

Abstract
The nucleus of the solitary tract (NTS) is the major site for termination of visceral sensory afferents contributing to homeostatic regulation of, for example, arterial pressure, gastric motility, and breathing. Whereas much is known about how different neuronal populations influence these functions, information about the role of glia remains scant. In this article, we propose that glia may contribute to NTS functions by modulating excitatory neurotransmission. We found that acidification (pH 7.0) depolarizes NTS glia by inhibiting K+-selective membrane currents. NTS glia also showed functional expression of voltage-sensitive glutamate transporters, suggesting that extracellular acidification regulates synaptic transmission by compromising glial glutamate uptake. To test this hypothesis, we evoked glutamatergic slow excitatory potentials (SEPs) in NTS neurons with repetitive stimulation (20 pulses at 10 Hz) of the solitary tract. This SEP depends on accumulation of glutamate following repetitive stimulation, since it was potentiated by blocking glutamate uptake with dl-threo-β-benzyloxyaspartic acid (TBOA) or a glia-specific glutamate transport blocker, dihydrokainate (DHK). Importantly, extracellular acidification (pH 7.0) also potentiated the SEP. This effect appeared to be mediated through a depolarization-induced inhibition of glial transporter activity, because it was occluded by TBOA and DHK. In agreement, pH 7.0 did not directly alter d-aspartate-induced responses in NTS glia or properties of presynaptic glutamate release. Thus acidification-dependent regulation of glial function affects synaptic transmission within the NTS. These results suggest that glia play a modulatory role in the NTS by integrating local tissue signals (such as pH) with synaptic inputs from peripheral afferents.
Keywords: glia, astrocyte, glutamate uptake, nucleus of the solitary tract, CNS acid-base influence, synaptic transmission, TBOA
besides providing metabolic and structural support for neurons, glia also modulate neuronal activity, synaptic transmission, and critical physiological functions (Angulo et al. 2004; Fellin et al. 2004; Gourine et al. 2010; Grosche et al. 1999; Oliet et al. 2001; Perea et al. 2009; Poskanzer and Yuste 2011; Wang and Bordey 2008). The caudal nucleus of the solitary tract (NTS), a major hub of visceral sensory input, mediates regulation of crucial homeostatic functions such as breathing, arterial pressure, gastric motility, and food intake (Alheid and McCrimmon 2008; Dean and Putnam 2010; Herman et al. 2009; Kanoski et al. 2012). Light and electron microscopy studies have shown that astrocytic processes cover excitatory synapses in the NTS, suggesting they can regulate neurotransmission (Chounlamountry and Kessler 2011). NTS glia respond to stimulation of afferent fibers in the solitary tract with an increase in intracellular calcium, and selective activation of astrocytes leads to glutamate-dependent activation of NTS neurons (Hermann et al. 2009; McDougal et al. 2011), implying that astrocytes are functionally integrated within NTS networks that modulate sensory processing. However, the role of glial function on neuronal synaptic transmission within the NTS remains unknown.
Glia throughout the brain buffer and uptake glutamate released during synaptic transmission through expression of high-affinity glutamate transporters (Anderson and Swanson 2000). This safeguards neurons from excitotoxicity by maintaining a low ambient concentration of glutamate (Rothstein et al. 1996), limits glutamate spillover to preserve synapse specificity (Arnth-Jensen et al. 2002; Asztely et al. 1997), and controls activation of extra- and presynaptic receptors (Chen and Diamond 2002; Huang and Bordey 2004; Mitchell and Silver 2000). In addition, glia are characterized by a particularly hyperpolarized resting membrane potential, which is set by background potassium channels (Wang and Bordey 2008; Zhou et al. 2009). Importantly, the activity of several of these channels is regulated by both extracellular and intracellular pH (Enyedi and Czirják 2010; Mulkey and Wenker 2011; Patel and Honoré 2001). Downregulation of background potassium channel activity depolarizes cortical glia, and since glutamate transport is electrogenic (Zerangue and Kavanaugh 1996), this leads to a decrease in glutamate clearance (Djukic et al. 2007; Kucheryavykh et al. 2007).
In this article, we present evidence supporting a novel mechanism for glia to modulate excitatory synaptic transmission within the NTS. We show that extracellular acidification depolarizes the glial membrane potential through an effect on pH-sensitive potassium channels. Acidification-induced glial depolarization limits glutamate uptake, thereby potentiating the glutamatergic slow excitatory potential previously described in the NTS (Ballanyi et al. 1993; Fortin et al. 1992). These findings suggest that glia may regulate the integration of peripheral synaptic afferents with local tissue signals (such as pH) to modulate NTS functions.
MATERIALS AND METHODS
All animals were handled in accordance with National Institutes of Health guidelines and protocols were approved by the Northwestern University Animal Care and Use Committee.
Acute slice preparation and patch-clamp recordings.
Brain stem slices were obtained from 14- to 25-day-old male Sprague-Dawley rats. Animals were deeply anesthetized with isoflurane and then decapitated. The brain stem was quickly removed into ice-cold low-Ca2+, high-Mg2+ artificial cerebrospinal fluid (ACSF; in mM: 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 25 glucose, 0.1 CaCl2, and 7 MgCl2) equilibrated with 95% O2 and 5% CO2.
Transverse slices (275–300 μm) containing the NTS (from between ∼925 and 100 μm caudal to the obex; i.e., the opening of the central canal into the 4th ventricle) were cut using a Vibratome (Thermo Scientific), and recordings were made from commissural and medial NTS. Slices were stored in sucrose-based ACSF (in mM: 87 NaCl, 25 NaHCO3, 25 glucose, 75 sucrose, 2.5 KCl, 1.25 NaH2PO4, 0.1 CaCl2, and 7 MgCl2; equilibrated with 95% O2 and 5% CO2) for ∼30 min at 33–35°C after cutting and at room temperature thereafter until used for recordings.
Slices were transferred to a recording chamber constantly superfused with regular bicarbonate-buffered ACSF (in mM: 125 NaCl, 2.5 KCl, 25 NaHCO3, 25 glucose, 2 CaCl2, 1 MgCl2, bubbled with 95% O2 and 5% CO2) at room temperature (22–24°C, unless stated otherwise). Recordings were made with an Axopatch 200B (Axon Instruments) and/or MultiClamp 700A (Axon Instruments) amplifier from up to two cells at once, digitized with a 1440A Digidata acquisition board (Molecular Devices), and displayed and saved on a digital computer with Clampex 10.2 software (Molecular Devices). Slices were visualized with an upright microscope (Olympus) with oblique infrared illumination and video microscopy using a digital camera (DVC). Patch pipettes were fabricated from borosilicate capillary tubes (Dagan: OD = 1.65 mm, ID = 0.75 mm; or Sutter instruments: OD = 1.5 mm, ID = 0.86 mm) using a horizontal (P97; Sutter Instruments) or vertical puller (PC-10; Narishige) and had tip resistances of 2–8 MΩ when filled with working solution. Traces were filtered at 10 kHz (current clamp) or 5 kHz (voltage clamp) using the built-in filter and digitized at 20 or 10 kHz, respectively.
Extracellular acidification (from pH 7.4 to 7.0) was obtained either by reducing the concentration of NaHCO3 from 25 to 9 mM (while NaCl concentration was increased from 125 to 141 mM) or by focal application of a HEPES-buffered ACSF titrated at pH 7.0 (see below). Recordings were performed at room temperature unless otherwise specified (such as in Fig. 8).
Fig. 8.
SEP potentiation by acidification is also observed at near physiological temperature. A and B: recordings performed at a bath temperature of 35°C. Bath acidification potentiated the SEP to 146 ± 28% of control (n = 19). *P < 0.05, Wilcoxon signed-rank test.
Patch-clamp recordings from neurons.
Synaptic recordings were made using a K-gluconate-based solution (in mM: 130 K-gluconate, 10 NaCl, 2 MgCl2, 2 Na2ATP, 0.2 NaGTP, 10 EGTA, and 10 HEPES, titrated to pH 7.3 with KOH) in the presence of 100 μM picrotoxin and 1 μM strychnine (to block GABAergic and glycinergic neurotransmission, respectively). Series resistances were <25 MΩ and were not compensated. Synaptic stimulation was done using a large (∼1–2 MΩ) pipette filled with HEPES-buffered ACSF (HB-ACSF; in mM: 138 NaCl, 2.5 KCl, 10 HEPES, 25 glucose, 2 CaCl2, and 1 MgCl2, titrated to pH 7.4 with 1 N NaOH) placed in the solitary tract. Responses were evoked using a 200-μs-long pulse delivered through an isolation unit (A365; World Precision Instruments) with a stimulus intensity of 50–200 μA. Correct placement of the stimulating pipette was verified through presence of synaptic currents in response to a single shock in voltage-clamp mode (at −60 mV). Slow excitatory potentials (SEPs) were recorded in the current-clamp mode, and negative DC current was injected to keep the baseline membrane potential around −70 mV (0 to −100 pA). SEPs were recorded in response to trains of 20 shocks delivered at a frequency of 10 Hz (see Fig. 4A). This train was repeated at a frequency of 0.02 Hz, and 3–10 traces were averaged for each condition. The baseline was zeroed, and the SEP amplitude was determined by averaging data points within a 20-ms window, 300 ms after the last stimulus in the train (dotted vertical line in Fig. 4A), relative to a similar baseline window preceding the train. The 300-ms time frame was chosen because fast excitatory postsynaptic potentials (EPSPs) showed a full decay to baseline within 300 ms. A K-thiocyanate-based internal solution was used for recording the response to focal application of d-aspartate (d-Asp) on neurons (see Fig. 5C; in mM: 160 K-thiocyanate, 10 EGTA, 10 HEPES, 2 Na2ATP, 0.2 NaGTP, and 1 MgCl2).
Fig. 4.

Repetitive stimulation evokes glutamatergic slow excitatory potentials in NTS neurons. A: current-clamp recording from a neuron in an acute NTS slice shows that stimulation with a train of stimuli (20 pulses at 10 Hz) evoked both fast excitatory postsynaptic potentials (EPSPs; top) and a slow excitatory potential (SEP; bottom) which decayed over several seconds. The vertical dotted line is placed 300 ms after the last stimulus; the peak of the SEP is calculated over a 20-ms window from this time point. B: the SEP was not mediated by group I/II metabotropic glutamate receptor (mGluR) signaling, since it was not modified by bath application of (RS)-α-methyl-4-carboxyphenylglycine (MCPG). C: NMDA receptor blockade by d-(−)-2-amino-5-phosphonopentanoic acid (d-AP5; 50 μM) attenuated the SEP. D: 6,7-dinitroquinoxaline-2,3-dione disodium salt (DNQX; 20 μM), a blocker of AMPA receptors, also inhibited the SEP. E: bar chart summarizing the effects of the drugs tested, normalized to control. SEPs were evoked 5 min apart in a time control (TC) experiment. Amplitude of the second SEP was 99.9 ± 10.8% of the first one. MCPG did not affect the SEP (it was 93.8 ± 22.4% of control, n = 6), whereas d-AP5 and DNQX reduced the SEP to 54.4 ± 21.1% (n = 8) and 26.5 ± 10.5% (n = 7) of control values. *P < 0.05, Wilcoxon signed-rank test; stimulus artifacts have been truncated.
Fig. 5.

Glutamate transporter blockade potentiates the SEP. A: blocking glial glutamate transporters with TBOA potentiates the SEP to 263 ± 40% of control (n = 9). B: focal application of d-Asp onto an NTS neuron voltage-clamped at −70 mV did not elicit transporter currents (8 cells). C: blocking the glia-specific glutamate transporter GLT-1 with DHK potentiated the SEP to 157 ± 37% of control (n = 8). *P < 0.05, Wilcoxon signed-rank test.
Patch-clamp recordings from glia.
To identify potential glia, we used the dye sulforhodamine-101 (SR-101), which in the NTS predominantly labels astrocytes (McDougal et al. 2011). Slices were incubated in sucrose-based ACSF containing 0.67 μM SR-101 for 25 min, and then for 15 min in dye-free sucrose ACSF at 34–35°C, followed by storage at room temperature. A KCl-based intrapipette solution was used for all glial whole cell patch-clamp recordings (in mM: 140 KCl, 2 MgCl2, 10 EGTA, 10 HEPES, 2 Na2ATP, 0.2 NaGTP, and 20 sucrose, titrated to pH 7.3 with 2 N KOH). Rhodamine-labeled cellular structures resembling glia were targeted for recordings while avoiding large-diameter structures resembling neurons. The glial phenotype of these cells was confirmed by the absence of action potentials in response to positive current injections of up to 6 nA (see Fig. 1A).
Fig. 1.

Nucleus of the solitary tract (NTS) glia are pH sensitive. A: current-clamp recording from an NTS glia showing the voltage response to positive and negative current injections in 0.5-nA steps. B: average current (I)-voltage (Vm) relationship of NTS glia (n = 13). C: focal application of pH 7.0 artificial cerebrospinal fluid (ACSF) onto an NTS glia from a baseline of pH 7.4 led to a reversible depolarization. D: similarly, isocapnic acidification of the bath with a solution containing 9 mM bicarbonate (HCO3) also depolarized NTS glia.
We used pressure applications (10–20 psi) through puffer pipettes pulled from single-barrel borosilicate glass tubing with resistances of ∼1–2 MΩ connected to a Picospritzer II (General Valve) for focal application. A HEPES ACSF solution titrated to pH 7.0 was used to focally acidify glia. d-Asp (100 or 500 μM) was dissolved in bath ACSF (which contained 5 mM kynurenic acid, 100 μM picrotoxin, and 1 μM strychnine; bubbled with 95% O2 and 5% CO2). Puffer pipettes for the current-voltage relationship of d-Asp-induced responses were pulled from single-barrel tubing, whereas pipettes for experiments requiring drug applications were pulled from theta glass tubing (Sutter Instruments). One side of the theta pipette was filled with a solution containing d-Asp, whereas the other contained the drug of interest in addition to d-Asp. Drugs were also preapplied in the bath. To eject solution from only one barrel at a time, a piece of polyethylene tubing was inserted in each of the barrels and the output line of the Picospritzer was manually switched from one outlet to another. Barium-containing solution was applied using a gravity-driven quartz pipe (OD = 360 μm, ID = 255 μm; Polymicro Technologies) placed near the patched glia.
All chemicals, as well as kynurenic acid and strychnine, were purchased from Sigma-Aldrich. d-(−)-2-Amino-5-phosphonopentanoic acid (d-AP5), picrotoxin, 6,7-dinitroquinoxaline-2,3-dione disodium salt (DNQX), (RS)-α-methyl-4-carboxyphenylglycine (MCPG), dl-threo-β-benzyloxyaspartic acid (TBOA), (3S)-3-[[3-[[4-(trifluoromethyl)benzoyl]amino]phenyl]methoxy]-l-aspartic acid (TFB-TBOA), and (2S,3S,4R)-2-carboxy-4-isopropyl-3-pyrrolidineacetic acid (DHK) were purchased from either Tocris (R & D Systems) or Ascent Scientific (Abcam Chemicals). Drugs were dissolved in stock solutions (kynurenic acid, 500 mM in 1 N NaOH; picrotoxin, 50 mM in DMSO; DNQX, 20 mM in dH2O; d-AP5, 50 mM in dH2O; MCPG, 100 mM in dH2O; TBOA, 100 mM in DMSO; TFB-TBOA, 1 mM in DMSO; DHK, 25 mM in dH2O; strychnine, 1 mM in dH2O) and stored at 2–3°C (kynurenic acid, picrotoxin, strychnine) or −20°C (d-AP5, DNQX, MCPG, TFB-TBOA, DHK). Working solutions were prepared daily. Acidified ACSF or drug were bath applied for ∼5 min.
Analysis of electrophysiological data.
Electrophysiological data were analyzed using Clampfit 10.2 software (Molecular Devices). The effect of acidification on glial membrane potential was measured by subtracting the peak potential reached during application of pH 7.0 (averaged over a 1- to 5-s window) from the preceding baseline. For voltage-clamp ramp experiments, the current at the peak of the ramp (0 mV) was measured during perfusion of control solution (pH 7.4) or focal application of pH 7.0. A hyperpolarizing pulse (300 ms, from −70 to −90 mV; not shown) was used to measure the input resistance during control and pH 7.0 application.
To determine the current-voltage relationship of the d-Asp-induced currents, we focally applied d-Asp at holding potentials ranging from −100 to −40 mV in 10-mV increments. Currents were normalized to the current evoked at −80 mV. We determined the net current amplitude for each potential by subtracting the peak amplitude from the preceding baseline. Currents were normalized to the current evoked at −80 mV. For experiments investigating the pharmacology of transport currents, we measured the charge transfer induced by a 3-s focal application of d-Asp by integrating over a 20- to 40-s window from the beginning of the application.
The effect of acidification and barium on glia was tested using paired t-tests. The effect of external K+ on the pH-sensitive current in glia was assessed using an unpaired t-test. Effects of the experimental treatments on the neuronal SEP were evaluated using the two-tailed Wilcoxon signed-rank test (unless stated otherwise). One-tailed paired t-tests were used for assessing the effects of pharmacological agents on transport currents, and two-tailed t-tests were used for all other analyses, unless stated otherwise. Data were normalized to the group average of control values for each respective experiment. All data are means ± SE; error bars in graphs represent ±SE.
RESULTS
NTS glia are pH sensitive.
We performed patch-clamp recordings from acute slices to investigate proton modulation of NTS glia and its possible contribution to NTS chemosensitivity. To identify glial cells, transverse brain stem slices containing the NTS were stained with 0.67 μM SR-101 (see materials and methods) and fluorescently labeled cellular structures resembling glial morphology were targeted for patch-clamp recordings. Glial phenotype of the patched cells was confirmed by their characteristic negative resting membrane potential (−81.6 ± 1.1 mV; n = 19) and a passive current-voltage relationship (n = 13; Fig. 1, A and B). Pressure application of HB-ACSF titrated to pH 7.0 from a pipette depolarized glia by 11.2 ± 3 mV (Fig. 1C; n = 6). Likewise, bath application of pH 7.0 bicarbonate-buffered ACSF (containing 9 mM NaHCO3, bubbled with 95% O2 and 5% CO2) caused a depolarization of 8.5 ± 1.5 mV (Fig. 1D; n = 5).
Next, we used voltage-clamp ramp recordings to elucidate the ionic mechanism underlying this depolarization (Fig. 2). Membrane currents were recorded in control conditions and during focal application of pH 7.0 (HB-ACSF) while the membrane potential was ramped from −130 to 0 mV (from a holding potential of −70 mV, Fig. 2A). Subtracting the current traces obtained in pH 7.0 from control revealed that the pH-sensitive current (Fig. 2A, bottom) has a linear current-voltage relationship and reverses at −89.1 ± 5.5 mV (n = 8), close to the theoretical K+ Nernst potential of −102 mV expected for our experimental conditions (2.5 mM extracellular K+). Acidification led to a significant reduction in the membrane current recorded at 0 mV (Fig. 2B; 3.35 ± 0.4 vs. 2.39 ± 0.3 vs. 3.06 ± 0.4 nA in pH 7.4, pH 7.0, and washout, respectively; n = 8; P < 0.05), which was accompanied by an increase in membrane resistance (Fig. 2B; 26.9 ± 3.9 vs. 39.9 ± 7.0 vs. 30.1 ± 5.7 MΩ in pH 7.4, pH 7.0, and washout, respectively; n = 8; P < 0.05).
Fig. 2.

Inhibition of K+-selective currents mediates the pH sensitivity of NTS glia. A: NTS glia were voltage-clamped at −70 mV. The voltage was stepped down to −130 mV, slowly ramped up to 0 mV (1-s duration), and then stepped back down to −70 mV (top trace). Middle traces, current response recorded in pH 7.4 and during focal application of pH 7.0 (HEPES-buffered ACSF; average of 5–7 traces) in the presence of 2.5 mM extracellular K+. Bottom trace, the pH-sensitive component obtained by digital subtraction of the current in pH 7.0 from the control current. In this cell, the pH-sensitive current reversed at −90 mV (Erev, reversal potential). B: bar chart summarizing results from 8 cells. Focal acidification reduced the peak current (I) obtained at 0 mV from 3.35 ± 0.4 to 2.39 ± 0.3 nA. This reduction was accompanied by an increase in membrane resistance (Rm) from 26.9 ± 3.9 to 39.9 ± 7.0 MΩ. C: the same voltage ramp as in A was used to measure membrane currents in control solution (pH 7.4) and during focal application of pH 7.0 in the presence of 10 mM external K+. Digital subtraction of the membrane current in pH 7.0 from control reveals the pH-sensitive component, which reversed at −64 mV in this cell. D: bar chart summarizing the Erev of pH-sensitive membrane currents measured in the presence of 2.5 (n = 8) or 10 mM (n = 5) external K+. *P < 0.05, 2-tailed paired Student's t-test. **P < 0.05, 2-tailed independent Student's t-test.
To test the K+ selectivity of the pH-sensitive current, we focally applied pH 7.0 (HB-ACSF) in the presence of 10 mM extracellular K+. Under this condition, the reversal potential of the pH-sensitive current was depolarized to −59.1 ± 3.3 mV (close to the predicted K+ Nernst potential of −67 mV; n = 5), compared with −89.1 ± 5.5 mV in the presence of 2.5 mM extracellular K+ (n = 8; Fig. 2, C and D; P < 0.05, 2-tailed independent Student's t-test). Increasing the extracellular K+ concentration from 2.5 to 10 mM led to an average ∼30-mV depolarizing shift in the reversal potential of the pH-sensitive current, close to the 35-mV shift predicted by the Nernst equation for a K+-selective conductance.
These experiments acidified the extracellular solution by switching from bicarbonate-buffered (pH 7.4) to HEPES-buffered (pH 7.0) ACSF. Hence, the observed reduction in the membrane current might be due to a reduction in the bicarbonate concentration, rather than acidification. To address this possibility, we repeated the acidification experiments in slices superfused with HEPES-buffered ACSF (pH 7.4, bubbled with 100% O2, 2.5 mM extracellular K+). Similar to previous experiments, focal acidification (HEPES-buffered titrated to pH 7.0) led to a significant increase in the membrane resistance (41.7 ± 6.8 vs. 51.5 ± 7.9 MΩ in pH 7.4 and pH 7.0, respectively; n = 5; P < 0.05; data not shown). As expected, the pH-sensitive current reversed at −90 ± 6.7 mV (n = 4), close to the K+ Nernst potential of −102 mV. Together, these results suggest that pH 7.0 depolarizes glia in the NTS by inhibiting a K+-selective conductance.
NTS glia show functional expression of glutamate transport currents.
Glia throughout the brain express high-affinity glutamate transporters and play a vital role in clearing synaptically released glutamate (Wang and Bordey 2008). Thus we tested for functional expression of glutamate transporters in glia to determine the role of glutamate uptake during synaptic transmission in the NTS. Since NTS glia have been shown to express AMPA receptors (McDougal et al. 2011), we used the substrate d-Asp (instead of l-glutamate) to minimize activation of ionotropic glutamate receptors (iGluRs). To further limit contamination from ionotropic receptor activation (in particular, NMDA receptors), we made these recordings in the presence of 5 mM kynurenic acid, 100 μM picrotoxin, and 1 μM strychnine. As shown in Fig. 3A, a 1-s focal application of 500 μM d-Asp (at a holding potential of −80 mV) led to a slowly activating inward current that declined over several seconds. To determine the current-voltage relationship of the transporter current, we applied d-Asp at holding potentials ranging from −100 to −40 mV in 10-mV increments. As expected for electrogenic glutamate-coupled transport currents (Zerangue and Kavanaugh 1996), depolarization from a holding potential of −100 mV led to a decrease in the peak amplitude of the transport current induced by d-Asp (Fig. 3B). If the d-Asp response is mediated by transporters, then 100 μM TBOA, a broad-spectrum blocker of glutamate transporters, should inhibit this response. Indeed, TBOA inhibited the 100 μM d-Asp induced charge transfer by 88.9 ± 9.0% (Fig. 3, C and D; P < 0.05, n = 4). Together, these results show that the voltage-dependent d-Asp-induced responses in NTS glia are due to activation of glutamate transporters.
Fig. 3.

NTS glia show functional expression of electrogenic glutamate transporters. A: focal application of d-aspartate (d-Asp) onto an NTS glia voltage-clamped at −80 mV resulted in an inward current. B: current-voltage relationship of d-Asp-induced currents. Transporter currents elicited by a 1-s focal application of d-Asp were recorded at voltage potentials ranging from −100 to −40 mV in 10-mV increments (n = 5–7 for each potential) and normalized to the substrate current evoked at −80 mV (I/I−80 mV). C: pharmacological profile of d-Asp-induced currents. dl-Threo-β-benzyloxyaspartic acid (TBOA) is a nonspecific, selective blocker of glutamate transporters, whereas (3S)-3-[[3-[[4-(trifluoromethyl)benzoyl]amino]phenyl]methoxy]-l-aspartic acid (TFB-TBOA), at the concentration used, blocks glial glutamate transporters (GLAST and GLT-1). Dihydrokainate (DHK) is a specific inhibitor of GLT-1 transporters. TBOA (100 μM), TFB-TBOA (100 nM), and DHK (300 μM) were preapplied in the bath. Application of each of these antagonists led to a marked reduction in the d-Asp-induced response. D: bar chart summarizing the effects of drugs tested on the d-Asp-induced charge transfer, normalized to control. TBOA, TFB-TBOA, and DHK reduced the d-Asp-evoked responses to 11.1 ± 6.4% (n = 4), 15.4 ± 11.1% (n = 4), and 26.6 ± 4.9% (n = 4) of control values, respectively. The d-Asp-induced response recovered to 36.9 ± 13.9% and 99.8 ± 38.7% on washout of TBOA and DHK, respectively. Washout data for TFB-TBOA is not available. *P < 0.05, 1-tailed paired Student's t-test.
Glia are known to express the GLAST and GLT-1 glutamate transporters (Chaudhry et al. 1995; Rothstein et al. 1996). Immunocytochemical and electron microscopy studies have suggested that NTS glia predominantly express the GLT-1 (EAAT2) glutamate transporter (Chounlamountry and Kessler 2011). To determine the identity of the transporters underlying the d-Asp-induced currents in NTS glia, we applied 100 μM d-Asp in the presence of 100 nM TFB-TBOA. At this concentration, TFB-TBOA selectively blocks glial glutamate transporters GLAST and GLT-1 (Shimamoto et al. 2004; Tsukada et al. 2005). Indeed, 100 nM TFB-TBOA attenuated the d-Asp induced response by 84.6 ± 11.1% (Fig. 3, C and D; P < 0.05, n = 4), similar to the proportion inhibited by TBOA. To further narrow down the identity of the transporter(s) involved, we tested the effect of 300 μM DHK, a selective blocker of GLT-1 subtype (Arriza et al. 1994). DHK reduced the charge transfer by 73.4 ± 4.9% (Fig. 3, C and D; P < 0.05, n = 4), suggesting that the majority of the transporter current is mediated by GLT-1 transporters.
Repetitive stimulation in transverse brainstem slices leads to slow excitatory potentials.
The strong pH sensitivity of glial background channels and the resulting potential inhibition of voltage-dependent glutamate uptake suggest that acidification may affect synaptic transmission in the NTS. This effect may be particularly important for the peculiarly slow excitatory potential (SEP) previously described in this brain stem area (Ballanyi et al. 1993; Fortin et al. 1992). To elicit SEPs, we repetitively stimulated the solitary tract (10 Hz, 20 pulses) and recorded the resulting excitatory postsynaptic potentials (in the presence of 100 μM picrotoxin and 1 μM strychnine) from NTS neurons. In addition to fast EPSPs (Fig. 4A, inset), this stimulation evoked SEPs that decayed exponentially over several seconds (decay time constant: 4.3 ± 0.4 s, n = 47). A time control experiment where SEPs were evoked 5 min apart demonstrated that they can be evoked reliably within this time frame (second SEP was 99.9 ± 10.9% of the first SEP; n = 4; P > 0.05; Fig. 4E); hence, drugs were applied for ∼5 min in all subsequent pharmacology experiments. Previous studies suggested that a portion of the SEP is mediated by glutamatergic signaling (Ballanyi et al. 1993; Fortin et al. 1992). Metabotropic glutamate receptor (mGluR) activation following repetitive stimulation has been shown to cause slow EPSPs in other brain areas (Hartmann et al. 2008). However, application of 500 μM MCPG, a nonselective group I/II mGluR antagonist, did not cause any significant change in the SEP amplitude (93.8 ± 22.4% of control, P > 0.05, n = 6; Fig. 4, B and E), suggesting that in the NTS mGluR signaling is not necessary for the SEP. To investigate the involvement of iGluRs, we tested the effect of 50 μM d-AP5 and 20 μM DNQX, selective antagonists for AMPA/kainate and NMDA receptors, respectively. Bath application of 50 μM d-AP5 led to a significant decrease in the SEP amplitude (to 54.4 ± 21.1% of control, P < 0.05, n = 8; Fig. 4, C and E). Application of 20 μM DNQX eliminated the fast EPSPs (not shown) and also caused a reduction in the SEP (to 26.5 ± 10.5% of control, P < 0.05, n = 7; Fig. 4, D and E). Together, these studies suggest that activation of iGluRs underlies a portion of the SEP elicited by repetitive stimulation in the NTS.
Glial glutamate transporters regulate the SEP.
Given that repetitive stimulation in the NTS leads to a SEP mediated via glutamatergic signaling, we reasoned that this process may be regulated by glial glutamate transporters. In keeping with this hypothesis, inhibiting glutamate uptake with 100 μM TBOA potentiated the SEP almost threefold (4.5 ± 1.1 mV vs. 11.7 ± 1.8 mV in control and TBOA, respectively; n = 9, P < 0.05; Fig. 5, A and B), suggesting that glutamate accumulation following repetitive stimulation causes the SEP. Because TBOA blocked a large fraction of the glial transport current (Fig. 3C), its effect on the SEP is likely mediated by glial glutamate transporters. To further rule out a contribution from neuronal glutamate uptake, we tested whether NTS neurons show functional expression of glutamate transporters. To increase our chances of detecting a response to d-Asp, we made recordings using a potassium thiocyanate-based internal solution to take advantage of the larger currents observed through the anion conductance of the transporters (Bergles and Jahr 1998). Focal application of 500 μM d-Asp (in the presence of 5 mM kynurenic acid, 100 μM picrotoxin, and 1 μM strychnine) onto neurons voltage-clamped at −70 mV led to no detectable substrate-induced currents (Fig. 5B; n = 8), further supporting the idea that TBOA affects the SEP through its effect on glia. Although these data clearly point toward an effect of TBOA on glial transporters, an effect on neuronal transporters could not be definitely ruled out. Therefore, we performed a set of recordings to investigate whether DHK, a blocker specific for the glial transporter GLT-1, also enhances the SEP amplitude. In keeping with the idea that the TBOA effect is mostly on glia transporters, DHK also significantly increased the size of the SEP (6.6 ± 2.1 vs. 10.4 ± 2.5 mV in control and DHK, respectively; n = 8, P < 0.05, 1-tailed Wilcoxon signed-rank test; Fig. 5C).
Glial background potassium channels regulate the neuronal SEP.
Given that glial glutamate transporters regulate the SEP, we hypothesized that hindering the function of these transporters would mimic the potentiating effect of TBOA and DHK on the SEP magnitude. NTS glia express pH-sensitive background potassium currents, and since barium inhibits most of them (Patel and Honoré 2001; Tang et al. 2010; Taverna et al. 2005), we tested its effect on glial membrane currents. Indeed, 1 mM Ba2+ reduced the peak current recorded in glia (at 0 mV it was reduced from 4.84 ± 0.6 to 2.74 ± 0.4 nA; n = 5; P < 0.05; Fig. 6, A and B) and led to a significant increase in the membrane resistance (15.1 ± 3.9 vs. 22.0 ± 7.0 MΩ in control and Ba2+, respectively; n = 5; P < 0.05; Fig. 6B). Because depolarizing the membrane potential reduces glutamate uptake (Fig. 3), we predicted that depolarizing the glia with barium should increase glutamate accumulation following repetitive stimulation and, therefore, potentiate the SEP. As expected, 1 mM Ba2+ potentiated the neuronal SEP to 430 ± 168% of control (n = 5, P < 0.05; Fig. 6, C and D).
Fig. 6.

Barium inhibits glial background K+ channels and potentiates the SEP. A, top trace: 1-s voltage ramp protocol used to test the effect of 1 mM Ba2+ on glial membrane currents. Middle traces, current response recorded in pH 7.4 and during local application of Ba2+. Bottom trace, Ba2+-sensitive component obtained by digital subtraction of the membrane current recorded in Ba2+ from control. In this cell, the Ba2+-sensitive current reversed at −92 mV. B: summary results from 5 cells. Ba2+ reduced the peak current obtained at 0 mV from 4.84 ± 0.6 to 2.74 ± 0.4 nA. Concurrently, Rm increased from 15.1 ± 3.9 to 22.0 ± 7.0 MΩ. C and D: bath application of 1 mM Ba2+ potentiated the SEP to 430 ± 168% of control (n = 5). *P < 0.05, paired Student's t-test. **P < 0.05, Wilcoxon signed-rank test; stimulus artifacts have been truncated.
pH regulates the SEP through glial glutamate transporters.
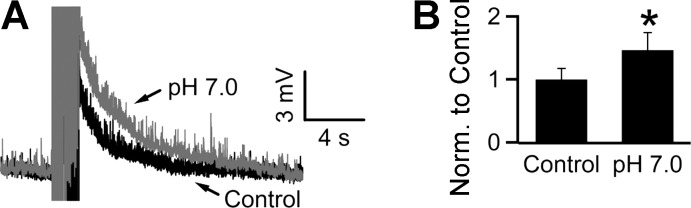
Since extracellular acidification depolarizes the glial membrane potential (Fig. 1), it should also potentiate the SEP. As expected, reducing the bath pH to 7.0 (by decreasing the NaHCO3 concentration in bicarbonate-buffered ACSF to 9 mM) potentiated the SEP to 126 ± 29% of control (7.2 ± 1.6 vs. 9.1 ± 2.1 mV in control and pH 7.0, respectively; n = 9, P < 0.05; Fig. 7A). In keeping with the hypothesis that the SEP potentiation by acidification is mediated through the depolarization-induced reduction of glial transporter activity, bath acidification (pH 7.0) did not potentiate the SEP when glial glutamate transport was blocked by either TBOA or DHK. In TBOA, pH 7.0 actually reduced the SEP to 43.7 ± 15% of control, although not significantly (16.8 ± 4.3 vs. 7.4 ± 2.5 mV in TBOA at pH 7.4 and pH 7.0, respectively; n = 6, P > 0.05; Fig. 7B). Since the SEP is mediated by iGluRs (Fig. 4), the observed decrease likely reflects the direct inhibitory effect of acidic pH on iGluRs (Dingledine et al. 1999; Mayer 2005; Mott et al. 2003; Traynelis and Cull-Candy 1990). Accordingly, the SEP response to pH 7.0 (normalized to control at pH 7.4) for all neurons tested in the absence of TBOA was significantly larger than the response to acidification in its presence (118 ± 14% vs. 53 ± 15% in the absence and presence of TBOA, respectively; n = 9 and 6, respectively; P < 0.05, unpaired Student's t-test). As expected, DHK also occluded the potentiating effect of acidification on the SEP (10.4 ± 2.5 vs. 7.8 ± 1.5 mV in DHK at pH 7.4 and pH 7.0, respectively; n = 6; P > 0.05; Fig. 7C).
Fig. 7.

Acidification potentiates the SEP through block of glutamate transporter activity. A: acidification with pH 7.0 potentiated the SEP to 126 ± 29% of control (pH 7.4). B: preapplication of TBOA in a pH 7.4 solution occluded the potentiating effect of pH 7.0 (43.7 ± 15% of the value in TBOA alone; n = 6; P > 0.05). C: similarly, preapplication of DHK also occluded the potentiating effect of acidification (84 ± 17% of the amplitude in DHK alone). D: SEPs were evoked at 10 Hz (20 pulses) or 50 Hz (50 pulses) in the presence of TBOA. The 50-Hz stimulation evoked a larger SEP than the 10-Hz stimulation (180 ± 38% of the SEP at 10 Hz; n = 6). *P < 0.05, Wilcoxon signed-rank test; stimulus artifacts have been truncated.
By inhibiting glutamate uptake, TBOA or DHK lead to an increase in glutamate accumulation following synaptic release. This may saturate the iGluRs mediating the SEP and can preclude the detection of a potentiating effect of pH 7.0 in the presence of these blockers. To address this possibility, we tested whether increasing presynaptic glutamate release (by stimulating with 50 pulses at 50 Hz) in the presence of TBOA potentiates the SEP compared with the stimulation protocol used for the occlusion experiments above (20 pulses at 10 Hz). SEPs elicited by the 50-Hz train had a significantly higher amplitude than those evoked by the 10-Hz stimulation (11.6 ± 2.5 vs. 20.9 ± 4.4 mV with 10-Hz and 50-Hz stimulation, respectively; n = 6; P < 0.05; Fig. 7C). Together, these results argue that the stimulation protocol used does not saturate the neuronal SEP and that the potentiating effect of acidification is mediated through glial glutamate transporters.
These recordings were done at room temperature, which allows for more stable recording conditions. The activity of glutamate transporters, however, is enhanced at higher temperature. Hence, we tested the pH-dependent modulation of the SEP near to normal body temperature. Acidifying the bath solution to pH 7.0 at 35°C potentiated the SEP to 146 ± 28% of control (6.0 ± 1.1 vs. 8.8 ± 1.7 mV; n = 19; P < 0.05; Fig. 8, A and B), similar to the increase observed at room temperature.
pH does not affect presynaptic glutamate release or glial glutamate uptake.
So far we have provided evidence that acidification potentiates the SEP by inhibiting glutamate uptake via glial glutamate transporters. However, acidification might have a direct impact on presynaptic glutamate release. Therefore, we tested the effect of acidification on release probability by measuring the paired-pulse ratio (PPR) of excitatory postsynaptic currents (EPSCs) evoked 20 ms apart (Fig. 9, A and B). To reliably rule out this possibility, we performed this experiment only on cells that showed a clear potentiation of the SEP with acidification at 35°C (acidification potentiated the SEP to 230 ± 80% of control in this set of cells, n = 6). EPSCs elicited in control or pH 7.0 had similar amplitudes (−178.4 ± 57 vs. −153.3 ± 63 pA, respectively; n = 6; P > 0.05) and PPR (0.60 ± 0.07 vs. 0.66 ± 0.13, respectively; n = 6; P > 0.05). These findings suggest that acidification does not significantly influence properties of presynaptic release.
Fig. 9.

Acidification does not affect synaptic paired-pulse ratio (PPR) in neurons or d-Asp-induced responses in NTS glia. A: synaptic responses were stimulated 20 ms apart in the presence of control (pH 7.4) or acidified (pH 7.0) bicarbonate-buffered ACSF. B: the normalized PPR in pH 7.0 was 109 ± 22% of control (n = 6). C: application of d-Asp induced similar responses whether focally applied in a control (pH 7.4) or pH 7.0 solution (pH 7.0 was also preapplied in the bath). D: during acidification the response was 109 ± 33% of control (n = 4).
Glutamate transporters cotransport a proton for every glutamate molecule (Zerangue and Kavanaugh 1996), suggesting that acidic pH may modulate the SEP by acting directly on the transporters, rather than through the glial membrane potential. To test this possibility, we elicited substrate currents by focal application of d-Asp dissolved in control (pH 7.4) or acidic (pH 7.0) ACSF. Since the glia membrane potential was voltage-clamped at −80 mV, any effect of acidification on the substrate current is attributable to a direct effect of pH on the transporters. The bath was acidified to pH 7.0 prior to focal application of d-Asp dissolved in acidic ACSF to avoid contamination from pH-sensitive background K+ currents (Fig. 2). d-Asp induced similar responses under both conditions (109 ± 33% of control in 9 mM NaHCO3; n = 4, P > 0.05; Fig. 9, C and D), suggesting that acidification influences glutamate uptake through an effect on the glial membrane potential.
DISCUSSION
We performed electrophysiological recordings from acute slices to test the hypothesis that glia play a modulatory role in excitatory synaptic transmission in the NTS. In keeping with this hypothesis, we found that depolarizing glia inhibits glutamate transport currents, suggesting reduced glutamate uptake, which is critical in regulating the magnitude of the SEP. As expected, the SEP is potentiated by both TBOA and DHK, blockers of glutamate uptake, and extracellular acidification, which depolarizes glia. Importantly, the potentiating effect of pH 7.0 is occluded by TBOA and DHK, suggesting that acidification works through glial glutamate transporters. Consistent with this interpretation, pH 7.0 did not exert a direct effect on substrate-coupled transporter currents in glia or on presynaptic glutamate release (Fig. 9).
What type of glia is involved?
We used the vital dye SR-101 to identify glia in the NTS. This dye has been shown to specifically label astrocytes when injected in vivo (Nimmerjahn et al. 2004) and in vitro (Kafitz et al. 2008; Takahashi et al. 2010; Uwechue et al. 2012). Importantly, in vivo injection of SR-101 into the NTS led to specific labeling of structures that were also glial fibrillary acidic protein (GFAP) immunoreactive, validating their astrocytic phenotype (McDougal et al., 2011). However, care must be taken in categorizing these cells as astrocytes based solely on the fact that they were labeled by SR-101, since our dye-loading procedure was performed on acute slices. Moreover, heterogeneity in SR-101 loading across brain regions has been described recently (Schnell et al. 2012). NTS SR-101-labeled cells displayed the typical biophysical properties associated with astrocytes (Wang and Bordey 2008). They are electrically silent (i.e., lack action potentials), have a hyperpolarized membrane potential, exhibit a low input resistance, and express large voltage-independent K+ currents (Fig. 2). In agreement with the electron microscopy studies showing expression of GLT-1 in NTS astrocytes (Chounlamountry and Kessler 2011), the SR-101-labeled cells show functional expression of glutamate transporter currents that are significantly inhibited by DHK, a GLT-1-specific inhibitor (Fig. 3C). Interestingly, studies from transgenic reporter mice show that GLT-1 promoter activity is almost completely overlapping with GFAP immunoreactivity in the brainstem, indicating that GLT-1 is preferentially expressed in astrocytes (Regan et al. 2007). Together, these observations suggest that the SR-101-labeled cells in the NTS are indeed astrocytes.
Glutamatergic signaling and slow excitatory potentials.
In agreement with studies published two decades ago (Ballanyi et al. 1993; Fortin et al. 1992), we found that synaptic stimulation of NTS neurons with a train of stimuli elicits a glutamatergic potential that decays slowly over several seconds and is mediated by both AMPA and NMDA receptors. A portion of the SEP is resistant to blockers of iGluRs and likely represents accumulation of extracellular K+ following repetitive synaptic stimulation, as described previously (Ballanyi et al. 1993). Given that neurons were held at −70 mV before synaptic stimulation, the contribution of NMDA channels in generating the SEP is surprising because of the voltage-dependent Mg2+ block (Dingledine et al. 1999; Nowak et al. 1984). However, the NR2D subunit, which is expressed by most NTS neurons (Liu and Wong-Riley 2010; Paarmann et al. 2005) shows a reduced sensitivity to blockade by Mg2+ (Kuner and Schoepfer 1996) and may mediate the NMDA receptor response. Alternatively, the depolarization induced by AMPA receptor activation might relieve a portion of the Mg2+ block, hence enabling NMDA receptor activation (Lee et al. 2007).
The presence of iGluR-mediated slow excitatory synaptic transmission is not unique to the NTS. Indeed, high-frequency stimulation of the parallel fiber to stellate cell synapse in the cerebellum results in relatively slow AMPA receptor-mediated synaptic currents that are under the control of glutamate transporters (Carter and Regehr 2000). What might be the structural basis for SEP in the NTS? Interestingly, only ∼60% of the synaptic perimeter is contacted by astrocytic processes in the NTS (Chounlamountry and Kessler 2011). Incomplete glial wrapping of the synaptic space can hinder glutamate clearance (Oliet et al. 2001), allowing the released glutamate to escape the synaptic cleft and activate extrasynaptic receptors, thus mediating a portion of the SEP. Accordingly, electron microscopy studies show that a majority of dendritic labeling for NMDA receptors in the NTS occurs at extrasynaptic sites (Aicher et al. 1999). Under normal conditions this activation would be limited by uptake of glutamate by transporters that are present in and around the synaptic space. However, reducing transporter activity with TBOA or glia depolarization (acidification) would increase the lifetime of glutamate in the extracellular space, thereby potentiating the SEP.
Our whole cell recordings also did not detect functional expression of glutamate transporters in NTS neurons (Fig. 5C), suggesting that the glial transporters are largely responsible for uptake of the released glutamate. Similar findings were obtained in the hippocampus, where outside-out patches from pyramidal neurons did not show functional expression of transporters, and synaptically evoked transporter currents were found only in glia (Bergles and Jahr 1998). However, it must be noted that our experiments were done using focal application of d-Asp onto the neuronal somata; thus transporters expressed in distal dendrites may have been missed. Moreover, neuronal transporters might be expressed at a low density that precludes detection by our method. Hence, we cannot completely rule out the expression of some types of glutamate transporters in NTS neurons; yet, we are quite confident that the observed modulation of the SEP is mostly mediated through the glial transporters.
Acidification potentiates slow excitatory potentials.
We show that extracellular acidification potentiates the SEP. Interestingly, 1 mM Ba2+, which also blocks leak potassium channels in the glia, mimics the potentiating effect of pH 7.0 (Fig. 6). Inhibition of background potassium channels by Ba2+ has been shown to reduce glutamate clearance by cultured cortical astrocytes (Kucheryavykh et al. 2007). Although these results are in agreement with our interpretation that pH 7.0 potentiates the SEP by depolarizing the glial resting potential, Ba2+ may also influence presynaptic release. However, this is unlikely since Ba2+ does not significantly affect the PPR of evoked synaptic events (Barral et al. 2003; Huang and Gean 1994; Ziakopoulos et al. 2000; but also see Choi et al. 2008).
We also found that acidification-induced potentiation of the SEP was not present to the same extent in every neuron tested, whereas TBOA and DHK potentiated the SEP in all neurons tested. Several explanations could account for this. Acidification depolarizes the glial membrane potential by ∼10 mV, which is expected to reduce glutamate uptake by ∼15% (Fig. 3B). Since TBOA inhibits ∼90% of the transporter currents in glia (Fig. 3, C and D), it is expected to have a larger effect on the neuronal SEP. As previously discussed, only ∼60% of the synaptic perimeter is contacted by astrocytic processes in the NTS, and it is likely that not all synapses are equally covered with transporter-expressing glial membrane. Therefore, any modulatory effect of pH on glutamate uptake would necessarily depend on the amount of glial wrapping at each individual synapse. In this context, it is also possible that part of this wrapping is lost during the slicing process; if that is the case, we expect that the amount of pH modulation described here represents a lower limit for what happens in the in vivo setting. Since glutamate uptake is enhanced at high temperature, it is surprising that we could not detect an effect of temperature on the acidification-induced potentiation of the SEP. However, since not all cells are responsive to acidification, the temperature sensitivity is decreased by the inclusion of nonresponsive cells in our analysis. Indeed, if only cells that show a clear potentiation with acidification are analyzed, acidification potentiates the SEP to a greater extent at 35°C than at room temperature (P < 0.05; data not shown).
Glutamate transport is directly affected by extracellular pH since it is fueled by cotransport of protons (Zerangue and Kavanaugh 1996). Although our results suggest that pH 7.0 does not significantly influence uptake (Fig. 9, C and D), glial glutamate transporters located in the vicinity of synapses might be exposed to more acidic pH since neurotransmitter-containing vesicles are usually quite acidic (Krishtal et al. 1987; Miesenbock et al. 1998). Neuronal activity following synaptic stimulation also leads to alterations in local concentrations of K+ (Ballanyi et al. 1993), another ion coupled to uptake of glutamate. Dynamic changes in the extracellular concentration of these ions, as well as the ambient level of glutamate, may influence the amount of glutamate that can be taken up by glial transporters. Hence, these variables will likely influence the extent of depolarization-induced reduction in transporter activity. Adding to this complexity is the direct effect of pH on iGluRs. For instance, AMPA, NMDA, and most kainate receptor subunits are inhibited by acidic pH (Dingledine et al. 1999; Mayer, 2005; Mott et al. 2003). In light of this, the potentiating effect of acidic pH observed in a subset of neurons likely reflects scenarios where the direct inhibitory effect of protons on iGluRs is outweighed by its inhibitory effect on glutamate uptake by glia and the ensuing increase in extracellular glutamate.
Taken together, these results strongly support the hypothesis that acidification potentiates the SEP by depolarizing glia, thereby inhibiting glutamate uptake. Although this is the most likely explanation for our data, we cannot rule out a contribution from other unknown mechanisms to the observed effects of acidification.
Implications for NTS function.
Selective chemical activation of NTS astrocytes with a proteinase-activated receptor 1 (PAR1) agonist leads to glutamate-mediated calcium signals in NTS neurons (Hermann et al. 2009). Similarly, our results suggest that NTS glia exert a modulatory influence on neuronal excitability by regulating the SEP. Glia express a wide array of receptors for various neurotransmitters and hormones (Heni et al. 2011; Lalo et al. 2011; Rao and Sikdar 2007; Spence et al. 2011). Together, these may allow glia to integrate relevant local chemical signals with incoming visceral sensory information from the periphery. In this regard, the pH-dependent modulation of glutamate uptake by glia may be particularly important for cardiorespiratory-related central chemoreception, a process that contributes importantly to pH homeostasis by regulating cardiorespiratory activity. The NTS is ideally positioned to integrate peripheral and central chemosensory information, since it receives afferent input from peripheral chemoreceptors and is also directly involved in central chemosensitivity (Dean and Putnam 2010; Huda et al. 2012; Nattie and Li 2002; Nichols et al. 2009). Moreover, NTS neurons send projections to regions in the ventral respiratory column where they provide synaptic drive to respiratory pattern- and rhythm-generating neurons as well as other chemosensitive regions such as the retrotrapezoid nucleus (Alheid et al. 2011; Huda et al. 2012; Takakura et al. 2006). Repetitive afferent input into the NTS would activate fast EPSPs and SEPs in relevant subpopulation of NTS neurons. Concurrently, acidification would lead to a depolarization of NTS glia, a decrease in glutamate uptake, and increase in the SEP (in neurons either directly or indirectly activated by peripheral chemoreceptor input). By increasing the excitability of these neurons, acidification-induced glial depolarization may potentiate activation of NTS neurons responsible for relaying chemosensory information to downstream targets of the respiratory network. Such a mechanism may contribute to the proposed central chemoreception-mediated potentiation of peripheral chemoreceptor inputs (Dempsey et al. 2012). Although speculative, this scenario highlights one way in which glia can serve as a signal-gain module of NTS circuitry that can integrate a local tissue signal (pH) with glutamatergic synaptic information.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants HL095731 (to M. Martina) and HL088580 (to D. R. McCrimmon). R. Huda was supported in part by NIH Grant F31 NS076201.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.H. and M.M. conception and design of research; R.H. performed experiments; R.H. analyzed data; R.H. and M.M. interpreted results of experiments; R.H. prepared figures; R.H. drafted manuscript; R.H., D.R.M., and M.M. edited and revised manuscript; R.H., D.R.M., and M.M. approved final version of manuscript.
REFERENCES
- Aicher S, Sharma S, Pickel V. N-Methyl-d-aspartate receptors are present in vagal afferents and their dendritic targets in the nucleus tractus solitarius. Neuroscience 91: 119–132, 1999 [DOI] [PubMed] [Google Scholar]
- Alheid GF, Jiao W, McCrimmon DR. Caudal nuclei of the rat nucleus of the solitary tract differentially innervate respiratory compartments within the ventrolateral medulla. Neuroscience 190: 207–227, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alheid GF, McCrimmon DR. The chemical neuroanatomy of breathing. Respir Physiol Neurobiol 164: 3–11, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CM, Swanson RA. Astrocyte glutamate transport: Review of properties, regulation, and physiological functions. Glia 32: 1–14, 2000 [PubMed] [Google Scholar]
- Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci 24: 6920–6927, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci 5: 325–331, 2002 [DOI] [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci 14: 5559–5569, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asztely F, Erdemli G, Kullmann DM. Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron 18: 281–293, 1997 [DOI] [PubMed] [Google Scholar]
- Ballanyi K, Branchereau P, Champagnat J, Fortin G, Velluti J. Extracellular potassium, glial and neuronal potentials in the solitary complex of rat brainstem slices. Brain Res 607: 99–107, 1993 [DOI] [PubMed] [Google Scholar]
- Barral J, Mendoza E, Galarraga E, Bargas J. The presynaptic modulation of corticostriatal afferents by μ-opioids is mediated by K+ conductances. Eur J Pharmacol 462: 91–98, 2003 [DOI] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Glial contribution to glutamate uptake at Schaffer collateral-commissural synapses in the hippocampus. J Neurosci 18: 7709–7716, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci 20: 4423–4434, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry FA, Lehre KP, Lookeren Campagne van M, Ottersen OP, Danbolt NC, Storm-Mathisen J. Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 15: 711–720, 1995 [DOI] [PubMed] [Google Scholar]
- Chen S, Diamond JS. Synaptically released glutamate activates extrasynaptic NMDA receptors on cells in the ganglion cell layer of rat retina. J Neurosci 22: 2165–2173, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi IS, Cho JH, Jeong SG, Hong JS, Kim SJ, Kim J, Lee MG, Choi BJ, Jang IS. GABAB receptor-mediated presynaptic inhibition of glycinergic transmission onto substantia gelatinosa neurons in the rat spinal cord. Pain 138: 330–342, 2008 [DOI] [PubMed] [Google Scholar]
- Chounlamountry K, Kessler JP. The ultrastructure of perisynaptic glia in the nucleus tractus solitarii of the adult rat: comparison between single synapses and multisynaptic arrangements. Glia 59: 655–663, 2011 [DOI] [PubMed] [Google Scholar]
- Dean JB, Putnam RW. The caudal solitary complex is a site of central CO2 chemoreception and integration of multiple systems that regulate expired CO2. Respir Physiol Neurobiol 173: 274–287, 2010 [DOI] [PubMed] [Google Scholar]
- Dempsey JA, Smith CA, Blain GM, Xie A, Gong Y, Teodorescu M. Role of central/peripheral chemoreceptors and their interdependence in the pathophysiology of sleep apnea. Adv Exp Med Biol 758: 343–349, 2012 [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev 51: 7–62, 1999 [PubMed] [Google Scholar]
- Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci 27: 11354–11365, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enyedi P, Czirják G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev 90: 559–605, 2010 [DOI] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 43: 729–743, 2004 [DOI] [PubMed] [Google Scholar]
- Fortin G, Velluti JC, Denavit-Saubié M, Champagnat J. Responses to repetitive afferent activity of rat solitary complex neurons isolated in brainstem slices. Neurosci Lett 147: 89–92, 1992 [DOI] [PubMed] [Google Scholar]
- Gourine AV, Kasymov V, Marina N, Tang F, Figueiredo MF, Lane S, Teschemacher AG, Spyer KM, Deisseroth K, Kasparov S. Astrocytes control breathing through pH-dependent release of ATP. Science 329: 571–575, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosche J, Matyash V, Moller T, Verkhratsky A, Reichenbach A, Kettenmann H. Microdomains for neuron-glia interaction: parallel fiber signaling to Bergmann glial cells. Nat Neurosci 2: 139–143, 1999 [DOI] [PubMed] [Google Scholar]
- Hartmann J, Dragicevic E, Adelsberger H, Henning HA, Sumser M, Abramowitz J, Blum R, Dietrich A, Freichel M, Flockerzi V, Birnbaumer L, Konnerth A. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron 59: 392–398, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heni M, Hennige AM, Peter A, Siegel-Axel D, Ordelheide AM, Krebs N, Machicao F, Fritsche A, Häring HU, Staiger H. Insulin promotes glycogen storage and cell proliferation in primary human astrocytes. PLoS One 6: e21594, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman MA, Cruz MT, Sahibzada N, Verbalis J, Gillis RA. GABA signaling in the nucleus tractus solitarius sets the level of activity in dorsal motor nucleus of the vagus cholinergic neurons in the vagovagal circuit. Am J Physiol Gastrointest Liver Physiol 296: G101–G111, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GE, Van Meter MJ, Rood JC, Rogers RC. Proteinase-activated receptors in the nucleus of the solitary tract: evidence for glial-neural interactions in autonomic control of the stomach. J Neurosci 29: 9292–9300, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Gean PW. Paired-pulse depression of the N-methyl-d-aspartate receptor-mediated synaptic potentials in the amygdala. Br J Pharmacol 113: 1029–1035, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Bordey A. Glial glutamate transporters limit spillover activation of presynaptic NMDA receptors and influence synaptic inhibition of Purkinje neurons. J Neurosci 24: 5659–5669, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huda R, Pollema-Mays SL, Chang Z, Alheid GF, McCrimmon DR, Martina M. Acid-sensing ion channels contribute to chemosensitivity of breathing-related neurons of the nucleus of the solitary tract. J Physiol 590: 4761–4775, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafitz KW, Meier SD, Stephan J, Rose CR. Developmental profile and properties of sulforhodamine 101-labeled glial cells in acute brain slices of rat hippocampus. J Neurosci Methods 169: 84–92, 2008 [DOI] [PubMed] [Google Scholar]
- Kanoski SE, Zhao S, Guarnieri DJ, DiLeone RJ, Yan J, De Jonghe BC, Bence KK, Hayes MR, Grill HJ. Endogenous leptin receptor signaling in the medial nucleus tractus solitarius affects meal size and potentiates intestinal satiation signals. Am J Physiol Endocrinol Metab 303: E496–E503, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishtal OA, Osipchuk YV, Shelest TN, Smirnoff SV. Rapid extracellular pH transients related to synaptic transmission in rat hippocampal slices. Brain Res 436: 352–356, 1987 [DOI] [PubMed] [Google Scholar]
- Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, Skatchkov SN, Eaton MJ. Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 55: 274–281, 2007 [DOI] [PubMed] [Google Scholar]
- Kuner T, Schoepfer R. Multiple structural elements determine subunit specificity of Mg2+ block in NMDA receptor channels. J Neurosci 16: 3549–3558, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalo U, Pankratov Y, Parpura V, Verkhratsky A. Ionotropic receptors in neuronal-astroglial signalling: what is the role of “excitable” molecules in non-excitable cells. Biochim Biophys Acta 1813: 992–1002, 2011 [DOI] [PubMed] [Google Scholar]
- Lee CJ, Mannaioni G, Yuan H, Woo DH, Gingrich MB, Traynelis SF. Astrocytic control of synaptic NMDA receptors. J Physiol 581: 1057–1081, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Wong-Riley MTT. Postnatal development of N-methyl-d-aspartate receptor subunits 2A, 2B, 2C, 2D, and 3B immunoreactivity in brain stem respiratory nuclei of the rat. Neuroscience 171: 637–654, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML. Glutamate receptor ion channels. Curr Opin Neurobiol 15: 282–288, 2005 [DOI] [PubMed] [Google Scholar]
- McDougal DH, Hermann GE, Rogers RC. Vagal afferent stimulation activates astrocytes in the nucleus of the solitary tract via AMPA receptors: evidence of an atypical neural-glial interaction in the brainstem. J Neurosci 31: 14037–14045, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394: 192–195, 1998 [DOI] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. Glutamate spillover suppresses inhibition by activating presynaptic mGluRs. Nature 404: 498–502, 2000 [DOI] [PubMed] [Google Scholar]
- Mott DD, Washburn MS, Zhang S, Dingledine RJ. Subunit-dependent modulation of kainate receptors by extracellular protons and polyamines. J Neurosci 23: 1179–1188, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey DK, Wenker IC. Astrocyte chemoreceptors: mechanisms of H+ sensing by astrocytes in the retrotrapezoid nucleus and their possible contribution to respiratory drive. Exp Physiol 96: 400–406, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattie EE, Li A. CO2 dialysis in nucleus tractus solitarius region of rat increases ventilation in sleep and wakefulness. J Appl Physiol 92: 2119–2130, 2002 [DOI] [PubMed] [Google Scholar]
- Nichols NL, Mulkey DK, Wilkinson KA, Powell FL, Dean JB, Putnam RW. Characterization of the chemosensitive response of individual solitary complex neurons from adult rats. Am J Physiol Regul Integr Comp Physiol 296: R763–R773, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Kerr JND, Helmchen F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Methods 1: 31–37, 2004 [DOI] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature 307: 462–465, 1984 [DOI] [PubMed] [Google Scholar]
- Oliet SHR, Piet R, Poulain DA. Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science 292: 923–926, 2001 [DOI] [PubMed] [Google Scholar]
- Paarmann I, Frermann D, Keller BU, Villmann C, Breitinger HG, Hollmann M. Kinetics and subunit composition of NMDA receptors in respiratory-related neurons. J Neurochem 93: 812–824, 2005 [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honoré E. Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci 24: 339–346, 2001 [DOI] [PubMed] [Google Scholar]
- Perea G, Navarrete M, Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci 32: 421–431, 2009 [DOI] [PubMed] [Google Scholar]
- Poskanzer KE, Yuste R. Astrocytic regulation of cortical UP states. Proc Natl Acad Sci USA 108: 18453–18458, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SP, Sikdar SK. Acute treatment with 17β-estradiol attenuates astrocyte-astrocyte and astrocyte-neuron communication. Glia 55: 1680–1689, 2007 [DOI] [PubMed] [Google Scholar]
- Regan MR, Huang YH, Kim YS, Dykes-Hoberg MI, Jin L, Watkins AM, Bergles DE, Rothstein JD. Variations in promoter activity reveal a differential expression and physiology of glutamate transporters by glia in the developing and mature CNS. J Neurosci 27: 6607–6619, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein J, Dykes-Hoberg M, Pardo C, Bristol L, Jin L, Kuncl R, Kanai Y, Hediger M, Wang Y, Schielke J, Welty D. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16: 675–686, 1996 [DOI] [PubMed] [Google Scholar]
- Schnell C, Hagos Y, Hulsmann S. Active sulforhodamine 101 uptake into hippocampal astrocytes. PLoS One 7: e49398, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto K, Sakai R, Takaoka K, Yumoto N, Nakajima T, Amara SG, Shigeri Y. Characterization of novel l-threo-β-benzyloxyaspartate derivatives, potent blockers of the glutamate transporters. Mol Pharmacol 65: 1008–1015, 2004 [DOI] [PubMed] [Google Scholar]
- Spence RD, Hamby ME, Umeda E, Itoh N, Du S, Wisdom AJ, Cao Y, Bondar G, Lam J, Ao Y. Neuroprotection mediated through estrogen receptor-α in astrocytes. Proc Natl Acad Sci USA 108: 8867–8872, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi DK, Vargas JR, Wilcox KS. Increased coupling and altered glutamate transport currents in astrocytes following kainic-acid-induced status epilepticus. Neurobiol Dis 40: 573–585, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura ACT, Moreira TS, Colombari E, West GH, Stornetta RL, Guyenet PG. Peripheral chemoreceptor inputs to retrotrapezoid nucleus (RTN) CO2-sensitive neurons in rats. J Physiol 572: 503–523, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Schmidt TM, Perez-Leighton CE, Kofuji P. Inwardly rectifying potassium channel Kir4.1 is responsible for the native inward potassium conductance of satellite glial cells in sensory ganglia. Neuroscience 166: 397–407, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna S, Tkatch T, Metz AE, Martina M. Differential expression of TASK channels between horizontal interneurons and pyramidal cells of rat hippocampus. J Neurosci 25: 9162–9170, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Cull-Candy SG. Proton inhibition of N-methyl-d-aspartate receptors in cerebellar neurons. Nature 345: 347–350, 1990 [DOI] [PubMed] [Google Scholar]
- Tsukada S, Iino M, Takayasu Y, Shimamoto K, Ozawa S. Effects of a novel glutamate transporter blocker, (2S,3S)-3-{3-[4-(trifluoromethyl)benzoylamino]benzyloxy}aspartate (TFB-TBOA), on activities of hippocampal neurons. Neuropharmacology 48: 479–491, 2005 [DOI] [PubMed] [Google Scholar]
- Uwechue NM, Marx MC, Chevy Q, Billups B. Activation of glutamate transport evokes rapid glutamine release from perisynaptic astrocytes. J Physiol 590: 2317–2331, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DD, Bordey A. The astrocyte odyssey. Prog Neurobiol 86: 342–367, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerangue N, Kavanaugh MP. Flux coupling in a neuronal glutamate transporter. Nature 383: 634–637, 1996 [DOI] [PubMed] [Google Scholar]
- Zhou M, Xu G, Xie M, Zhang X, Schools GP, Ma L, Kimelberg HK, Chen H. TWIK-1 and TREK-1 are potassium channels contributing significantly to astrocyte passive conductance in rat hippocampal slices. J Neurosci 29: 8551–8564, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziakopoulos Z, Brown MW, Bashir ZI. GABAB receptors mediate frequency-dependent depression of excitatory potentials in rat perirhinal cortex in vitro. Eur J Neurosci 12: 803–809, 2000 [DOI] [PubMed] [Google Scholar]