

Abstract
Recent data revealed that protein kinase C-potentiated myosin phosphatase inhibitor of 17 kDa (CPI-17), a myosin phosphatase inhibitory protein preferentially expressed in smooth muscle, is upregulated/activated in several diseases but whether this CPI-17 increase plays a causal role in pathologically enhanced vascular smooth muscle contractility and blood pressure remains unclear. To address this possibility, we generated a smooth muscle-specific CPI-17 transgenic mouse model (CPI-17-Tg) and demonstrated that the CPI-17 transgene was selectively expressed in smooth muscle-enriched tissues, including mesenteric arteries. The isometric contractions in the isolated second-order branch of mesenteric artery helical strips from CPI-17-Tg mice were significantly enhanced compared with controls in response to phenylephrine, U-46619, serotonin, ANG II, high potassium, and calcium. The perfusion pressure increases in isolated perfused mesenteric vascular beds in response to norepinephrine were also enhanced in CPI-17-Tg mice. The hypercontractility was associated with increased phosphorylation of CPI-17 and 20-kDa myosin light chain under basal and stimulated conditions. Surprisingly, the protein levels of rho kinase 2 and protein kinase Cα/δ were significantly increased in CPI-17-Tg mouse mesenteric arteries. Radiotelemetry measurements demonstrated that blood pressure was significantly increased in CPI-17-Tg mice. However, no vascular remodeling was detected by morphometric analysis. Taken together, our results demonstrate that increased CPI-17 expression in smooth muscle promotes vascular smooth muscle contractility and increases blood pressure, implicating a pathological significant role of CPI-17 upregulation.
Keywords: phosphatase inhibitory protein, protein kinase C-potentiated myosin phosphatase inhibitor of 17 kilodaltons, hypercontractility
protein kinase c-potentiated myosin phosphatase inhibitor of 17 kDa (CPI-17), a myosin phosphatase inhibitory protein, is predominantly expressed in mature smooth muscle, with especially high levels in arteries (8, 38). Under physiological conditions, CPI-17 plays a critical role in regulating smooth muscle contraction (16, 17, 20). It is established that the reversible phosphorylation of CPI-17 at Thr38 regulates its activity (8, 34). Phosphorylated CPI-17 directly binds to myosin phosphatase, inhibits its activity, and consequently causes the accumulation of phosphorylated 20-kDa myosin light chain (MLC20) and smooth muscle contraction (7). In isolated enzyme systems, multiple kinases have been demonstrated to phosphorylate CPI-17 (7). In mature vascular smooth muscle tissue, rho kinase (ROCK) 2 is believed to be primarily responsible for slow, sustained CPI-17 phosphorylation, whereas protein kinase C (PKC) is believed to be mainly responsible for fast CPI-17 phosphorylation (6).
Recently, accumulating evidence indicated that, in addition to reversible phosphorylation, the expression levels of CPI-17 are altered under various pathological conditions. CPI-17 mRNA or protein is increased in airway smooth muscle in antigen-induced airway hyperresponsive rats (33), in the bladder detrusor muscle of alloxan-induced type 1 diabetic rabbits (3), and in the aorta of type 2 diabetic db/db mice (40). CPI-17 protein is diminished in intestinal smooth muscle during intestinal inflammation (29) and in the neointima of wire-injured rat aorta (15). CPI-17 upregulation is often associated with increased smooth muscle contractility, whereas CPI-17 downregulation is often associated with decreased smooth muscle contractility. Together, these data implicate a role for CPI-17 expression levels in the alteration of smooth muscle contractility under various disease conditions. Moreover, we have demonstrated that CPI-17 upregulation/activation in the type 2 diabetic db/db vasculature is associated with a significant blood pressure increase (36), further suggesting a possible role for CPI-17 activation/upregualtion in increasing blood pressure. However, in addition to CPI-17, numerous alterations are present in smooth muscle under such pathological conditions. Consequently, it is unclear whether increased CPI-17 expression contributes to these observed changes in smooth muscle contractility and blood pressure. To address this issue, we have generated a smooth muscle-specific CPI-17 transgenic mouse model (CPI-17-Tg). Using this model and a combination of physiological, pharmacological, and biochemical approaches, the current study tests the hypothesis that CPI-17 upregulation in vascular smooth muscle increases both vascular smooth muscle contractility and blood pressure.
EXPERIMENTAL PROCEDURES
Generation of smooth muscle-specific CPI-17 transgenic mice.
A 557-bp mouse CPI-17 cDNA (11) containing a Kozak sequences was amplified by RT-PCR and ligated into the transgenic vector. The CPI-17 expression is under the control of rabbit smooth muscle myosin heavy chain (SM-MHC) promoter. The procedures for cloning the SM-MHC promoter and generating the smooth muscle-specific transgenic vector were previously described (25). As shown in Fig. 1A, the SMC-specific CPI-17 transgenic vector contains a rabbit SM-MHC promoter, a mouse CPI-17 cDNA, a chimeric intron, and a SV40 late poly(A). The linear SMC-specific CPI-17 transgenic vector was microinjected into zygotes by the University of Kentucky Transgenic Mouse Facility. The zygotes are in a mixed genetic background of C57BL/6 and C3H/HeN. Pups derived from the microinjected embryos were screened for the presence of the CPI-17 transgene by mouse-tail genotyping PCR using two sets of primers: the first set of primers, Trans-CPI-17-up (TGTCCACTCCCAGTTCAATTACAG) and Trans-CPI-17-down (GACCTCGTCCGGCATGTCT), was used to amplify a fragment from intron to the CPI-17 gene; the second set of primers, SM-MHC-up (CACTGCTCTTGGATTAGC) and (GGAGCCCTGAAAGAGAAAGG)-down, was used to amplify a fragment in the SM-MHC promoter. Five independent founder mice were found to be positive in both sets of PCR. Pups derived from the five founders were then subjected to Western blot analysis using CPI-17 antibodies. Two of the five founders were found to express CPI-17 protein in vascular smooth muscle tissues; these animals were retained in the laboratory and backcrossed with C57BL/6J mice at least nine generations for the current studies. All animal studies were performed in accordance with the American Association for Accreditation of Laboratory Animal Care “Guidelines for the Care and Use of Experimental Animals” and were approved by the Institutional Animal Care and Use Committee at the University of Kentucky.
Fig. 1.

Characterization of smooth muscle-selective protein kinase C-potentiated myosin phosphatase inhibitor of 17 kDa (CPI-17) transgenic (Tg) mice. A: schematic diagram demonstrating the CPI-17 transgene plasmid construct. Note that CPI-17 expression is driven by the rabbit smooth muscle myosin heavy chain (SM-MHC) promoter. B: representative Western blots of CPI-17 in a variety of tissues from wild-type (WT) and CPI-17-Tg mice. As controls, the expression of GAPDH and β-actin was not different between WT and CPI-17-Tg mice. The amounts of total protein loaded for each of the tissues are as follows: mesenteric artery and bladder: 40 μg; heart, brain, and kidney: 50 μg; and skeletal muscle: 100 μg. C: quantification of CPI-17 overexpression in CPI-17-Tg mouse mesenteric arteries; n = 7. D: representative immunofluorescent images of CPI-17 (a and d), smooth muscle α-actin (b and e), and merge of CPI-17 and smooth muscle α-actin (c and f) in cryosections of femoral arteries from WT control and CPI-17-Tg mice. ***P < 0.001.
Isometric tension measurements in isolated mesenteric artery helical strip.
Age- and gender-matched CPI-17-Tg and littermate control wild-type (WT) mice were anesthetized with isoflurane and killed, as approved by the Animal Research Committee. The mesenteric artery beds were removed immediately and placed in 24°C HEPES-buffered Krebs solution; one secondary order branch from each vascular bed was dissected free from surrounding fat and cut into small spiral strips (∼3 mm in length and 0.4 mm in width). The endothelium was denuded by gentle scraping with a razor blade, and the successful denudation was verified by losses of maximal-dose acetylcholine (1 μM)-induced relaxation. The two ends of the strips were tied to two tungsten wire hooks with monofilament thread and stretched to 1.3× resting length, which has been determined in pilot length-tension studies to be the optimal or near-optimal length in the control and CPI-17-Tg mice. The length-tension curve was not constructed in each strip due to the relative rapid “run-down” of the contractions in mouse mesenteric artery tissue. One of the tungsten wires was attached to a force transducer (AE 801; SensoNor, Horten, Norway), and isometric contractions were measured at 24°C in a well on a “bubble” plate (9, 14). The solution was changed completely by immersing the muscle strip into an adjacent bubble by sliding the bubble plate. The muscle strips were stimulated with 154 mM high-potassium solution three to four times until a stable response was obtained. ANG II (100 nM) or cumulative doses of phenylephrine (PE), serotonin (5-HT), or U-46619 were added in a random order. The strips were then incubated in a Ca2+-free solution and permeabilized with 17.5 μg/ml Staphylococcus aureus α-toxin for 40 min. To deplete the sarcoplasmic reticulum of calcium, all permeabilized strips were treated with A-23187 (10 μM; Calbiochem) as described (19). The pCa-tension curves were obtained by cumulative addition of increased doses of free calcium using a 10 mM EGTA-buffered solution as described previously (19).
Mesenteric vascular bed perfusion.
Mesenteric vascular bed perfusion was carried out using the HSE Perfusion System (PS1 Type 834/7; Harvard Apparatus, Gruenstrasse, Germany). Eight pairs of male WT and CPI-17-Tg mice were killed, and the abdominal cavity was opened. The superior mesenteric artery was identified, cleaned of connective tissue, cannulated, and perfused at 1 ml/min for 5 min with Krebs solution containing heparin (100 U/ml). The composition of the Krebs solution was as follows (mmol/l): 113 NaCl, 4.8 KCl, 1.2 MgSO4, 1.2 NaH2PO4, 1.2 CaCl2, 25 NaHCO3, and 5.5 glucose. Following this initial perfusion period, the mesentery was separated from the intestine by carefully cutting close to the intestinal border, and the preparation was placed in Krebs solution at 37°C. The first three branches of the superior mesenteric artery were tied under stereomicroscope. The preparation was then perfused at a constant flow rate of 1 ml/min using a peristaltic pump (REGLO Digital 4 channels ISM 833). The solution was maintained at 37°C and continually gassed with a 95% O2-5% CO2 gas mixture. Mesenteric vascular responses were detected as changes in perfusion pressure (mmHg). This was monitored continuously using a pressure transducer and recorded using a PowerLab. The preparation was allowed to equilibrate for 30 min before the dose-dependent contractile responses to exogenous norepinephrine were obtained.
Immunoblot analyses.
The isolated mesenteric arteries were cleaned from surrounding fat, equilibrated in normal Krebs solution for 30 min, and then frozen in liquid nitrogen-chilled acetone containing 10% trichloroacetic acid. Proteins were separated by SDS-polyacrylamide gel electrophoresis. Specific proteins were detected using immunoblot analysis as described previously (30, 31, 39, 40). The signals were captured by the Kodak Image Station 4000 MM and quantified by Kodak Molecular Image Software. The sources of specific antibodies were as follows. The antibodies against total and phosphorylated CPI-17 were generated in our laboratory as described previously (30). The antibodies against GAPDH, β-actin, and phosphorylated MLC20 (Ser19) were purchased from Cell Signaling (Danvers, MA). The antibody against total MLC20, α-actin, caldesmon, and calponin were purchased from Sigma-Aldrich. The antibodies against PKCα, SM22α (transgelin), RhoA, and total and phosphorylated MYPT1 were purchased from Santa Cruz Biotech (Santa Cruz, CA). The antibodies against PKCδ, ROCK1, and ROCK2 were purchased from BD Biosciences (San Jose, CA).
Measurement of MLC20 phosphorylation with glycerol gel electrophoresis.
The isolated mesenteric arteries were cleaned from surrounding fat and equilibrated in normal Krebs solution for 30 min. The mesenteric arteries were then flash-frozen in liquid nitrogen-chilled acetone containing 10% trichloroacetic acid either directly or after incubation with 5-HT (10 μM) plus PE (100 μM) for 2 s. After the acetone was removed, the samples were dissolved in 8 M urea sample buffer and subjected to urea/glycerol-PAGE to separate nonphosphorylated, monophosphorylated, and diphosphorylated MLC20 as previously described (32).
Measurement of blood pressure with radiotelemetry.
Each mouse was anesthetized with isoflurane, the left common carotid artery was isolated, and a catheter was inserted. The body of the telemetry transmitter unit (TA11PA-C10; Data Sciences International, St. Paul, MN) was inserted under the skin and into a dissected free “pocket” along the flank as close to the right hindlimb as possible. The neck incision was closed using silk suture and then further sealed with tissue adhesive. The mice were kept warm on a heating pad and monitored closely until they recovered from anesthesia. After surgery, mice were singly caged under normal environmental conditions in 12:12-h light-dark cycles with lights on at 0600. All mice were allowed a 7- to 10-day recovery from surgery before baseline blood pressure, heart rate, and locomotor activity were assessed. Data from the TA 11PA-C10 device were transmitted via radiofrequency signals to a receiver below the home cage (model RPC-1) and thereafter collected using the Dataquest A.R.T. system, version 4.0 (Data Sciences International). Data were sampled continuously day and night for a total of 72 h with a sampling rate of 500 Hz. For calculation of the basal systolic, diastolic, and mean arterial pressure, data collected over 72 h were averaged. The rhythmicity of blood pressure was analyzed by means of the nonlinear least-squares fitting program PHARMFIT (27), as described previously (36).
Mesenteric vessel structure studies.
One secondary order branch was isolated from each mesenteric vascular bed, dissected free from surrounding fat, and mounted onto a pressure myograph system with automated edge detection (Living Systems Instrumentation, Burlington, VT). Arteries were equilibrated at 60 mmHg luminal pressure under no-flow conditions for 30 min in 2 mM EGTA and 100 μM sodium nitroprusside containing Ca2+-free physiological saline solution. A passive pressure-diameter curve was generated by 20-mmHg stepwise increases in intraluminal pressure from 0 to 120 mmHg. Both internal diameter and wall thickness were measured under each step of pressure. After completing the pressure-diameter curve, the vessel segments were maintained under 60 mmHg pressure and fixed in 10% formalin, paraffin-embedded, sectioned, and stained with Van Gieson Elastin stain. Morphometric analysis of vascular remodeling was carried out using Olympus MicroSuit-B3 software. The media area was determined by measuring the area between the internal and external elastic lamina. The luminal area was calculated based on area = πr2, where the radius r is calculated according to r = l/2π and the circumference l is determined by measuring the length of the internal elastic lamina. The media-to-lumen ratio was calculated based on the measured lumen and media areas.
Immunofluorescence detection of CPI-17 and smooth muscle α-actin.
The femoral arteries from WT control and CPI-17-Tg mice were exposed, and the position of the arteries was marked by tying a suture at the anterior tibial artery branch point. After isolation, the femoral arteries were embedded in optimum cutting temperature compound and cut into 10-μm sections. Sections from the same distances of the suture knot were fixed with 4% paraformaldehyde, and nonspecific binding sites were blocked by incubation in 5% normal goat serum in PBS (30 min). To detect CPI-17, the sections were sequentially incubated with a rabbit anti-CPI-17 antibody (overnight, 4°C, 1:100 dilution; Upstate Biotechnology) and then Alexa Fluo 594 goat anti-rabbit IgG (30 min, room temperature, 1:100 dilution). To detect smooth muscle α-actin, the nonspecific binding sites were blocked by incubation with mouse blocking reagent (1 h, room temperature; Vector Lab MOM kit) and then sequentially incubated with a mouse anti-smooth muscle α-actin antibody (30 min, room temperature) and the Alexa Fluor 488 goat anti-mouse IgG antibody with MOM (10 min, room temperature). The slides were photographed with an Olympus IS70 microscope equipped with an Olympus DP70 digital camera.
Statistic analyses.
All data are expressed as means ± SE. Unless specified otherwise, statistical analysis was performed using the Student's t-test, and differences were considered significant when P < 0.05. For comparison of the contractions between the CPI-17-Tg and control mice across various agonist concentrations, statistical analysis was performed using a two-way ANOVA. A post hoc Bonferroni analysis was performed when appropriate, and differences were considered significant when P < 0.05.
RESULTS
Generation and characterization of smooth muscle-specific CPI-17-Tg mice.
To determine whether CPI-17 upregulation in smooth muscle is sufficient to enhance smooth muscle contraction and blood pressure, we generated the transgenic mouse model CPI-17-Tg in which exogenous CPI-17 expression is driven by a SM-MHC promoter (Fig. 1A). Immunoblot analysis of various tissues from CPI-17-Tg mice demonstrated that CPI-17 expression was selectively increased in smooth muscle-enriched mesenteric arteries and bladder with undetectable levels in heart, skeletal muscle, brain, and kidney (Fig. 1B). Overexpression of CPI-17 did not affect the expression levels of GAPDH and β-actin. CPI-17 expression levels in mesenteric arteries were 2.3 ± 0.25-fold higher (Fig. 1C) and in bladder were 8.8 ± 1.18-fold higher (data not shown) in CPI-17-Tg mice compared with WT controls. The moderate overexpression of CPI-17 in mesenteric arteries is comparable to the level reached in type 2 diabetic db/db mouse aorta (40). Moreover, immunofluorescence staining of CPI-17 indicated that the CPI-17 transgene colocalized with the smooth muscle α-actin, indicating CPI-17 was expressed within smooth muscle cells (Fig. 1D).
Contractile responses were enhanced in CPI-17-Tg mouse mesenteric arteries.
To test whether the contractile responses are enhanced in the vascular smooth muscle tissue from CPI-17-Tg mice, we measured isometric contractions to various stimuli in endothelium-denuded, second-order branch mesenteric artery strips isolated from CPI-17-Tg and WT mice. The contractile responses to the α1-adrenergic agonist PE (Fig. 2, A, D, and G), thromboxane A2 receptor agonist U-46619 (Fig. 2, B, E, and H), 5-HT (Fig. 2, C, F, and I), 100 nM ANG II (Fig. 2J), and high-potassium depolarization (143 mM; Fig. 2K) were significantly increased in CPI-17-Tg mice compared with controls. The enhanced contractions primarily manifested as increases in maximum contractile responses (Table 1).
Fig. 2.

Contractile responses are enhanced in mesenteric arteries isolated from CPI-17-Tg mice. Second-order branches of mesenteric arteries were isolated from 12- to 14-wk-old male CPI-17-Tg and WT control mice, cut into helical strips, and denuded of endothelia. Isometric contractions in response to cumulative doses of phenylephrine (PE, A, D, and G), U-46619 (B, E, and H), serotonin (5-HT, C, F, and I), 100 nM ANG II (J), or 143 mM high-potassium solution (K) were determined. In addition, some of the strips were permeabilized by Staphylococcus aureus α-toxin, and cumulative concentrations of calcium were added to obtain the pCa-tension curve (L). *P < 0.05 and **P < 0.01.
Table 1.
Effects of smooth muscle-selective CPI-17 overexpression on maximum contraction of mesenteric artery in response to various agonists
| WT | CPI-17-Tg | P Value | |
|---|---|---|---|
| PE mg | 22.7 ± 2.08 (11) | 32.6 ± 3.54 (10) | 0.0239* |
| U-46619 mg | 12.5 ± 1.35 (7) | 21.9 ± 3.85 (6) | 0.0257* |
| 5-HT mg | 4.6 ± 0.68 (11) | 10.34.6 ± 1.36 (10) | 0.0001*** |
Values are means ± SE; nos. in parentheses indicate the no. of mice used. CPI-17, protein kinase C-potentiated myosin phosphatase inhibitor of 17 kDa; WT, wild type; CPI-17-Tg, CPI-17 transgenic mouse model; PE, phenylephrine; 5-HT, serotonin.
P < 0.05 and
P < 0.001.
Agonists induce smooth muscle contraction via increasing cytoplasmic Ca2+ concentrations as well as increasing the sensitivity of myofilaments toward Ca2+. To further test whether the sensitivity of myofilaments toward Ca2+ is increased in CPI-17-Tg mice, we determined calcium dose-response curves in α-toxin-permeabilized mesenteric artery tissues. As shown in Fig. 2L, the maximal contractions induced by Ca2+ significantly increased from 22.5 ± 1.52 mg (n = 11) to 28.9 ± 1.96 mg (n = 10, P < 0.05).
To test a possible role of increased smooth muscle mass in the observed enhanced contractile responses, we examined the thicknesses and cross-sectional areas of the strips after finishing the isometric contraction studies. Neither the thickness [8.7 ± 0.31 μm (n = 14) vs. 8.9 ± 0.24 μm (n = 12), P = 0.58] nor the cross-sectional area [2,524 ± 163 μm2 (n = 14) vs. 2,639 ± 166 μm2 (n = 12), P = 0.63] showed significant differences between the CPI-17-Tg and WT control mice.
To verify that the observed enhancement in contractile response is caused by the selective increase in CPI-17 expression rather than an effect of random gene insertion, we determined the contractile responses of mesenteric arteries from an independent CPI-17-Tg founder line. Similar increases in contractile responses to PE, 5-HT, and calcium were observed (data not shown), suggesting that the enhanced contractile responses observed were not some random gene insertion artifacts but direct or indirect consequences of the CPI-17 expression.
Perfusion pressure responses were enhanced in CPI-17-Tg mouse mesenteric vascular beds.
To investigate whether the enhanced contractile responses in the strips of second-order branch mesenteric artery translate into a perfusion pressure increase, we measured the pressor responses in mesenteric vascular beds isolated from CPI-17-Tg and WT mice. The results demonstrated that the pressure increases in response to cumulative doses of norepinephrine were significantly enhanced in the CPI-17-Tg mice (Fig. 3). No significant difference was detected in baseline perfusion pressure (20 ± 1.9 vs. 25 ± 2.6 mmHg, n = 6–8, P = 0.1765).
Fig. 3.

The increases of perfusion pressure in response to norepinephrine (NE) are enhanced in mesenteric vascular bed isolated from CPI-17-Tg mice. Mesenteric vascular beds were isolated from 21-wk-old male CPI-17-Tg and control WT mice and perfused under constant flow. Cumulative doses of norepinephrine were added, and the perfusion pressure changes were recorded, n = 6–8. *P < 0.05.
Phosphorylation of CPI-17 and MLC20 was increased in association with enhanced contractile responses.
To begin to elucidate the mechanisms underlying the enhanced contractile responses in the vascular smooth muscle tissues of CPI-17-Tg mice, we first determined whether the active, phosphorylated CPI-17 is increased under basal conditions and in response to stimulation. As shown in Fig. 4, A and B, increased expression of CPI-17 protein provoked a proportional increase in active, phosphorylated CPI-17 under basal nonstimulated conditions. In response to stimulation (2 s, 10 μM 5-HT plus 100 μM PE), the CPI-17 phosphorylation was further significant increased in CPI-17-Tg tissue, whereas no significant increase was detected in WT tissue (Fig. 4, A and B). When the phosphorylated CPI-17 was normalized to total CPI-17 (Fig. 4C), under basal conditions, no difference between the CPI-17-Tg and WT was detected, suggesting the increase in phosphorylated CPI-17 is largely attributed to the increase in total CPI-17. However, when stimulated, the phosphorylated CPI-17 was increased even when normalized to total CPI-17, suggesting, in addition to the increase in CPI-17 expression, other mechanism(s) that respond to the stimulation have changed in the CPI-17-Tg tissues (Fig. 4C). To further investigate whether the increased active phosphorylated CPI-17 is associated with increased MLC20 phosphorylation, we used glycerol gel electrophoresis to determine the stoichiometry of MLC20 phosphorylation under the basal condition and in response to stimulations. The results demonstrated that, under the basal nonstimulated condition, phosphorylated MLC20 significantly increased from 1.9 ± 0.47% in WT mice to 4.5 ± 0.95% in CPI-17-Tg mice (n = 5–9 each, P < 0.05; Fig. 4, D and E). Moreover, stimulation with 5-HT (10 μM) plus PE (100 μM) increased MLC20 phosphorylation in both strains of mice, reaching 8.3 ± 1.69% in WT and 40.7 ± 1.31% in CPI-17-Tg mice. Importantly, the magnitude of increase in MLC20 phosphorylation in response to stimuli was significantly higher in the CPI-17-Tg mice than in WT controls (n = 5–9 each, P < 0.001). Together, these data indicate that the increased phosphorylation of CPI-17 and MLC20 account for, at least in part, the enhanced contractile responses in the vascular smooth muscle tissue in CPI-17-Tg mice.
Fig. 4.
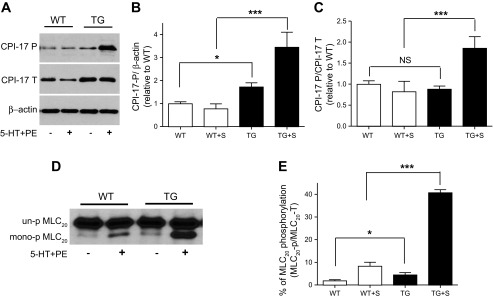
Phosphorylation of CPI-17 and 20-kDa myosin light chain (MLC20) is increased in mesenteric arteries of CPI-17-Tg mice. Mesenteric arteries were isolated from 12- to 14-wk-old male CPI-17-Tg and control WT mice. After carefully removing the surrounding fat, the mesenteric arteries were equilibrated in Krebs solution for 30 min. Next, the tissues were frozen at rest or after stimulation with 5-HT (10 μM) plus PE (100 μM) for 2 s as indicated. Proteins were separated by SDS-PAGE (A) or by glycerol gel electrophoresis (D). CPI-17 and MLC20 phosphorylation was determined by Western blot using antibodies against total CPI-17 (CPI-17-T), phospho-CPI-17 (CPI-17-P), or total-MLC20 (MLC20-T). A and D: representative Western blots. B and C are the quantification of CPI-17 phosphorylation normalized to β-actin or total CPI-17 protein, respectively. E: quantification of the percentage of the MLC20 that are phosphorylated under resting and after stimulation by 5-HT plus PE by using urea/glycerol gel electrophoresis; n = 4–10. S, stimulated with 5-HT plus PE; NS, not significant. *P < 0.05 and ***P < 0.001.
ROCK2 and PKCα/δ protein were increased in CPI-17-Tg vascular smooth muscle.
PKCα/δ and ROCK1/2 are important in the physiological phosphorylation of CPI-17; we therefore investigated whether their expression levels are altered in CPI-17-Tg mouse mesenteric arteries. The results showed that, surprisingly, PKCα (Fig. 5, A and B), PKCδ (Fig. 5, C and D), and ROCK2 (Fig. 5, G and H) but not ROCK1 (Fig. 5, E and F) protein were significantly increased. In contrast, no significant increase was observed in rhoA protein (Fig. 5, K and L) or other smooth muscle marker proteins, including α-actin (Fig. 5, M and N), SM22α (Fig. 5, O and P), caldesmon (Fig. 5, Q and R), and calponin (Fig. 5, S and T). To further test whether increased ROCK2 protein expression is associated with an increase in its activity, we immunoblotted phosphorylated MYPT-1 (Thr853) as an indicator of ROCK2 activity. The result demonstrated that MYPT-1 phosphorylation was significantly increased (Fig. 5, I and J).
Fig. 5.
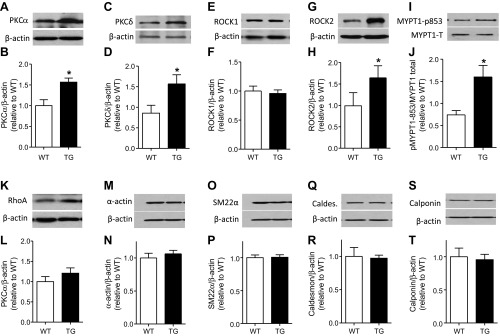
Protein kinase C (PKC) α/δ and rho kinase (ROCK) 2 proteins are increased in CPI-17-Tg mouse mesenteric arteries. Mesenteric arteries were isolated from 12- to 14-wk-old male CPI-17-Tg and control WT mice. After carefully removing the surrounding fat, the mesenteric arteries were homogenized, and the expression levels of PKCα/δ, rhoA, ROCK1/2 protein, pMYPT1-853, α-actin, SM22α, caldesmon, and calponin were determined by Western blot, respectively, shown in A, C, E, G, I, K, M, O, Q, and S. B, D, F, H, J, L, N, P, R, and T, respectively, are the quantifications of the blots of 3–11 independent experiments. *P < 0.05.
To verify that the selective increased protein expression of PKCα/δ and ROCK2 in smooth muscle tissue of CPI-17-Tg mice is not the result of genetic variation in the clone of CPI-17-Tg mouse, we blotted and compared the PKCα, PKCδ, ROCK1, and ROCK2 protein expressions in the brain tissue where the ectopic CPI-17 is not expressed. There was no significant difference detected in any of these proteins between WT and CPI-17-Tg mice (data not shown), suggesting the upregulation of PKCα, PKCδ, and ROCK2 in CPI-17-Tg mouse smooth muscle is likely a consequence of increased CPI-17 expression.
Blood pressure is moderately but significantly increased in CPI-17-Tg mice.
To determine the effects of enhanced CPI-17 expression and the resulting vascular hypercontractility on blood pressure, we measured blood pressure in active, free-moving CPI-17-Tg and WT mice by radiotelemetry. The 24-h average systolic pressure (Fig. 6A), diastolic pressure (Fig. 6B), and mean arterial pressure (Fig. 6C) were significantly increased, although the amplitude of the increase was moderate, 5–7 mmHg. In WT and CPI-17-Tg mice, respectively, systolic pressure was 124 ± 1.2 mmHg (n = 15) and 129 ± 1.3 mmHg (n = 16, P < 0.05), diastolic pressure was 92 ± 1.3 mmHg (n = 15) and 99 ± 2.0 mmHg (n = 16, P < 0.01), and mean arterial pressure was 108 ± 1.0 mmHg (n = 15) and 114 ± 1.34 mmHg (n = 16, P < 0.01). The increase in blood pressure is not a consequence of enhanced locomotor activity since there was no significant difference detected between the CPI-17-Tg and WT mice in locomotor activity (4.6 ± 0.43 vs. 4.9 ± 0.48 counts/min, P = 0.64). In addition, no difference in heart rate (570 ± 8.72 vs. 564 ± 6.35 beats/min, P = 0.58) or the ratio of left ventricle weight and body weight (2.1 ± 0.05 vs. 2.1 ± 0.086 mg/g, n = 8 for each group, P = 0.8334) was detected between the WT control and CPI-17-Tg mice.
Fig. 6.
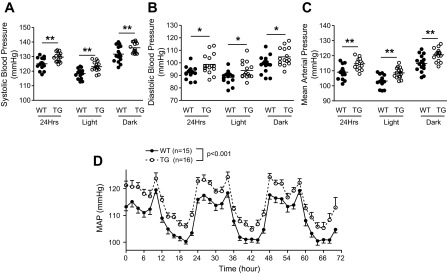
Smooth muscle-selective CPI-17 overexpression moderately but significantly enhances basal blood pressure with no apparent disruption of blood pressure circadian rhythm. Male CPI-17-Tg and littermate control mice (12–16 wk of age) were implanted with radiotelemetry transmitters in the left common carotid artery. After 7 to 10 days of recovery, basal blood pressure data were collected for three consecutive days under a 12:12-h light-dark cycle, and the 2-h average mean arterial pressure (MAP) was plotted (D). The 24-h, 12-h light, or 12-h dark phase systolic pressure (A), diastolic pressure (B), and mean arterial pressure (C) are presented; n = 15 for control and n = 16 for CPI-Tg mice. *P < 0.05 and **P < 0.01.
We found that enhanced CPI-17 expression and vascular smooth muscle hypercontractility in type 2 diabetic db/db mice were associated with severe disruption in blood pressure circadian rhythm (36); we therefore analyzed the blood pressure circadian rhythm in CPI-17-Tg mice to investigate the possibility that CPI-17 upregulation in smooth muscle contributes to this disruption. As shown in Fig. 6D, no significant difference was detected in any of the blood pressure circadian rhythm parameters tested, including the amplitude (8.0 ± 0.44, n = 15 vs. 8.4 ± 0.41 mmHg, n = 16, P = 0.49), acrophase (0.49 ± 0.19, n = 15 vs. 0.20 ± 0.21 o'clock, n = 16, P = 0.33), or period length (23.7 ± 0.09, n = 15 vs. 23.6 ± 0.19 h, n = 16, P = 0.52).
To investigate whether vascular remodeling occurred in CPI-17-Tg mice, we constructed a passive pressure-diameter curve in a cannulated 1.5-mm segment of a second-order branch of mesenteric arteries isolated from WT control and CPI-17-Tg mice by using a pressure myograph system. No significant difference between the two strains of mice was detected in inner diameter (Fig. 7A), external diameter (Fig. 7B), medial cross-sectional areas (Fig. 7C), and wall-to-lumen ratio (Fig. 7D) in all of the pressures tested (0 to 120 mmHg with 20-mmHg step). Moreover, when the vessels were fixed under the Ca2+-free condition and in the presence of 60 mmHg pressure, morphometric analysis demonstrated that there were no significant differences in luminal area, medial area, and the wall-to-lumen ratio (Fig. 7, E–H). Together, these results indicate that there is no detectable vascular structural remodeling in these 14-wk-old CPI-17-Tg mice relative to WT control mice.
Fig. 7.
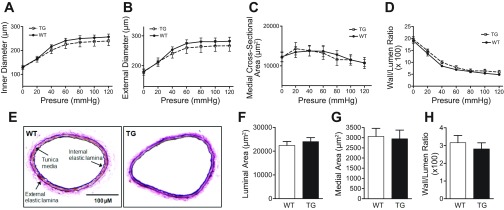
Lack of detectable remodeling in the second-order mesenteric arteries isolated from CPI-17-Tg mice. A passive pressure-diameter curve was generated in a segment of second-order branch of mesenteric artery by 20-mmHg stepwise increases in intraluminal pressure from 0 to 120 mmHg from each WT control or CPI-17-Tg mouse. The changes in internal diameter (A), external diameter (B), medial cross-sectional area (C), and wall-to-lumen ratio (D) were measured or calculated under each step of pressure. After the pressure-diameter curve was completed, the vessel segments were maintained under 60 mmHg pressure and fixed in 10% formalin, paraffin-embedded, sectioned, and stained with elastin. Representative images of the vessels from WT control and CPI-17-Tg mice were shown in E, and morphometric analysis of these images was shown in F, G, and H.
DISCUSSION
Major findings of the current study are that the smooth muscle-specific CPI-17-Tg mice exhibit vascular smooth muscle hypercontractility and blood pressure increase. Moreover, sustained CPI-17 upregulation increases ROCK2 and PKCα/δ protein expression, suggesting a previously unrecognized positive feedback loop between CPI-17 and its upstream kinases ROCK2 and PKCα/δ.
Under physiological conditions, CPI-17 plays an important role in the regulation of smooth muscle contraction (7). Recent evidence demonstrates an association between alterations in CPI-17 expression level and changes in smooth muscle contractile responses. For example, CPI-17 upregulation in asthmatic airway smooth muscle (33), or in diabetic vascular or bladder detrusor smooth muscle (3, 40), are associated with smooth muscle hypercontractility. However, numerous alterations in addition to CPI-17 upregulation are present in smooth muscle tissues under these disease conditions. Consequently, it is unclear whether upregulated CPI-17 plays a causal role in observed smooth muscle hypercontractility under these conditions. With the use of a novel smooth muscle-selective CPI-17 transgenic mouse model, the current study demonstrates that smooth muscle-specific overexpression of CPI-17 causes vascular smooth muscle hypercontractility. Importantly, the hypercontractility is obtained with a 2.3-fold increase in CPI-17 expression (Figs. 1C and 2), a level reached in the diabetic db/db mouse vasculature (40), stressing the potential pathological significance of CPI-17 upregulation.
Enhanced total CPI-17 protein expression in CPI-17-Tg mice is associated with a proportional increase in phosphorylated CPI-17 under the basal condition (Fig. 4, A and B). Giving the fact that about 40% of the total CPI-17 becomes phosphorylated at the peak of PE stimulation (6), it is unlikely that CPI-17 protein level is a rate-limiting factor in determining the level of CPI-17 phosphorylation. It is possible that increased CPI-17 content simply increased the substrate concentration in the phosphorylation reaction and thus results in proportional increased phosphorylated CPI-17. Alternatively, CPI-17 upregulation may cause activation/upregulation of the upstream kinase(s) that in turn phosphorylates more CPI-17. The following evidence supports the latter. First, when stimulated, the CPI-17 phosphorylation was significantly increased even when normalized to the total CPI-17 level (Fig. 4C), suggesting additional mechanism(s) that are activated upon agonist stimulation are changed when the CPI-17 expression was upregulated. Second, Stevenson et al. (35) showed that the addition of recombinant CPI-17 protein in permeabilized CPI-17-deficient chicken amnion smooth muscle caused a significant increase in active GTP-bound rhoA, suggesting that CPI-17 can regulate rhoA and, thus, ROCK2 activity. The current study, while identified no significant change in rhoA protein expression, found that the protein levels of PKCα/δ and ROCK2, three kinases known to phosphorylate and activate CPI-17 (16, 21–23), were upregulated in the vasculature of CPI-17-Tg mice (Fig. 5). This identifies a previously unrecognized function of CPI-17: it not only serves as a target that is phosphorylated by PKCα/δ and ROCK2 but also regulates their expression, directly or indirectly.
It is surprising that, in WT tissue, no increase in CPI-17 phosphorylation was detected 2 s after agonist stimulation in the current study (Fig. 4, A–C) since CPI-17 phosphorylation has been demonstrated to increase rapidly in response to agonist stimulation (6, 18). This discrepancy may be because the very early time point of 2 s stimulation was tested in the current study. In previous studies, the earliest time points investigated were 7 or 10 s after stimulation (6, 18).
The hypercontractility of the mesenteric arteries isolated from CPI-17-Tg mice was observed in the absence of enhanced smooth muscle mass but was associated with enhanced MLC20 phosphorylation compared with WT mice (Fig. 4), suggesting that increased CPI-17 protein inhibits myosin phosphatase activity and consequently increases MLC20 phosphorylation as expected. Because phosphatase inhibition and MLC20 phosphorylation are steps at the very end of the signaling cascade that mediates various stimuli-induced force productions, it is expected that the enhancement of the contractile response is general. Indeed, contraction in response to PE, 5-HT, U-46619, ANG II, and high-potassium stimulation as well as to Ca2+ in permeabilized preparations is significantly enhanced in vascular tissues isolated from CPI-17-Tg mice compared with those from controls (Fig. 2). It has been demonstrated that neither high-potassium depolarization in intact tissue (6) nor calcium in α-toxin-permeabilized tissue (16) is sufficient to increase endogenous CPI-17 phosphorylation in isolated vascular tissues. However, in CPI-17-Tg mice, both total CPI-17 protein and active phosphorylated CPI-17 are significantly increased. Such increased active CPI-17 is therefore expected to enhance both high-potassium- and calcium-induced as well as other stimuli-induced contractions. Indeed, this is what we observed, as shown in Fig. 2. Such heightened contractile responses in vascular smooth muscle can cause increased vascular tone in vivo when the sympathetic activity and vasoactive hormonal levels are the same, which could result in an increase in blood pressure. Indeed, this is what we observed in the CPI-17-Tg mice.
Telemetry measurements of blood pressure in large numbers of conscious, free-moving mice revealed a significant increase in 24-h averages of systolic, diastolic, and mean arterial pressures in CPI-17-Tg mice compared with controls. Among the multiple systems contributing to the homeostasis of blood pressure, the kidney plays a central role (12). However, emerging evidence suggests that primary changes in vascular morphology and tone can lead to sustained increases in peripheral resistance and blood pressure (2, 10, 13, 28, 37). A kidney cross-transplantation approach in ANG II receptor AT1a knockout mice demonstrated distinct and virtually equivalent contributions of AT1 receptor actions in the kidney and extra renal tissues to determine the level of blood pressure (5). The results of the current study demonstrate that a modest and selective increase in CPI-17 by 2.3-fold in smooth muscle is sufficient to significantly increase blood pressure, supporting the notion that primary vascular smooth muscle alterations are sufficient to cause blood pressure changes. However, such blood pressure increases can result from increased systemic vascular resistance or an indirect effect of kidney hemodynamic disruption due to renal vasculature hyperreactivity. Regarding the role of a specific signaling pathway, a study by Wirth et al. demonstrated the Gq/11 pathway in smooth muscle is essential for normal basal blood pressure (37). Our results extend their findings by demonstrating that CPI-17 is likely a downstream target by which the Gq/11 pathway regulates vascular tone and basal blood pressure.
The amplitude of blood pressure increases in CPI-17-Tg mice is modest: a 5-mmHg increase in systolic blood pressure and a 7-mmHg increase in diastolic blood pressure. Such modest blood pressure increases were insufficient to cause detectable vascular remodeling by 14 wk of age (Fig. 7). However, such modest blood pressure increases sustained for a long time period can have significant detrimental consequences. Blood pressure values clearly bear a linear relation with the incidence of cardiac and cerebrovascular events (4, 26). It has been demonstrated that, for each 10-mmHg decrease in systolic blood pressure, there is a reduced (by 11%) risk of myocardial infarction and microvascular complications (by 13%), resulting in a 15% decreased risk of death from diabetes (1, 24).
No alterations in the blood pressure circadian rhythm were noted in CPI-17-Tg mice, suggesting enhanced vascular constrictive responses driven by the sustained expression of CPI-17 under the control of the SM-MHC promoter do not contribute to the blood pressure circadian rhythm. However, this does not exclude the possibility that endogenous CPI-17 may contribute to the blood pressure circadian rhythm via circadian-dependent oscillations in CPI-17 activity and/or expression.
In summary, using a novel smooth muscle-selective CPI-17 transgenic mouse model, the current study demonstrates that increases in CPI-17 expression cause vascular smooth muscle hypercontractility and an increase in blood pressure. In addition, we demonstrated that CPI-17 expression level can regulate ROCK2 and PKCα/δ expression. These results implicate a potentially significant role for CPI-17 upregulation in a variety of disease conditions that manifest enhanced smooth muscle contractility and/or hypertension.
GRANTS
This work was supported in whole or in part by National Heart, Lung, and Blood Institute Grants HL-082791 to M. C. Gong and HL-088389 and HL-088389-02S1 to Z. Guo.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: W.S., Z.X., L.S., Z.G., and M.C.G. conception and design of research; W.S., Z.X., L.S., and L.E.C. performed experiments; W.S., Z.X., L.S., L.E.C., Z.G., and M.C.G. analyzed data; W.S., Z.X., L.S., L.E.C., Z.G., and M.C.G. interpreted results of experiments; W.S., Z.X., L.S., and M.C.G. prepared figures; W.S., Z.X., L.S., L.E.C., Z.G., and M.C.G. approved final version of manuscript; Z.G. and M.C.G. edited and revised manuscript; M.C.G. drafted manuscript.
ACKNOWLEDGMENTS
We thank Ming Zhang for technical assistance in breeding of CPI-17-Tg mice and WT littermates and thank Dr. Wendy Katz for technical assistance in embedding the vascular tissues for morphometric studies.
REFERENCES
- 1.Adler AI, Stratton IM, Neil HA, Yudkin JS, Matthews DR, Cull CA, Wright AD, Turner RC, Holman RR. Association of systolic blood pressure with macrovascular and microvascular complications of type 2 diabetes (UKPDS 36): prospective observational study. Br Med J 321: 412–419, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang L, Villacorta L, Zhang J, Garcia-Barrio MT, Yang K, Hamblin M, Whitesall SE, D'Alecy LG, Chen YE. Vascular smooth muscle cell-selective peroxisome proliferator-activated receptor-gamma deletion leads to hypotension. Circulation 119: 2161–2169, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang S, Hypolite JA, Disanto ME, Changolkar A, Wein AJ, Chacko S. Increased basal phosphorylation of detrusor smooth muscle myosin in alloxan-induced diabetic rabbit is mediated by upregulation of Rho-kinase beta and CPI-17. Am J Physiol Renal Physiol 290: F650–F656, 2006 [DOI] [PubMed] [Google Scholar]
- 4.Collins R, Peto R, MacMahon S, Hebert P, Fiebach NH, Eberlein KA, Godwin J, Qizilbash N, Taylor JO, Hennekens CH. Blood pressure, stroke, and coronary heart disease. Part 2. Short-term reductions in blood pressure: overview of randomised drug trials in their epidemiological context. Lancet 335: 827–838, 1990 [DOI] [PubMed] [Google Scholar]
- 5.Crowley SD, Gurley SB, Oliverio MI, Pazmino AK, Griffiths R, Flannery PJ, Spurney RF, Kim HS, Smithies O, Le TH, Coffman TM. Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin-angiotensin system. J Clin Invest 115: 1092–1099, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dimopoulos GJ, Semba S, Kitazawa K, Eto M, Kitazawa T. Ca2+-dependent rapid Ca2+ sensitization of contraction in arterial smooth muscle. Circ Res 100: 121–129, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eto M. Regulation of cellular protein phosphatase-1 (PP1) by phosphorylation of the CPI-17 family, C-kinase-activated PP1 inhibitors. J Biol Chem 284: 35273–35277, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eto M, Senba S, Morita F, Yazawa M. Molecular cloning of a novel phosphorylation-dependent inhibitory protein of protein phosphatase-1 (CPI17) in smooth muscle: its specific localization in smooth muscle. FEBS Lett 410: 356–360, 1997 [DOI] [PubMed] [Google Scholar]
- 9.Gong MC, Cohen P, Kitazawa T, Ikebe M, Masuo M, Somlyo AP, Somlyo AV. Myosin light chain phosphatase activities and the effects of phosphatase inhibitors in tonic and phasic smooth muscle. J Biol Chem 267: 14662–14668, 1992 [PubMed] [Google Scholar]
- 10.Guilluy C, Bregeon J, Toumaniantz G, Rolli-Derkinderen M, Retailleau K, Loufrani L, Henrion D, Scalbert E, Bril A, Torres RM, Offermanns S, Pacaud P, Loirand G. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat Med 16: 183–190, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Guo Z, Su W, Ma Z, Smith GM, Gong MC. Ca2+-independent phospholipase A2 is required for agonist-induced Ca2+ sensitization of contraction in vascular smooth muscle. J Biol Chem 278: 1856–1863, 2003 [DOI] [PubMed] [Google Scholar]
- 12.Guyton AC, Coleman TG, Cowley AV, Jr, Scheel KW, Manning RD, Jr, Norman RA., Jr Arterial pressure regulation Overriding dominance of the kidneys in long-term regulation and in hypertension. Am J Med 52: 584–594, 1972 [DOI] [PubMed] [Google Scholar]
- 13.Halabi CM, Beyer AM, de Lange WJ, Keen HL, Baumbach GL, Faraci FM, Sigmund CD. Interference with PPAR gamma function in smooth muscle causes vascular dysfunction and hypertension. Cell Metab 7: 215–226, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horiuti K. Mechanism of contracture on cooling of caffeine-treated frog skeletal muscle fibres. J Physiol 398: 131–148, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim JI, Young GD, Jin L, Somlyo AV, Eto M. Expression of CPI-17 in smooth muscle during embryonic development and in neointimal lesion formation. Histochem Cell Biol 132: 191–198, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitazawa T, Eto M, Woodsome TP, Brautigan DL. Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. J Biol Chem 275: 9897–9900, 2000 [DOI] [PubMed] [Google Scholar]
- 17.Kitazawa T, Eto M, Woodsome TP, Khalequzzaman M. Phosphorylation of the myosin phosphatase targeting subunit and CPI-17 during Ca2+ sensitization in rabbit smooth muscle. J Physiol 546: 879–889, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitazawa T, Kitazawa K. Size-dependent heterogeneity of contractile Ca2+ sensitization in rat arterial smooth muscle. J Physiol 590: 5401–5423, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitazawa T, Kobayashi S, Horiuti K, Somlyo AV, Somlyo AP. Receptor-coupled, permeabilized smooth muscle. Role of the phosphatidylinositol cascade, G-proteins, and modulation of the contractile response to Ca2+. J Biol Chem 264: 5339–5342, 1989 [PubMed] [Google Scholar]
- 20.Kitazawa T, Polzin AN, Eto M. CPI-17-deficient smooth muscle of chicken. J Physiol 557: 515–528, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitazawa T, Takizawa N, Ikebe M, Eto M. Reconstitution of protein kinase C-induced contractile Ca2+ sensitization in triton X-100-demembranated rabbit arterial smooth muscle. J Physiol 520: 139–152, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koyama M, Ito M, Feng J, Seko T, Shiraki K, Takase K, Hartshorne DJ, Nakano T. Phosphorylation of CPI-17, an inhibitory phosphoprotein of smooth muscle myosin phosphatase, by Rho-kinase. FEBS Lett 475: 197–200, 2000 [DOI] [PubMed] [Google Scholar]
- 23.Li L, Eto M, Lee MR, Morita F, Yazawa M, Kitazawa T. Possible involvement of the novel CPI-17 protein in protein kinase C signal transduction of rabbit arterial smooth muscle. J Physiol 508: 871–881, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindholm LH. The outcome of STOP-Hypertension-2 in relation to the 1999 WHO/ISH hypertension guidelines. Blood Press Suppl 2: 21–24, 2000 [PubMed] [Google Scholar]
- 25.Liu S, Xie Z, Zhao Q, Pang H, Turk J, Calderon L, Su W, Zhao G, Xu H, Gong MC, Guo Z. Smooth muscle-specific expression of calcium-independent phospholipase A2beta (iPLA2beta) participates in the initiation and early progression of vascular inflammation and neointima formation. J Biol Chem 287: 24739–24753, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacMahon S, Peto R, Cutler J, Collins R, Sorlie P, Neaton J, Abbott R, Godwin J, Dyer A, Stamler J. Blood pressure, stroke, and coronary heart disease. Part 1. Prolonged differences in blood pressure: prospective observational studies corrected for the regression dilution bias. Lancet 335: 765–774, 1990 [DOI] [PubMed] [Google Scholar]
- 27.Mattes A, Witte K, Hohmann W, Lemmer B. PHARMFIT–a nonlinear fitting program for pharmacology. Chronobiol Int 8: 460–476, 1991 [DOI] [PubMed] [Google Scholar]
- 28.Michael SK, Surks HK, Wang Y, Zhu Y, Blanton R, Jamnongjit M, Aronovitz M, Baur W, Ohtani K, Wilkerson MK, Bonev AD, Nelson MT, Karas RH, Mendelsohn ME. High blood pressure arising from a defect in vascular function. Proc Natl Acad Sci USA 105: 6702–6707, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohama T, Hori M, Momotani E, Iwakura Y, Guo F, Kishi H, Kobayashi S, Ozaki H. Intestinal inflammation downregulates smooth muscle CPI-17 through induction of TNF-α and causes motility disorders. Am J Physiol Gastrointest Liver Physiol 292: G1429–G1438, 2007 [DOI] [PubMed] [Google Scholar]
- 30.Pang H, Guo Z, Su W, Xie Z, Eto M, Gong MC. RhoA-Rho kinase pathway mediates thrombin- and U-46619-induced phosphorylation of a myosin phosphatase inhibitor, CPI-17, in vascular smooth muscle cells. Am J Physiol Cell Physiol 289: C352–C360, 2005 [DOI] [PubMed] [Google Scholar]
- 31.Pang H, Guo Z, Xie Z, Su W, Gong MC. Divergent kinase signaling mediates agonist-induced phosphorylation of phosphatase inhibitory proteins PHI-1 and CPI-17 in vascular smooth muscle cells. Am J Physiol Cell Physiol 290: C892–C899, 2006 [DOI] [PubMed] [Google Scholar]
- 32.Persechini A, Kamm KE, Stull JT. Different phosphorylated forms of myosin in contracting tracheal smooth muscle. J Biol Chem 261: 6293–6299, 1986 [PubMed] [Google Scholar]
- 33.Sakai H, Chiba Y, Misawa M. Augmentation of endothelin-1-induced phosphorylation of CPI-17 and myosin light chain in bronchial smooth muscle from airway hyperresponsive rats. Biol Pharm Bull 29: 1897–1899, 2006 [DOI] [PubMed] [Google Scholar]
- 34.Senba S, Eto M, Yazawa M. Identification of trimeric myosin phosphatase (PP1M) as a target for a novel PKC-potentiated protein phosphatase-1 inhibitory protein (CPI17) in porcine aorta smooth muscle. J Biochem (Tokyo) 125: 354–362, 1999 [DOI] [PubMed] [Google Scholar]
- 35.Stevenson AS, Matthew JD, Eto M, Luo S, Somlyo AP, Somlyo AV. Uncoupling of GPCR and RhoA-induced Ca2+-sensitization of chicken amnion smooth muscle lacking CPI-17. FEBS Lett 578: 73–79, 2004 [DOI] [PubMed] [Google Scholar]
- 36.Su W, Guo Z, Randall DC, Cassis L, Brown DR, Gong MC. Hypertension and disrupted blood pressure circadian rhythm in type 2 diabetic db/db mice. Am J Physiol Heart Circ Physiol 295: H1634–H1641, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind JS, Offermanns S. G12–G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med 14: 64–68, 2008 [DOI] [PubMed] [Google Scholar]
- 38.Woodsome TP, Eto M, Everett A, Brautigan DL, Kitazawa T. Expression of CPI-17 and myosin phosphatase correlates with Ca(2+) sensitivity of protein kinase C-induced contraction in rabbit smooth muscle. J Physiol 535: 553–564, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie Z, Gong MC, Su W, Turk J, Guo Z. Group VIA phospholipase A2 (iPLA2beta) participates in angiotensin II-induced transcriptional up-regulation of regulator of g-protein signaling-2 in vascular smooth muscle cells. J Biol Chem 282: 25278–25289, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie Z, Su W, Guo Z, Pang H, Post SR, Gong MC. Up-regulation of CPI-17 phosphorylation in diabetic vasculature and high glucose cultured vascular smooth muscle cells. Cardiovasc Res 69: 491–501, 2006 [DOI] [PubMed] [Google Scholar]