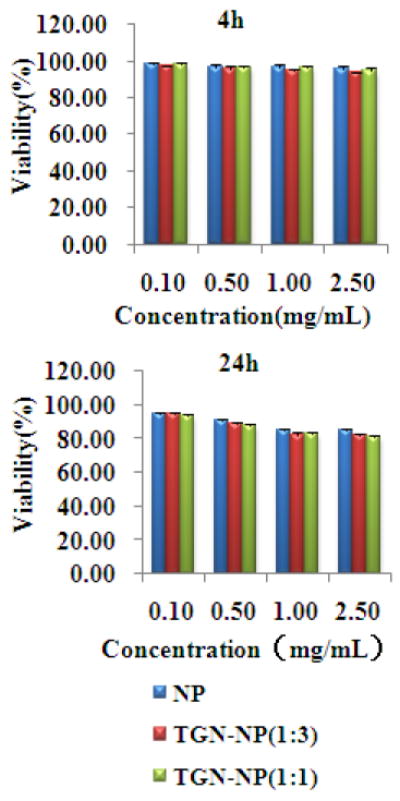
Cytotoxicity of NP, TGN-NP (1:3) and TGN-NP (1:1) incubated with bEnd.3 cells for (A) 4h, (B) 24h at 37°C. Cells without exposure to samples were used as control.
3.5. Cellular uptake
It was proved that free coumarin 6 released from PLGA nanoparticles accounted for only about 2% of the total uptake of the dye after 2h incubation (Fig S3), which was consistent with the previous report [31]. Thus, coumarin 6 detected in the cells dominantly reflected the nanoparticles behavior.
To better compare the intensity of fluorescence among the cells treated with various NP formulations, the images were taken by keeping the parameters such as sensitivity, gain and offset constant throughout the cell imaging process. The rate and the extent to which NP or TGN-NP was uptaken differed. TGN-NP was found within the cells as early as 30min. It could be observed that the green fluorescence, which corresponded to coumarin-6-loaded particles, was much stronger for TGN-NP (1:1) (Fig 5I, K, M) than that for the NP (Fig 5A, C, and E) at each time point, suggesting that the Pep TGN conjugation on the surface of nanoparticles resulted in higher cellular uptake.
Fig 5. In vitro uptake of NP and TGN-NP.
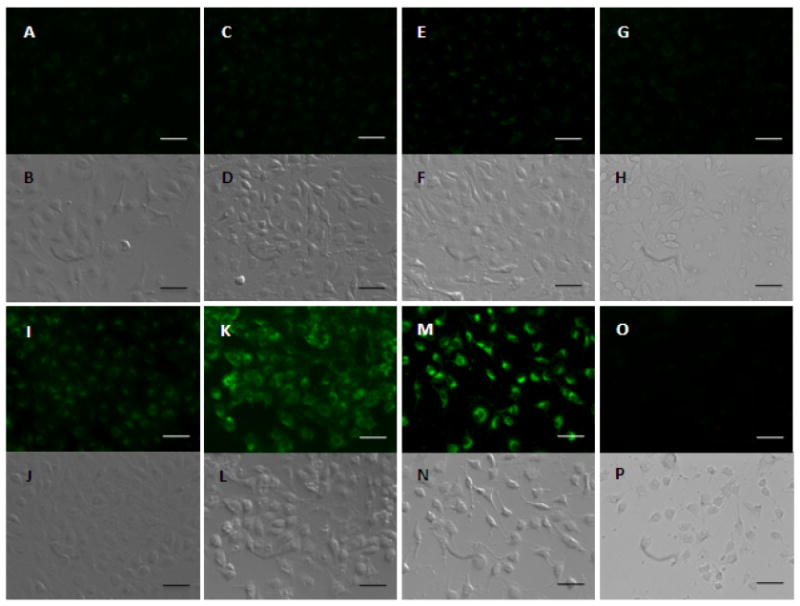
bEnd.3 cells uptake 50μg/mL NP at 37°C for 30min(A/B), 60min(C/D) and 120min(E/F) and 50μg/mL TGN-NP(1:1) for 30min(I/J), 60min(K/L) and 120min(M/N), respectively. bEnd.3 cells were incubated with free Pep TGN 1h before 50μg/mL NP (G/H) or 50μg/mL TGN-NP (O/P). Phase contrast photo were attached below each corresponding fluorescent photo. The bar is 25μm.
We observed a competitive effect when the cells were incubated with excess free Pep TGN 1h before the cells were incubated with the TGN-NP suspension (Fig 5O). There was no obvious difference when the cells were incubated with Pep TGN before the incubation with untargeted NP (Fig 5G). This confirmed that the enhanced uptake of TGN-NP compared to unmodified NP was contributed by Pep TGN. We inferred that the excess free Pep TGN reduced the binding sites available for the TGN-NP, thus specifically inhibiting the uptake of TGN-NP by bEnd.3 cells. The molecules or sites on the BBB which the Pep TGN interacted with need to be further identified.
3.6. Optical images in vivo
Optical in vivo images were taken 1h after injection. An obvious accumulation of DiR signal was detected in the brain of nude mice administered with DiR-loaded TGN-NP (1:1) (Fig 6A (III)) compared with that in those treated with DiR-loaded NP (Fig 6A (II)). The TGN-NP (1:3) of which surface was modified with low density Pep TGN (Fig 6A (IV)) displayed weaker fluorescence in the brain than TGN-NP (1:1). 2h after administration, main organs of nude mice were harvested. Compared with nude mice which injected with unmodified nanoparticles, higher brain accumulation efficiency was observed in those intravenously injected with both kinds of Pep TGN conjugated NP while there was lower accumulation of nanoparticles in liver and spleen (Fig 6B). The non-targeted nanoparticles were distributed mainly in liver and spleen, which might be attributed to the non-specific capture by the reticuloendothelial system. After modification, more TGN-NP entered the brain and less were captured by the liver and spleen, indicating modification of Pep TGN onto the surface can improve the organ selectivity of nanoparticles.
Fig 6. In vivo distribution of TGN-NP.
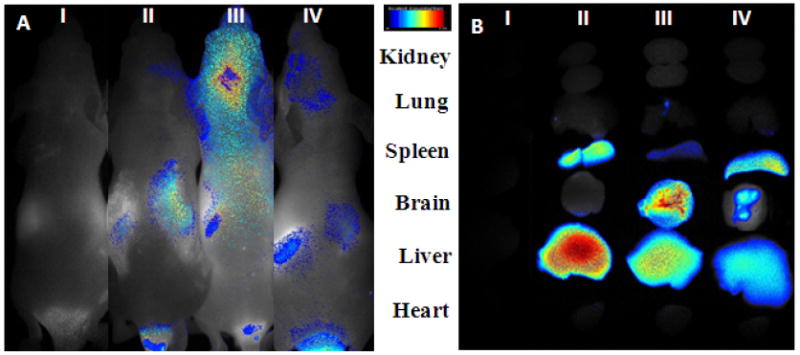
(A) Optical in vivo imaging of nude mouse vein administrated (II) NP; (III) TGN-NP (1:1); (IV) TGN-NP (1:3) respectively. (I) blank nude mice as blank control. Images were taken 1h after injection. (B) Ex vivo imaging of NPs in main organs. Nude mice were tail intravenously injected with (II) NP; (III) TGN-NP (1:1) ; (IV) TGN-NP (1:3) respectively. (I) blank nude mice as blank control. Organs were harvested 2h after administration.
3.7. Brain transport
Coumarin 6 was employed as a fluorescent marker to quantitatively compare the brain distribution between the unmodified nanoparticles and Pep TGN conjugated nanoparticles. Untargeted NP, TGN-NP (1:3) and TGN-NP (1:1) exhibited similar concentration-time profiles during the brain uptake process (Fig 7). An initial rapid and extensive increase occurred during the beginning 1h post-injection. During the first 2h following the administration, a significantly higher brain uptake of coumarin 6 was detected in mice administrated with TGN-NP (1:3) or TGN-NP (1:1) than that in those administrated with unmodified NP. At 1h, the brain uptake of coumarin 6 for TGN-NP (1:3) and TGN-NP (1:1) was 55.2ng/g and 95.1ng/g respectively, about 2.1 and 3.6-fold higher than that for non-targeted nanoparticles (26.3ng/g). At 24h, the brain uptake of TGN-NP (1:3) and TGN-NP (1:1) was 2 and 4-fold higher than the unmodified nanoparticles, showing the enhanced brain accumulation. The Drug Targeting Index (DTI) of coumarin 6 in the brain by TGN-NP (1:3) and TGN-NP (1:1) was about 2.84 and 3.78 folds compared with that of coumarin 6 associated to NP, respectively (Table 3.). The Pep TGN played a critical role in the brain distribution of TGN-NP.
Fig 7. Concentrations-time curves of coumarin 6 in cerebrum.
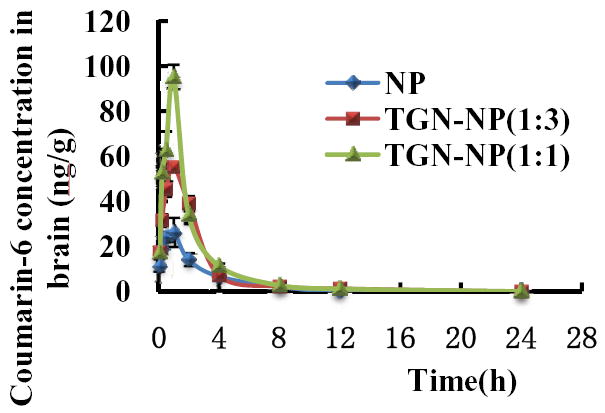
ICR mice were i.v. injected of coumarin-6-loaded NP, TGN-NP (1:3) and TGN-NP (1:1) at a dose of 30μg/kg in mice, respectively. Data represented the mean±S.D. n=4.
Tab 3.
AUC0-t comparisons of biodistribution curves of NP, TGN-NP (1:3) and TGN-NP (1:1) in brain versus blood
| AUC0-t | NP | TGN-NP (1:3) | TGN-NP (1:1) |
|---|---|---|---|
| AUC0-tBrain | 85.53 | 167.54 | 214.13 |
| AUC0-tBlood | 205.77 | 141.72 | 136.15 |
| Ratio brain/blood | 0.42 | 1.18 | 1.57 |
| DTI* | - | 2.84 | 3.78 |
As indicated in the study, the accumulation of nanoparticles in the brain enhanced with the increased modification of Pep TGN on the nanoparticle surface. We speculated that the more Pep TGN on the surface, the bigger chance that Pep TGN will have to interact with the BBB, thus leading to enhanced brain transport efficiency. This demonstrates the great prospect of Pep TGN as ligand in the brain targeting and these characteristics could be optimized by adjusting the density of Pep TGN on the surface.
4. Conclusions
A consensus sequence of TGNYKALHPHNG (denoted as Pep TGN) was obtained by rounds of in vivo phage display screening. Phage Clone 12-2 displaying Pep TGN revealed a significant superiority on brain transport efficiency compared with native M13 phage. When conjugated on the surface of PLGA nanoparticles, Pep TGN facilitated the targeted delivery of nanoparticles across the BBB, leading to significant higher bEnd.3 cells uptake and in vivo brain accumulation. To best of our knowledge, the Pep TGN is a brain-target motif never been reported before and could be applied to the design of drug delivery system homing to the brain. Further studies are right under way to identify the molecules or sites to which the Pep TGN binds on the BBB. Since the sequences of the binding motifs do not reveal any similarities with known receptor or markers, the Pep TGN may be useful in the discovery of new receptors or markers and in the brain-targeted drug delivery.
Supplementary Material
1 × 1011 pfu of M13 phages from Ph.D.-12™ phage display peptide library were injected into ICR mice. The blood samples were harvested at the indicated time intervals. Data represented the mean±SD. n = 3.
Distribution of Clone 12-2 in lung (A1-3), spleen (B1-3), heart (C1-3), liver (D1-3) and kidney (E1-3) and distribution of native M13 phages in lung (F1-3), spleen (G1-3), heart (H1-3), liver (ID1-3) and kidney (J1-3) visualized 1h after i.v. administration in mouse caudal vein. Red(A1, B1, C1, D1, E1, F1,G1, H1, I1, J1): a secondary Cy3 conjugated Anti-mouse IgG; Blue (A2, B2, C2, D2, E2, F2, G2, H2, I2, J2): cell nuclei stained with 1μg/mL DAPI for 10min. The magnification bar represents 100μm.
In vitro leakage of coumarin 6 from coumarin-6-loaded NP or coumarin-6-loaded TGN-NPs in PBS buffer of pH 4.0 and pH 7.4 Data represented the mean±SD. n = 3.
Acknowledgments
This work was supported in part by grants from the National Basic Research Program of China (973 Program) (2007CB935800), National Science and Technology Major Project 2009ZX09310-006, Doctorial Innovation Fund of Fudan University, National Institutes of Health (NIH) R01 Grants NS066945, and a Hartwell Foundation Biomedical Research Award. This work was also partially sponsored by Grant R31-2008-000-10103-01 from the WCU project of South Korea. Victor C. Yang is currently a participating faculty member in the Department of Molecular Medicine and Biopharmaceutical Sciences, Seoul National University, South Korea.
We thank Xiaomei Wang (University of Science & Technology of China, School of Life Sciences) for technological support in phage display screening. We thank Haorang Zhang (Peking University School of Pharmaceutical Sciences) for assistance with quantitative analysis of brain uptake of NPs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Miller G. Drug targeting. Breaking down barriers. Science. 2002;297:1116–8. doi: 10.1126/science.297.5584.1116. [DOI] [PubMed] [Google Scholar]
- 2.Praveen B, Alex B, Maiken N. The blood-brain barrier: an overview: structure, regulation and clinical implications. Neurobiol Dis. 2004;16:1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 3.Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pardridge WM. Blood-brain barrier delivery. Drug Discov Today. 2007;12:54–61. doi: 10.1016/j.drudis.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 5.Gabathuler R. Approaches to transport therapeutic drugs across the blood-brain barrier to treat brain diseases. Neurobiol Dis. 2010;37:48–57. doi: 10.1016/j.nbd.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 6.Begley DJ. Delivery of therapeutic agents to the central nervous system: the problems and the possibilities. Pharmacol Thel. 2004;104:29–45. doi: 10.1016/j.pharmthera.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Liu X, Chen C. Strategies to optimize brain penetration in drug discovery. Curr Opin Drug Disc. 2005;8:505–12. [PubMed] [Google Scholar]
- 8.Béduneau A, Saulnier P, Benoit JP. Active targeting of brain tumors using nanocarriers. Biomaterials. 2007;28:4947–67. doi: 10.1016/j.biomaterials.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 9.Calvo P, Gouritin B, Chacun H, Desmaële D, D’Angelo J, Noel JP, et al. Long circulating PEGylated polycyanoacrylate nanoparticles as new drug carrier for brain delivery. Pharm Res. 2001;18:1157–66. doi: 10.1023/a:1010931127745. [DOI] [PubMed] [Google Scholar]
- 10.Pardridge WM. Molecular biology of the blood-brain barrier. Mol Biotechnol. 2005;30:57–70. doi: 10.1385/MB:30:1:057. [DOI] [PubMed] [Google Scholar]
- 11.Huwyler J, Wu D, Pardridge WM. Brain drug delivery of small molecules using immunoliposomes. Proc Natl Acac Sci. 1996;93:14164–9. doi: 10.1073/pnas.93.24.14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Pardridge WM. Delivery of β-galactosidase to mouse brain via the blood–brain barrier transferrin receptor. J Pharmaco Exp Ther. 2005;313:1075–108. doi: 10.1124/jpet.104.082974. [DOI] [PubMed] [Google Scholar]
- 13.Hu KL, Li JW, Shen YH, Gao XL, Zhang QZ, Jiang XG. Lactoferrin-conjugated PEG–PLA nanoparticles with improved brain delivery: In vitro and in vivo evaluations. J Control Rel. 2009;134:55–61. doi: 10.1016/j.jconrel.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 14.Demeule M, Regina A, Che C, Poirier J, Nguyen T, Gabathuler R, et al. Identification and design of peptides as a new drug delivery system for the brain. Pharmacol Exp Ther. 2008;324:1064–72. doi: 10.1124/jpet.107.131318. [DOI] [PubMed] [Google Scholar]
- 15.Smith GP, Petrenko VA. Phage Display. Chem Rev. 1997;97:391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- 16.Rodi DJ, Janes RW, Sanganee HJ, Holton RA, Wallace BA, Makowski L. Screening of a library of phage-displayed peptides identifies human Bcl-2 as a taxol-binding protein. J Mol Biol. 1999;285:197–203. doi: 10.1006/jmbi.1998.2303. [DOI] [PubMed] [Google Scholar]
- 17.Samoylova TI, Petrenko VA, Morrison NE, Globa LP, Baker HJ, Cox NR. Phage probes for malignant glial cells. Mol Cancer Ther. 2003;2:1129–37. [PubMed] [Google Scholar]
- 18.Meyers SR, Khoo XJ, Huang X, Walsh EB, Grinstaff MW, Kenan DJ. The development of peptide-based interfacial biomaterials for generating biological functionality on the surface of bioinert materials. Biomaterials. 2009;30:277–86. doi: 10.1016/j.biomaterials.2008.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pasqualini R, Ruoslahti E. Organ targeting in vivo using phage display peptide libraries. Nature. 1996;380:364–6. doi: 10.1038/380364a0. [DOI] [PubMed] [Google Scholar]
- 20.Haubner R, Wester HJ, Burkhart F, Senekowitsch SR, Weber W, Goodman SL, et al. Glycosylated RGD-containing peptides, tracer for tumor targeting and angiogenesis imaging with improved biokinetics. J Nucl Med. 2001;42:326–36. [PubMed] [Google Scholar]
- 21.Arap W, Haedicke W, Bernasconi M, Kain R, Rajotte D, Krajewski S, et al. Targeting the prostate for destruction through a vascular address. Proc Natl Acad Sci. 2002;99:1527–31. doi: 10.1073/pnas.241655998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolonin MG, Sun J, Do KA, Vidal CI, Ji Y, Baggerly KA, et al. Synchronous selection of homing peptides for multiple tissues by in vivo phage display. FASEB J. 2006;20:E99–107. doi: 10.1096/fj.05-5186fje. [DOI] [PubMed] [Google Scholar]
- 23.Wan XM, Chen YP, Xu WR, Yang WJ, Wen LP. Identification of nose-to-brain homing peptide through phage display. Peptides. 2009;30:343–50. doi: 10.1016/j.peptides.2008.09.026. [DOI] [PubMed] [Google Scholar]
- 24.Rooy IV, Tascioglu SC, Couraud PO, Romero IA, Weksler B, Storm G, et al. Identification of peptide ligands for targeting to the blood-brain barrier. Pharm Res. 2010;27:673–82. doi: 10.1007/s11095-010-0053-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao XL, Tao WX, Lu W, Zhang QZ, Zhang Y, Jiang XG. Lectin-conjugated PEG-PLA nanoparticles: Preparation and brain delivery after intranasal administration. Biomaterials. 2006;27:3482–90. doi: 10.1016/j.biomaterials.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 26.Salles II, Tucker AE, Voth DE, Ballard JD. Toxin-induced resistance in Bacillus anthracis lethal toxin-treated macrophages. Proc Natl Acad Sci. 2003;100:12426–12431. doi: 10.1073/pnas.2134042100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou J, Dickerson MT, Owen NK, Landon LA, Deuscher SL. Biodistribution of filamentous phage peptide libraries in mice. Mol Biol Rep. 2004;31:121–9. doi: 10.1023/b:mole.0000031459.14448.af. [DOI] [PubMed] [Google Scholar]
- 28.Rajotte D, Arap W, Hagedorn M, Koivunen E, Pasqualini R, Ruoslahti E. Molecular heterogeneity of the endothelium revealed by in vivo phage display. J Clin Invest. 1998;102:430–7. doi: 10.1172/JCI3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicklin SA, White SJ, Watkins SJ, Hawkins RE, Baker AH. Selective targeting of gene transfer to vascular endothelial cells by use of peptides isolated by phage display. Circulation. 2000;102:231–7. doi: 10.1161/01.cir.102.2.231. [DOI] [PubMed] [Google Scholar]
- 30.Smith MW, Gumbleton M. In vivo phage display to identify peptides that target the brain. Drug Discov Today. 2010;15:1113. [Google Scholar]
- 31.Desai MP, Labhasetwar V, Wwalter E, Levy RJ, Amidon GL. The mechanism of uptake of biodegradable microparticles in Caco-2 cells is size dependent. Pharm Res. 1997;14:1568–73. doi: 10.1023/a:1012126301290. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1 × 1011 pfu of M13 phages from Ph.D.-12™ phage display peptide library were injected into ICR mice. The blood samples were harvested at the indicated time intervals. Data represented the mean±SD. n = 3.
Distribution of Clone 12-2 in lung (A1-3), spleen (B1-3), heart (C1-3), liver (D1-3) and kidney (E1-3) and distribution of native M13 phages in lung (F1-3), spleen (G1-3), heart (H1-3), liver (ID1-3) and kidney (J1-3) visualized 1h after i.v. administration in mouse caudal vein. Red(A1, B1, C1, D1, E1, F1,G1, H1, I1, J1): a secondary Cy3 conjugated Anti-mouse IgG; Blue (A2, B2, C2, D2, E2, F2, G2, H2, I2, J2): cell nuclei stained with 1μg/mL DAPI for 10min. The magnification bar represents 100μm.
In vitro leakage of coumarin 6 from coumarin-6-loaded NP or coumarin-6-loaded TGN-NPs in PBS buffer of pH 4.0 and pH 7.4 Data represented the mean±SD. n = 3.