

Abstract
Probing the intracellular proteome of Thermotoga maritima and Caldicellulosiruptor saccharolyticus in pure and co-culture affords a global investigation into the machinery and mechanisms enduring inside the bacterial thermophilic cell at the time of harvest. The second of a two part study, employing GeLC-MS2 a variety of proteins were confidently identified with <1% false discovery rate, and spectral counts for label-free relative quantification afforded indication of the dynamic proteome as a function of environmental stimuli. Almost 25% of the T. maritima proteome and 10% of the C. saccharolyticus proteome were identified. Through comparison of growth temperatures for T. maritima, a protein associated with chemotaxis was uniquely present in the sample cultivated at the non-optimal growth temperature. It is suspected that movement was induced due to the non-optimal condition as the organism may need to migrate in the culture to locate more nutrients. The inventory of C. saccharolyticus proteins identified in these studies and attributed to spectral counting, demonstrated that two CRISPR-associated proteins had increased expression in the pure culture versus the co-culture. Further focusing on this relationship, a C. saccharolyticus phage-shock protein was identified in the co-culture expanding a scenario that the co-culture had decreased antiviral resistance and accordingly an infection-related protein was present. Alterations in growth conditions of these bacterial thermophilic microorganisms offer a glimpse into the intricacy of microbial behavior and interaction.
Keywords: Thermotoga maritima, Caldicellulosiruptor saccharolyticus, GeLC-MS2, Co-cultivation, Intracellular proteome, Spectral counting
Introduction
Changes in stimuli (e.g., nutrients, temperature, proximity to other organisms, and onset of stress/trauma) alter the proteome of organisms both in identity and quantity. For example, the presence of two different organisms growing in concert produces varying proteomic results. Transcriptional analysis of the co-cultivation of two thermophilic bacterial microorganisms, Thermotoga maritima and Methanococcus jannaschii, demonstrated variations of culture growth in comparison to the monoculture of T. maritima [1]. Although the investigation did not specifically detect proteins, inferring translation through cDNA microarray analysis provides conclusions concerning the cultivation. One particular T. maritima gene encoding a small peptide was up-regulated 13-fold in the co-culture as compared to the pure culture, and further investigations revealed that as the mature protein was dosed into a monoculture, exopolysaccharide formation occurred whereas the opposite was true for the control. In another example, Dorrestein and coworkers observed interspecies communication of two bacterial microorganisms inoculated 5 mm apart on an agar plate analyzed by MALDI-imaging mass spectrometry (MS) [2]. Investigating metabolic activity, they were able to demonstrate that in co-culture Bacillus subtilis produces a bioactive compound which inhibits the production of the peptide SapB consequently preventing some downstream development. Inducing and manipulating proteomic changes as a function of exposure to differ stimuli may provide opportunity to control and regulate particular moieties such as technologically valuable biomolecules. Considering the possibilities of the biomolecules, stability at high temperature would afford great opportunity for industrial use, and thus, probing the proteomes of thermophilic microorganisms is extremely advantageous.
Thermophilic bacterial microorganisms existing and growing at conditions otherwise harsh and extreme for mesophiles and species such as Homo sapiens, house uniquely suited thermostable biomolecules, many of which have only been limitedly defined. Attributable to past and very recent investigations of bacterial microorganisms, evidence demonstrates or highly suggests that thermophiles harbor peptides and proteins diverse in functions including antimicrobial potential [3, 4], interspecies signaling [2, 5], carbohydrate degradation [6–8], and toxin–antitoxin systems [9, 10]. These features render thermophiles as ideal candidates for biomass conversion, and industrial and biopharmaceutical processes such as those necessitating thermostable biocatalysts. Herein, two thermophilic bacterial microorganisms, T. maritima and Caldicellulosiruptor saccharolyticus offer a glimpse into the individual and microbial communities both in identity and abundance of proteins. As with environmental changes, challenges, and obstacles, organisms adapt accordingly in an effort to survive inducing proteomic changes. Speculating variation in protein translation (i.e., identified proteins and expression levels) evidence will contribute to the generation of a microorganism or group of microorganisms tailored for a specific function(s) opposed to generating an identical or equivalent function by means of several resources.
Found in marine sediment in Vulcano Island, Italy, T. maritima is the most studied of the order Thermotogales [11]. T. maritima, a rod-shaped, gram-negative, anaerobic bacterium is surrounded by a sheath-like form or “toga”, and grows between 55 and 90 °C although optimally at 80 °C [12]. The genome encodes for the most glycoside hydrolases, enzymes that hydrolyze glycosidic bonds between two carbohydrates or a carbohydrate and another moiety, of sequenced thermophiles [13, 14] and nearly 7% of open reading frames (ORFs) of known function encode carbohydrate degrading enzymes [15]. These features and others afford T. maritima to ferment, or convert simple, complex, and non-glucose sugars into alcohol under anaerobic conditions with a preference for complex carbohydrates. C. saccharolyticus, briefly described in Part I of this study, exists at extreme temperatures as well; however, originating from a different high-temperature environment, a thermal spring in New Zealand, also has a similar gene profile generating proteins for carbohydrate degradation [16]. Of the ORFs of known function, it is estimated that 20% encode for carbohydrate degradation and related functions, thus C. saccharolyticus is attributed with very active and stable celluloytic enzymes, cellulases [11, 15]. Capable of growth at 45–80 °C and optimally 75 °C, this thermophile takes the shape of an oval-ended rod, is gram-positive, and anaerobic [16]. Noteworthy, C. saccharolyticus can employ an assortment of carbohydrate molecules for production of H2 [11, 17]. Although H2 is known to suppress the growth of these microorganisms, C. saccharolyticus can produce up to 4 moles per mole of glucose while tolerating the hydrogenated environment better than other thermophiles such as T. maritima [15]. Growth of these microbes individually in monoculture and together in co-culture provides opportunity in defining the cellular proteome by MS as a function of stimuli.
Subsequent to protein characterization, protein quantification measurements prove significant for proteomic science through which to measure changes in expression at the molecular level. Capable of absolute and relative quantification, these MS measurements afford determination of protein expression levels resultant from changes in stimuli (e.g., media nutrients, temperature, the presence of additional microorganisms) or incurred due to genetic makeup (e.g., gene mutations), for example. Protein quantification techniques have evolved into two different classes, label-free quantification and labeling strategies incorporating stable isotope nuclei. Requiring no additional entities or sample preparation for analysis, label-free quantification requires the independent interrogation of samples of interest by MS. Two label-free approaches are generally employed, the evaluation of integrated peak areas from the extracted ion chromatogram of one or more peptides mapping to a specific protein and assessment of the number of peptides interrogated by MS2 from a given protein and comparing the number of spectral counts (SpC) [18]. Spectral counting is merited the most straightforward method as it relatively quantifies proteins by the number of MS2 spectra from peptides corresponding to the particular protein [19–23]. Yates and coworkers investigated the relationship between protein abundance and SpC by spiking standard proteins into a soluble yeast lysate fraction and demonstrated uniform linear correlation of SpC as a function of percentage of protein added [20].
Due to varying protein lengths and consequently, varying numbers of tryptic peptides per protein, the normalized spectral abundance factor (NSAF) was described to account for these differences as well as attempting to correct for varying sample complexities when evaluating protein expression [23].
| (1) |
The NSAF (Eq. 1) for a given species k is proportional to the total number of SpC or total number of MS2 spectra from peptides identifying k divided by the length (L) of the species, this quotient divided by the sum SpC per species length (SpC/L) of all species (N) within the experiment. This calculation was performed for two proteins in Part I; however, the formula was not explicitly defined until now due to the extensive use for analysis in Part II. The practicality of label-free quantification can be highlighted by several measures; low cost and less sample preparation resultant of no need for isotopic reagents, no strict limit of the number of experimental conditions for comparison, no increase in MS complexity, and a suggested higher dynamic range [18, 20].
As described, C. saccharolyticus and T. maritima originate from dissimilar environments, wood found near a thermal spring and marine mud, respectively; however, growth of the two microbes in concert or a co-culture presents opportunity for a multitude of potential. Presence of specific proteins and differing expression of gene products offer insight into promising applications in biopharmaceuticals and biomass degradation. Extracting qualitative and quantification details offers avenues to protein expression in various environments and comparisons between individual and co-culture as well as a range of carbon sources for growth in future investigations. Here, we describe for the first time, growth of T. maritima and C. saccharolyticus in co-culture. These seemingly dissimilar thermophiles are capable of simultaneous growth and through evaluation of the gene products by gel electrophoresis followed by nano-flow liquid chromatography coupled to electrospray ionization Fourier transform ion cyclotron resonance tandem mass spectrometry (GeLC-MS2) of both the individual and co-culture thermophiles, the cellular proteomes will be characterized. Furthermore, through label-free quantification, spectral counting will afford comparison of protein expression levels.
Experimental
Thermophilic bacterial growth and cell lysis
C. saccharolyticus DSM 8903 (ATCC 43494), T. maritima DSM 3109 (JCM 10099), and a co-culture of the two microorganisms where grown in 1,600 mL volumes and were maintained under anaerobic conditions. Each culture medium included 5 g/L glucose as the carbon source and 1 g/L yeast extract proteins and peptides ≤1 kDa (Fisher Scientific, Waltham, MA). Additionally, C. saccharolyticus was grown as described previously in Part I of this investigation with 640 DSMZ media (DSMZ—Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Braunschweig, Germany) supplemented with 1 g/L tryptone at 75 °C. T. maritima was grown in a sea salt media consisting of the following: 40 g/L sea salts (Sigma-Aldrich, St. Louis, MO), 3.1 g/L piperazine-N,N’-bis(2-ethanesulfonic acid) buffer (Sigma-Aldrich), 5 g/L glucose, and 10 mL/L vent sulfothermophiles medium (VSM) trace elements solution. The VSM solution includes the following components in 1 L, 15 g nitrilotriacetic acid, 5 g FeCl2·6H2O, 3 g Na2WO4·2H2O, 4 g MnCl2·4H2O, 2 g NiCl2·6H2O, 1 g ZnSO4·7H2O, 1 g CoSO4·7H2O, 0.10 g CuSO4·5H2O, and 1 g Na2MoSO4·5H2O [24]. The pH of the final media was adjusted to 6.8 with potassium hydroxide (Fisher Scientific) and was autoclaved before inoculation. Two T. maritima cultures were cultivated, one at 75 °C and the other 80 °C. A co-culture of the two microbes was grown at 75 °C in the modified 640 media as described above with additional 1.4 g/L NaCl.
Cultures were harvested at mid-exponential phase which was determined by an estimated cell density of 5×107 cells/mL. Cells quantities were approximated using oil immersion microscopy in which cells were counted in ten random fields and averaged. To separate the cells from the culture media, samples were filtered as described in Part I thru 0.22 μm filters (Fisher Scientific). The microbial cells were lysed both chemically and physically to ensure that the bacterial membranes were sufficiently ruptured. Cells were reconstituted 10,000:1 μl 8 M urea (Fisher Scientific) in 50 mM ammonium biocarbonate (Fisher Scientific). This solution was vortexed and then sonicated with an unltrasonic processor (Misonix Inc., Newton, CT) for 10 s. Centrifugation for 10 min at 5,000 rpm allowed the cellular membrane and debris to pellet while the supernatant contained the cellular proteome. The supernatant was collected and an additional centrifugation step at 10,000 rpm for 30 min was performed to further remove debris. A modified Bradford Assay method, the Coomassie Plus Assay (Thermo Scientific, Rockford, IL), was used to approximate total protein concentration. The cellular lysates were fractionated into 50 μg, dried under reduced pressure, and stored at −20 °C until use.
GeLC-MS2
Similar to the C. saccharolyticus secretome samples in Part I, GeLC-MS2 was performed as follows, 50 μg of the thermophilic cellular lysates were reconstituted in 20 mM Tris–HCl pH 8.0 (Fisher) and one-dimensional sodium dodecyl sulfate polyacrylamide gel electrophoresis (1D SDS-PAGE) experiments were performed. A Criterion 10–20% Tris–HCl gel (BioRad, Hercules, CA) was employed and the buffer system was as follows, 25 mM Tris–HCl, 192 mM glycine, and 0.1% SDS at pH 8.3 (BioRad). The experiment was performed at 200 V for 55 min prior to fixing, staining overnight with Sypro Ruby (Invitrogen, Carlsbad, CA), and rinsing as indicated by the manufacturer’s protocol. Subsequent to gel imaging, each lane was divided into four fractions consisting of eight gel bands (2 mm×7 mm) excised with a disposable gridcutter (Gelcompany, San Francisco, CA). The gel bands were further sliced into 1 mm×2–3 mm pieces with a razor blade to afford greater opportunity for reagents and solvents used for the in-gel digestion to penetrate and saturate the gel pieces. The tryptic digestion was executed as described by Shevchenko et al. [25] with some modifications as performed in Part I of thermophilic investigations. The samples were rinsed with 100 mM ammonium bicarbonate (Fisher Scientific):HPLC-grade acetonitrile (Burdick and Jackson, Muskegon, MI; 1:1) for 30 min and then dehydrated with acetonitrile for 10 min. The solution was removed and 10 mM dithiotheritol (BioRad) reduced the samples incubating for 30 min at 56 °C. The solution was removed prior to a second incubation with acetonitrile for 10 min. Acetonitrile was removed and the samples alkylated with 55 mM iodoacetamide (Sigma-Aldrich) for 20 min at room temperature in the dark. The solution was removed and acetonitrile added for approximately 10 min before removal. Protein digestion was induced by the addition of a 200 μl aliquot of 3 ng/μl trypsin (Sigma-Aldrich) and incubated at 37 °C overnight. To stop the digestion and begin extraction of peptides from the gel pieces, a solution of 5% formic acid (Sigma-Aldrich): acetonitrile (1:2) was added and the mixture incubated at 37 °C for 15 min. The supernatant was collected and the samples dehydrated with acetonitrile which was also collected. Additional extraction steps of hydrating with 100 mM ammonium biocarbonate:acetonitrile (2:1), collecting the supernatant, and dehydrating with acetonitrile were repeated. The supernatants were pooled then dried under reduced pressure prior to storage at −20 °C.
Samples were reconstituted in 20 mM Tris–HCl pH 8.0 for triplicate analysis by nanoLC-ESI-FT-ICR MS2. The LC platform consisted of a nanoLC-2D system from Eksigent (Dublin, CA) in the continuous, vented column configuration as previously described [26], and modifications as well as the chromatographic separation was performed identical to the analysis of the extracellular proteome of C. saccharolyticus [27] and summarized here. Incorporated in the system between the transfer line and trap column, a stainless steel (SS) mesh screen (50 μm) was positioned between the transfer line and trap column by a zero dead volume SS fitting (VICI Valco Instruments Co. Inc., Houston, TX). A 100 μm i.d. IntegraFrit capillary packed 3.2 cm operated as the trap column, while a 75 μm i.d PicoTip capillary packed 15 cm functioned as the analytical column (New Objective, Woburn, MA). The columns were packed with Magic C18AQ packing material (Michrom BioReasources, Auburn, CA) utilizing a pressurized cell.
Solvents used for mobile phase A and B were water/acetonitrile/formic acid (98/2/0.1%) and (2/98/0.1%), respectively, and acquired from Burdick and Jackson. Into the sample loop, approximately 1 μg of protein was introduced prior to subjection to the trap column and washing for 5 min at 3 μl by the high-flow pump (Channel 1) with 2% B. The analytical separation was initiated following the metered injection when the valve switched from the high-flow pump to the low-flow pump (Channel 2) with a flow-rate of 300 nl/min and the trap and analytical column in-line. The gradient continued 75 min beginning and ending with 2% B for 3 and 5 min, respectively. Following initial conditions, 10% B was established in 2 min before two subsequent linear gradients set conditions ramping to 25% B over 40 min and 50% B over 15 min. Ninety-five percent B was instated over 3 min and held 2 min before restoring initial conditions.
The LC platform was in-line with a linear quadrupole FT Ultra MS (Thermo Scientific Inc., San Jose, CA) furnished with a 7-T superconducting magnet (Oxford Instruments, Concord, MA). The MS sequence included seven scan events in which the first scan was performed in the ICR cell followed by six data-dependent scans in the ion trap of the first thru sixth most abundant ≥2+ charge state ions analyzed by the initial survey scan. Dynamic exclusion was set to exclude those ions selected for MS2 twice within 30 s and remained on the list for 180 s. At m/z 400 the mass resolving power was 100,000fwhm and the AGC limits for the ICR cell and ion trap were set at 1×106 and 1×104, respectively. As advised by the instrument manufacturer, the MS was externally calibrated.
Data analysis
The resultant RAW files were processed through MASCOT Daemon (Matrix Science Ltd., London, UK) in which they were first subjected to MASCOT Distiller generating peak lists creating MGF files. The MGF files were searched by MASCOT version 2.2.04 producing lists of identified proteins. The FASTA file used for searching all data consisted of a concatenated database compiled of C. saccharolyticus, T. maritima, reverse databases of each microbe, Saccharomyces cerevisiae, Bos taurus trypsin sequence, and H. sapiens keratin and keratin-related protein sequences (177 proteins). All sequences were extracted from the UniProt database and a Perl script generated the reverse database sequences for a total of 15,765 entries. Search parameters for MASCOT protein identification consisted of ≤2 missed tryptic cleavages allowed, and protein modifications, carbamidomethylation (Cys) as fixed, and oxidation (Met) and deamidation (Asn, Gln) as variable. Tolerance for tryptic peptide mass and tandem MS mass measurements were set at ±5 ppm and ±0.6 Da, respectively. The MASCOT bioinformatic platform was coupled with PROTEOIQ 1.2.01 (BIOINQUIRE, Athens, GA), software enabling comparative and quantitative analysis. DAT files generated by MASCOT were uploaded and a protein project was created by PROTEOIQ. Statistically significant identifications were selected by first arranging the protein list by protein score and selecting those proteins having <1% false discovery rate (FDR) [28, 29], and, if for instance C. saccharolyticus and/or S. cerevisiae proteins were listed for the T. maritima sample or vice versa, all proteins above this first incorrect match were selected for the final protein identification inventory (e.g., the final inventory would be solely T. maritima proteins). SpC and related quantification data was generated and extracted for comparative analysis from the PROTEOIQ project created evaluating the thermophilic bacterial proteomes. Those proteins specifically described within the text or figures identified as putative, uncharacterized proteins, were evaluated by Protein Basic Local Alignment Search Tool 2.2.23 (Blastp) and Superfamily [30] prediction tools to predict function and are included in the description of the proteins following the phrase “putative, probable”.
Results and discussion
Cultivation temperature and the intracellular proteome of T. maritima
Following gel electrophoresis of the thermophilic cellular proteome and illustrated by Fig. 1, it was demonstrated that no additional sample preparation was necessary after cell lysis, whereas Part I investigations of the secretome of one of the microbes, C. saccharolyticus, emphasizes the essentiality of protein sample purification prior to 1D SDS-PAGE. This is resultant of limited interferences in the cellular proteome environment as opposed to the array of salts and small molecules present in the liquid media used for cultivation. The two rightmost lanes in Fig. 1 represent the cell lysate of T. maritima grown at two different temperatures. Although literature refers to the optimal growth temperature of T. maritima at 80 °C [12], the co-culture was grown at 75 °C due to compatibility with C. saccharolyticus. To directly compare the co-culture proteome with the individual microbes, it was imperative to demonstrate whether differences were incurred due to the cultivation temperature of the individual culture prior to investigating the proteomic differences in the co-culture proteome. Very few visual differences in the gel image are illustrated between the T. maritima cultures, and furthermore, MS analysis determined 368 proteins in common (see Fig. 2a), which amounts to greater than 10% of the T. maritima proteome and approximately 80% of T. maritima proteins identified in this study. Although the reproducibility of the proteomic expression profiles was not defined in these initial experiments, this demonstrates homology at two temperature points of thermophilic growth although only differing by 5 °C.
Fig. 1.
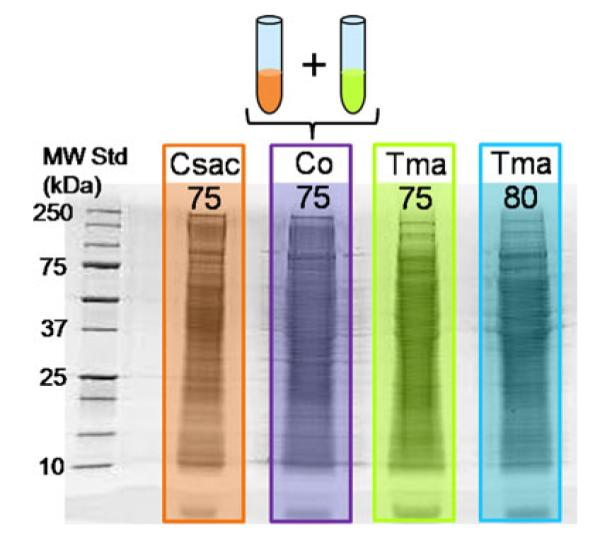
1D SDS-PAGE experiment of individual and co-cultured thermophile samples in which 50 μg were loaded into each sample well. Abbreviations are as follows; Csac75 C. saccharolyticus grown at 75 °C, Co75 a co-culture of the two microbes grown at 75 °C, Tma75 and Tma80 T. maritima cultivated at 75 °C and 80 °C, respectively
Fig. 2.
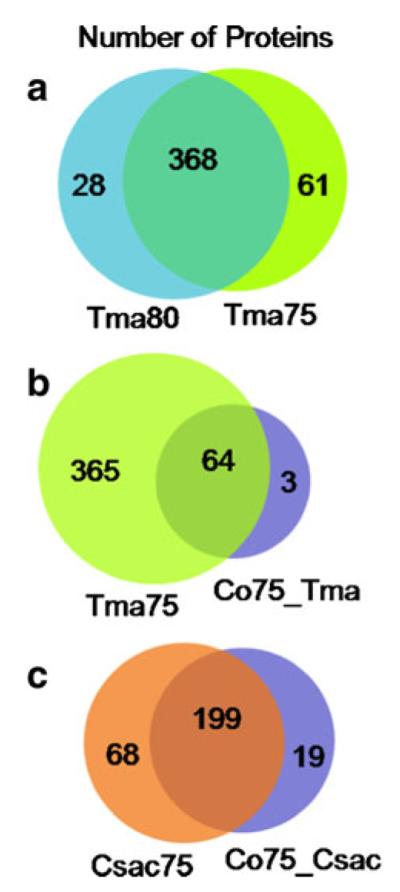
Venn diagrams summarizing the thermophilic bacterial proteins with <1% FDR between a the T. maritima samples grown at 75 °C and 80 °C, b the T. maritima culture grown at 75 °C versus the co-culture sample, and c the C. saccharolyticus sample versus the co-culture sample
A variety of proteins were identified in this first global look into the intracellular proteome of T. maritima. The bacterial proteome consists of three different regions that proteins harbor (in and outside the cell and anchored to the membrane), and although a stringent inventory of proteins confirmed to be in each region does not exist, we can expect certain proteins to be located inside the cell. Inquiring protein function affords a way to confirm or reinforce that the identified species are intracellular proteins. Many ribosomal proteins were identified (40 in common between the two temperature cultures), as well as proteins related to ATP synthase, ATPase subunits, DNA-binding, nucleotide associated proteins, periplasmic oligopeptide-binding proteins and transcriptional regulator proteins all associated with intracellular operations. This initial exploration by GeLC-MS2 provides insight into the proteins present inside the thermophilic cell with functional annotation corroborating the protein location.
Further exploring the proteins present in both T. maritima monocultures, label-free quantification by spectral counting provides comparison of relative protein expression levels and will indicate how significant cultivation temperature alters the thermophilic bacterial proteome as well as other alterations to the growth environment. As discussed by Old et al., a threshold of ≥4 SpC per protein was set for high confidence in differential protein expression [21]. It is demonstrated that when comparing SpC between different samples, those proteins detected with ≥4 require fewer differences in SpC to detect significant proteomic changes. Of those proteins identified in both T. maritima monocultures, 251 proteins were detected with high confident relative quantification and are illustrated in Fig. 3a. Due to weakness in correlation through failure of a linear relationship (R2=0.7988), it was demonstrated that a difference of 5 °C in cultivation temperature of T. maritima alters relative protein expression. Further probing these species, 35 proteins identified in both the 75 °C and 80 °C T. maritima samples (10% of the proteins in common) differed in relative protein expression by more than twofold change as described in Fig. 4a. Seven ribosomal proteins were up-regulated by more than twofold in T. maritima grown at 75 °C whereas no ribosomal proteins had significant down regulation. The opposite relationship would have been expected as the optimal growth temperature of T. maritima is noted at 80 °C where the microbe is thriving and growing most rapidly. This suggests more translation events occur in T. maritima at 75 °C, as these proteins in combination with rRNA are the framework for ribosomes, key components in protein production. Evidenced by the overall number of proteins identified in the 75 °C culture, 429 proteins versus 396 proteins in the culture grown at 80 °C as illustrated in Fig. 2a, the increased expression of ribosomal proteins may contribute to an increase in proteins present in the intracellular proteome. Exploring the gene products more than twofold up-regulated in the T. maritima culture grown at 80 °C versus the 75 °C culture, the majority included seven proteins lacking functional annotation. This was also an unexpected outcome as we would anticipate finding more proteins lacking function at non-optimal growth conditions.
Fig. 3.
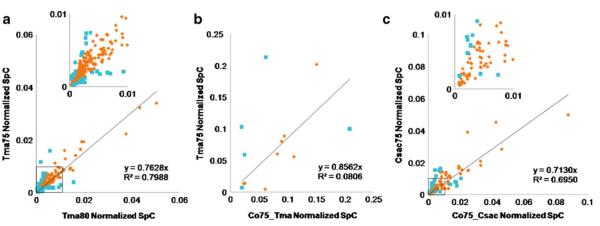
Plots of normalized SpC for comparison of relative protein quantification between thermophilic samples. These plots represent those proteins identified with ≥4 SpC from the corresponding sample; relative quantification of a 251 proteins from T. maritima grown at 75 °C versus T. maritima grown at 80 °C, b 13 proteins from T. maritima grown at 75 °C versus T. maritima proteins identified in the co-culture (Co75_Tma), and c 104 proteins from C. saccharolyticus versus C. saccharolyticus identified in the co-culture (Co75_Csac). The y-intercept was set to 0 and the insets in a and c are representative of the square box on the respective plots for magnification. The blue square markers indicate an increase or decrease of greater than twofold change in normalized SpC for relative protein quantification in which the following number of proteins are represented; a 35, b 5, and c 19
Fig. 4.

Bar graphs displaying greater than twofold change in relative expression of proteins identified with ≥4 SpC in both compared samples. Positive values indicate an increase in protein expression while negative values indicate a decrease in protein expression of a Tma75 with respect to Tma80 (35 proteins), b Tma75 with respect to Co75_Tma (5 proteins), and c Csac75 with respect to Co75_Csac (19 proteins)
Analysis also revealed that of those T. maritima proteins cultivated at 80 °C, almost 7% (28 proteins) were exclusive to the temperature, and from the 75 °C culture, approximately 14% (61 proteins) were unique (see Fig. 2a). The majority of those proteins uniquely present in the cultures grown at different temperatures included several putative uncharacterized and ribosomal proteins, and those with ≥4 SpC are displayed in Table 1a, b. A T. maritima protein of interest identified only in the 75 °C, chemotaxis protein cheY, is necessary in the bacterial chemotaxis pathway in which bacteria are influenced to migrate due to the chemical environment [31–33]. This movement may be induced by regions in the culture or bacterial environment having more favorable levels of glucose for energy, for example. It is probable that the microbe requires more nutrition as a function of the non-optimal temperature or varying concentrations of glucose exist in different regions in the culture media vessel at 75 °C. This situation stimulates the chemotaxis pathway, and thus translation of the cheY protein. Overall, the T. maritima intracellular proteome cultivated at the two different temperatures reveals only modest changes and future investigations warrant biological and analytical replicates for assessment.
Table 1.
List of unique proteins having >4 SpC for comparisons with the co-culture and relative monocultures
| MW, kDa | SpC | Normalized SpC | |
|---|---|---|---|
| (a) Tma Proteins Unique to Tma80 with ≥4 SpC, with respect to Tma75 | |||
| Q9X0X8 Phosphoribosylformylglycinamidine cyclo-ligase | 34.4 | 7 | 0.06989 |
| RL21 50S ribosomal protein L21 | 12.2 | 6 | 0.17742 |
| HSLU ATP-dependent hsl protease ATP-binding subunit hslU | 53.0 | 6 | 0.04024 |
| Q9X2G5 Putative uncharacterized protein | 7.2 | 5 | 0.26313 |
| LDH l-lactate dehydrogenase | 35.0 | 4 | 0.03893 |
| (b) Tma Proteins Unique to Tma75 with ≥4 SpC, with respect to Tma80 | |||
| SYGB Glycyl-tRNA synthetase beta subunit | 77.7 | 20 | 0.02343 |
| CHEY Chemotaxis protein cheY | 13.2 | 18 | 0.11806 |
| RS18 30 S ribosomal protein S18 | 9.0 | 17 | 0.17841 |
| Q9WXZ4 Putative, probable iojap-like protein | 12.8 | 12 | 0.08586 |
| SYT Threonyl-tRNA synthetase | 74.5 | 11 | 0.01353 |
| RL13 50 S ribosomal protein L13 | 17.3 | 9 | 0.04754 |
| HFQ Protein hfq | 10.5 | 7 | 0.05989 |
| Q9X176 Putative, probable DRTGG domain-containing protein | 12.8 | 6 | 0.04071 |
| RL18 50 S ribosomal protein L18 | 14.0 | 5 | 0.03226 |
| Q9X076 Putative, probable DRTGG domain-containing protein | 13.1 | 5 | 0.03335 |
| Q9WXN1 Laminarinase | 72.5 | 4 | 0.00490 |
| Q9WXM7 NADP-reducing hydrogenase, subunit A | 19.9 | 4 | 0.01789 |
| Q9X2B1 Putative, probable CRISPR-associated RAMP Cmr6 family protein | 27.9 | 4 | 0.01290 |
| Q9X2F4 Cyclomaltodextrinase, putative | 55.1 | 4 | 0.00666 |
| (c) Tma Proteins Unique to Co75_Tma with ≥4 SpC | |||
| Q9X0E8 Glutamate synthase-related protein | 55.9 | 6 | 0.63235 |
| (d) Tma Proteins Unique to Tma with ≥4 SpC, with respect to Co75_Tma | |||
| 34 Ribosomal proteins | |||
| 51 Putative uncharacterized proteins | |||
| 102 Enzyme, enzyme like proteins (i.e., synthetase, syntase, kinase, dehydratase, etc.) | |||
| 53 Other (i.e., chemotaxis, ABC transporter, binding proteins) | |||
| *See Supplemental Table 1 for full list | |||
| (e) Csac Proteins Unique to Co75_Csac with ≥4 SpC | |||
| A4XJ94 Ferredoxin-dependent glutamate synthase | 58.8 | 16 | 0.08850 |
| A4XI39 NUDIX hydrolase | 16.0 | 6 | 0.12612 |
| A4XHT6 Putative, probable YjbQ-like protein | 15.9 | 5 | 0.10662 |
| A4XG53 Monosaccharide-transporting ATPase | 34.1 | 5 | 0.04486 |
| A4XLT3 Putative, probable beta-lactamase-like protein | 5.9 | 4 | 0.22209 |
| A4XHL3 Lipolytic enzyme, G-D-S-L family | 26.9 | 4 | 0.05052 |
| A4XFT4 phage-shock protein A, PspA | 25.6 | 4 | 0.05302 |
| (f) Csac proteins unique to Csac75_with ≥4 SpC | |||
| A4XI09 Cell division protein FtsZ | 38.7 | 14 | 0.04415 |
| A4XI51 ABC-type tungstate transport system permease component-like protein precursor | 31.3 | 9 | 0.03770 |
| A4XI54 Molybdopterin molybdochelatase | 44.4 | 9 | 0.02529 |
| A4XKY2 Beta-lactamase domain protein | 46.3 | 8 | 0.02248 |
| A4XLC4 Putative, probable alkaline shock protein | 13.7 | 6 | 0.05322 |
| A4XL18 Mannose-1-phosphate guanylyltransferase | 41.0 | 6 | 0.01897 |
| A4XMK1 Appr-1-p processing domain protein | 20.3 | 6 | 0.03722 |
| RL19 50 S ribosomal protein L19 | 14.9 | 6 | 0.05322 |
| A4XHR8 Branched chain amino acid aminotransferase | 32.7 | 6 | 0.02325 |
| A4XLL5 Putative, probable SP0561-like | 7.7 | 5 | 0.07995 |
| KAD Adenylate kinase | 24.0 | 5 | 0.02640 |
| Y2087 UPF0296 protein Csac_2087 | 11.2 | 4 | 0.04496 |
| A4XG97 Hemerythrin-like metal-binding protein | 16.6 | 4 | 0.03291 |
| A4XJT0 Putative, probable YhbY-like protein | 10.8 | 4 | 0.04780 |
| A4XH18 Glutamate synthase | 41.3 | 4 | 0.01248 |
There was a reduction in the number of identified proteins as a function of SpC. The following describes this reduction in the number of proteins identified (unique proteins with <1% FDR→*** unique proteins with <1% FDR and ≥4 spectral counts); (a) 28→5, (b) 61→14, (c) 3→1, (d) 365→240 which is not described in detail due to the great amount of homology of the T. maritima proteins, (e) 19→7, and (f) 68→15
Co-culture media weakness
Consistent with the media makeup, which was derived from the DSMZ recommended C. saccharolyticus media with additional salt to account for nutritional needs of T. maritima described in the experimental section, C. saccharolyticus thrived and dominated over T. maritima. Two hundred and eighty-five proteins were identified in the co-culture cellular proteome, 76.5% (218 proteins) were of C. saccharolyticus species and 23.5% (67 proteins) from T. maritima. A visual representation of the level of protein overlap is shown in Fig. 2b, c. This same relationship was clearly demonstrated in comparison of SpC between C. saccharolyticus and T. maritima proteins identified in the co-culture (see Table 2). An exploration of the identified proteins will afford probable trends suggesting future investigations; however, no definitive conclusions can be made regarding the T. maritima proteins in the pure versus co-culture due to this non-optimal media formula. Only three T. maritima proteins unique to the co-culture were identified and only one of these having ≥4 SpC (see Table 1c). Also, of the T. maritima proteins identified, 240 were unique to the pure culture grown at 75 °C and discussed in Table 1d. Figure 3b illustrates that 13 T. maritima proteins having ≥4 SpC were in common between the pure and co-culture. Indicated by the plot and depicted in Fig. 4b, five proteins, about 40% of those with confident relative quantification, were differentially expressed by more than twofold. As a result of altering two variables, the culture media and the addition of a second microbe, the cause of change in expression cannot be differentiated.
Table 2.
Summary indicating the number of confident proteins identified having<1% FDR, and further, those proteins having≥4 SpC
| Sample | Protein IDs <1% FDR | Total spectral counts (SpC) | Protein IDs ≥4 SpC | # SpC of proteins ≥4 SpC |
|---|---|---|---|---|
| Tma80 | 396 | 6827 | 287 | 6636 |
| Tma75 | 429 | 7146 | 301 | 6903 |
| Co75_Tma | 67 | 212 | 14 | 127 |
| Csac75 | 267 | 3085 | 160 | 2866 |
| Co75_Csac | 218 | 2681 | 124 | 2499 |
Total number of SpC was calculated as the sum of individual SpC from each protein group. The co-culture sample was divided into two protein groups corresponding to T. maritima (Co75_Tma) or C. saccharolyticus (Co75_Csac) proteins identified in the co-culture
Furthermore, no clear interaction between T. maritima and C. saccharolyticus can be established by our exploration of the intracellular proteome. It is common for bacteria to exist cooperatively [34], for example, in anaerobic environments hydrogen is produced [35], and increased amounts can inhibit bacterial growth [12]; however, the presence of a methanogenic species behaves as a hydrogen sink [36]. It has been evidenced in the co-culture of T. maritima and the methanogenic M. jannaschii that the cell density increased by fivefold and moieties enhancing microbial interactions were present as compared to the monoculture [1]. While only features of T. maritima were monitored in these past studies, it can be suggested that a relationship of mutualism, in which both species benefit, or commensalism, where one species benefits while the other is unharmed, describes the interaction. In our investigations, 67 T. maritima proteins were confidently identified in the co-culture and 95.5% were in common with the pure culture. Housekeeping proteins such as glyceraldehyde-3-phosphate dehydrogenase, ribosomal proteins, and putative uncharacterized proteins were represented alluding to a relationship of mutualism or commensalism.
Comparison of C. saccharolyticus intracellular proteome in pure and co-culture
Although the number of ORFs in the C. saccharolyticus proteome exceeds that of the T. maritima proteome, 2,679 and 1,858 respectively, only 10.6% of the C. saccharolyticus proteome was confidently identified in this study and 11.6% of the proteome when including the proteins identified in Part I of this investigation, whereas almost 25% of the T. maritima proteome was identified. This outcome could be due to several factors such as insufficient lysis of the C. saccharolyticus cells, unsuccessful extraction of proteins from the 1D gel, as well as populations of low abundant proteins suppressed by highly abundant species, and thus inherently undetectable by GeLC-MS2. Nevertheless, a variety of C. saccharolyticus proteins was represented and demonstrated partiality for location in and outside the cell. Proteins associated with transcriptional regulation, ribosomes, and DNA and RNA function were identified, and inherent in their function, supported their existence inside the cell. Additionally, several proteins appear to function externally or outside the cell such as glycoside hydrolases and extracellular solute-binding proteins. This suggests several scenarios; for example, the protein may not be fully secreted into the extracellular space, but remain inside or anchored to the cell membrane due to growth and development of the microbe at the particular time of harvest.
Of the C. saccharolyticus proteins identified in both the pure and co-culture and having ≥4 SpC, Fig. 3c plots normalized SpC as a function of culture. Poor correlation (R2=0.6950) demonstrates that protein regulation between cultures is quite varied, and further probing those proteins with greater than twofold change in expression, Fig. 4c displays the differential regulation of the corresponding proteins. Interestingly, two proteins present having increased expression by more than twofold in the pure C. saccharolyticus culture are associated with CRISPR or clustered regularly interspaced short palindromic repeats associated proteins [37]. The presence of these proteins up-regulated in the pure C. saccharolyticus culture suggests an antiviral mechanism providing resistance to phages [38]. It appears that this resistance is mediocre if any in the co-culture as evidenced by the presence of a phage-shock protein unique to the co-culture and listed in Table 1e. This phage-shock protein implies the onset of an external stress or infection to the bacterial cell [39]. This is an example demonstrating that the addition of a second microbe to a pure culture triggers events causing the presence, absence, and differential expression of protein species within the system.
C. saccharolyticus extracellular and intracellular proteome evaluation
Merging data from the analysis of the gene products both in and outside the C. saccharolyticus thermophilic bacterial cell (Part I and II of this investigation) provides a compilation and comparative glimpse into the techniques, technologies, and analysis contributed from cell growth to protein preparation to bioinformatics for this microbe. Figure 5 demonstrates the compilation of confidently identified C. saccharolyticus proteins from each investigation. As described in Table 3 and displayed in Fig. 5, 53 proteins with <1% FDR were identified in both the intra- and extracellular thermophilic proteome of C. saccharolyticus, and this computes to 18.5% and 74.6% of the proteins identified, respectively. Indicated in Table 3, nine proteins were predicted for secretion and seven were predicted to have transmembrane domains anchoring them to the cell membrane from evaluations with prediction servers in Part I. The sequence coverage for each protein from each sample fraction was examined and compared to its counterpart for any obvious indications of cleavage. Of those proteins identified in both the cell lysis and culture medium as well as predicted for secretion, it would be expected that a cleaved signal peptide would remain adhered to the cell while the mature protein secreted into the extracellular space. Thus, sequence coverage would vary as a function of sample type.
Fig. 5.
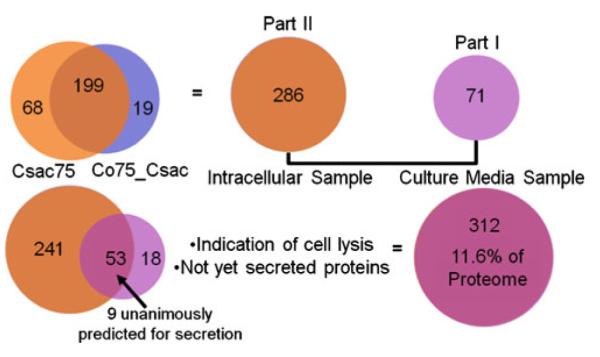
Schematic of the number of C. saccharolyticus proteins identified in Part I and Part II of this study. Of the proteins overlapping, nine were predicted for secretion and seven were predicted to have transmembrane domains. See Part I of this investigation for a detailed description of prediction servers and analysis
Table 3.
Combination of overlapping protein identifications from both parts of the thermophilic investigation (Part I and II data shown here)
| Protein Sequence ID and Function | PS | PTD | Intracellular | Extracellular |
|---|---|---|---|---|
| A4XG54 Periplasmic binding protein/Laci transcriptional regulator precursor | x | |||
| A4XG54 Pulative, probable transglutaminase domain protein | x | |||
| A4XGB3 5-methyttetrahydropteroyttriglutamate-homocysteine S-methyttransferase | ||||
| A4XGF7 d-isomer specific 2-hydroxyacid dehydrogenase, NAD-binding | Not C-terminus | C-terminus | ||
| A4XGF5 Extracellular solute-binding protein, family 1 precursor | x | |||
| A4XGNZ2 Glutamate synthase or A4XH19 Glutamate synthase | Throughout | 2nd half | ||
| A4XHF8 Pyruvate carboxyttransferase | Throughout | Middle | ||
| A4XH10 Pyruvate carboxyttransferase | Not middle | Middle | ||
| A4XH14 Aconitase | Throughout | Middle | ||
| A4XHL2 Transcriptional regulator/glucokinase | ||||
| A4XHV9 TIG Trigger factor | 2nd half | 1st half | ||
| A4XI26 Ser-tRNA(Thr) hydrolase/threonyl-tRNA synthetase | ||||
| A4XI36 EFG Elongation factor G | ||||
| A4XI37 EFTU Elongation factor Tu | ||||
| A4XI48 Aldehyde ferredoxin oxidoreductase | ||||
| A4XIF5 Glycoside hydrolase, family 48 precursor | x | |||
| A4XIF6 Glycoside hydrolase, family 5 precursor | x | 1st half | Throughout | |
| A4XIF7 Cellulose 1, 4-beta-cellobiose phosphorylase | x | |||
| A4XIG9 Cellobiose phosphorylase | ||||
| A4XIF8 2 isopropylmalate synthase | Throughout | Middle | ||
| A4XIL9 2-isopropylmalate synthase | Throughout | Middle | ||
| A4XIQ5 Mannose-6-phosphate isomerase/glucose-6-phosphate isomerase | Throughout | Middle | ||
| A4XIQ7 Fructose-bisphosphate aldolase | ||||
| A4XJ08 CH10 10 kDa chaperonin | ||||
| A4XJ09 CH60 60 kDa chaperonin | ||||
| A4XJ20L-aspartate aminotransferase/phosphoserine aminotransferase | Throughout | 1st half | ||
| A4XJH3 pyruvate ferredosin oxidoredectase, gamma subunit | x | |||
| A4XJH5 pyruvate ferredosin oxidoredectase, alpha subunit | ||||
| A4XJH6 Thiamine pyrophosphate enzyme domain protein TPP-binding | x | Throughout | Middle | |
| A4XJW3 Acyl carrier protein | Throughout | 2nd half | ||
| A4XJW4 3-oxoacyl-[acyl-carrier-protein] synthase II | x | |||
| A4XJY3 Putative, probable phophoenolpyruvate carboxykinase | x | Throughout | 2nd half | |
| A4XK10 6-phosphofructokinase | ||||
| A4XKL3 NADH dehydrogenase | ||||
| A4XKL4 Hydrogenase, Fe-only | ||||
| A4XKR2 SYI Isoleucyl-tRNA synthetase | Throughout | 2nd half | ||
| A4XKV2 Phosphoglycerate kinase/triosephosphate isomerase | ||||
| A4XKV3 Glyceraldehyde-3-phosphate dehydrogenase | Throughtout | 2nd half | ||
| A4XKV5 Pyruvate phospahte dikinase | ||||
| A4XKX0ATPB ATP synthase subunit beta | Throughout | 2nd half | ||
| A4XL36TAL Probable transaldolase | x | Throughout | 1st half | |
| A4XL36 TAL Probable transaldolase | x | Throughout | 1st half | |
| A4XM01 EFTS Elongation factor Ts | ||||
| A4XLL5 Putative, probable conservative domain with unknown function | Throughout | 1st half | ||
| A4XM70 Alpha amylase, catalytic region | ||||
| A4XM72 Rubrerythrin | ||||
| A4XM87 S-layer domain protein precursor | x | |||
| A4XM93 S-layer domain protein precursor | x | |||
| A4XMD5 Extracellular solute-binding protein, family 1 presursor | x | |||
| A4XMZ6 Ferritin, Dps family protein |
Following the protein description “x” defines if the protein was predicted for secretion (PS) or containing a transmembrane domain (PTD) from Part I of this investigation. Also, sequence information was examined to observe possible patterns corroborating the predictions made in Part I. Those proteins with comments in the two rightmost lanes define sequence coverage of the protein sequenced by MS as a function of sample (intracellular fraction or extracellular fraction). Those without indication of sequence coverage indicate no relative difference between the two samples
Probing the sequence coverage no definitive patterns were recognized suggesting signal peptides or transmembrane domains. In general, the proteins identified in Part I of the study were detected with fewer peptides and thus having lower sequence coverage, which is attributed to necessary sample handling of the secretome samples; it is essential to purify and cleanup the secretome sample prior to analysis. This processing appears to have resulted in protein degradation. The d-isomer-specific 2-hydroxyacid dehydrogenase, NAD-binding protein (A3XGF7) was demonstrated with sequence coverage throughout the protein with exception of the C-terminus for detection in the intracellular sample, and only the C-terminal peptide for detection in the extracellular proteome. Although this was the only uniquely observed pattern, a signal peptide is most commonly amino-terminal [40], thus the contrast. While a smaller protein overlap was anticipated, many factors may contribute to the identification of these species both in and outside the cell. Cell lysis presents the most likely case for identification of intracellular proteins in the culture media sample, and proteins not yet secreted present the most likely case for identification of extracellular proteins in the intracellular fraction. There is no explicit list delineating proteins secreted into the extracellular space versus those remaining inside the cell, here speculation based on function and experimental data compile predicted location.
Conclusions
Here, it was demonstrated thru GeLC-MS2 and spectral counting for label-free relative quantification that the thermophiles of interest are dynamic organisms as changes in the growth temperature and the addition of a second microbe induce the presence or absence of gene products and differential expression of these products. While no momentous changes were noted, several probable scenarios evolve based on protein expression. Exploration of the T. maritima proteome grown at its optimal growth temperature and that of the co-culture revealed significant homology of up to 80% of T. maritima proteins identified in this study amounting to approximately 25% of the proteome. Thirty-five proteins identified in both T. maritima cultures demonstrated greater than twofold change in protein expression; however, it would be expected that more ribosomal proteins would be up-regulated at the optimal growth temperature when the contrary was true. The majority of proteins unique to each culture with confident relative quantification of ≥4 SpC, encompassed ribosomal and putative uncharacterized proteins. One protein in particular, chemotaxis protein cheY, was identified only in the T. maritima culture grown at the co-culture temperature and is associated with movement of the organism as a function of the chemical surroundings. This suggests that T. maritima cultivated at its non-optimal temperature requires increased movement in attempt to thwart off a hostile environment or to locate additional food sources.
Analysis of the intracellular proteome sample of the two thermophiles grown in co-culture exhibited an unequal distribution of proteins. Over 75% of the proteins identified in the co-culture were of C. saccharolyticus origin and this is presumably in response to the media makeup which was almost identical to the C. saccharolyticus growth media. Unanticipated, this deficiency in number of identifications results in very few postulates based on protein expression of T. maritima in the co-culture. In addition, the type of relationship between the two microbes in co-culture cannot be clearly deciphered. In contrast, C. saccharolyticus proteins were in abundance leading to an interesting comparison with the pure culture. Label-free relative quantification demonstrated an up-regulation in two C. saccharolyticus proteins associated with infection resistance in the pure culture. Harboring the co-cultured sample, an infection-related protein was expressed. The presence of this phage protein corroborates with the decreased expression of the antiviral associated proteins in the co-culture and may provide evidence of microbial interaction. C. saccharolyticus may not have been fully aware of T. maritima and could not completely control the antiviral mechanism with an additional organism; the co-cultivation may have prevented the antiviral resistance and with the onset of a possible infection, the phage protein was translated. Monitoring protein expression by GeLC-MS2 and spectral counting affords a snapshot of the thermophilic bacterial culture at a variety of situations. Further modifying the environmental stimuli, such as altering the microbial community, will instigate differential protein expression expanding proteomic investigations for thermostable biomolecules having technological value.
Supplementary Material
Acknowledgments
We would like to acknowledge the financial support of the National Institutes of Health (Grant 5T32GM00-8776-08), which supports GLA in the North Carolina State University Molecular Biotechnology Training Program, and the W. M. Keck Foundation. RMK acknowledges support from the US National Science Foundation (CBT0617272) and the Bioenergy Science Center (BESC), a U.S. DOE Bioenergy Research Center supported by the Office of Biological and Environmental Research. DLL acknowledges support from a US Department of Education GAANN Fellowship.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00216-010-3929-8) contains supplementary material, which is available to authorized users.
Contributor Information
David Muddiman, Chemistry, North Carolina State University, Raleigh, NC 27695, USA; W.M. Keck FT-ICR Mass Spectrometry Laboratory, North Carolina State University, Raleigh, NC 27695, USA.
Genna Andrews, Chemistry, North Carolina State University, Raleigh, NC 27695, USA.
Derrick Lewis, Chemical and Biomolecular Engineering, North Carolina State University, Raleigh, NC 27695, USA.
Jaspreet Notey, Chemical and Biomolecular Engineering, North Carolina State University, Raleigh, NC 27695, USA.
Robert Kelly, Chemical and Biomolecular Engineering, North Carolina State University, Raleigh, NC 27695, USA.
References
- 1.Johnson MR, Montero CI, Conners SB, Shockley KR, Bridger SL, Kelly RM. Population density-dependent regulation of exopolysaccharide formation in the hyperthermophilic bacterium Thermotoga maritima. Mol Microbiol. 2005;55:664–674. doi: 10.1111/j.1365-2958.2004.04419.x. [DOI] [PubMed] [Google Scholar]
- 2.Yang YL, Xu YQ, Straight P, Dorrestein PC. Translating metabolic exchange with imaging mass spectrometry. Nat Chem Biol. 2009;5:885–887. doi: 10.1038/nchembio.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muhammad SA, Ahmad S, Hameed A. Antibiotic production by thermophilic bacillus specie Sat-4. Pak J Pharm Sci. 2009;22:339–345. [PubMed] [Google Scholar]
- 4.Esikova TZ, Temirov YV, Sokolov SL, Alakhov YB. Secondary antimicrobial metabolites produced by thermophilic Bacillus spp. strains VK2 and VK21. Appl Biochem Microbiol. 2002;38:226–231. [PubMed] [Google Scholar]
- 5.Shank EA, Kolter R. New developments in microbial interspecies signaling. Curr Opin Microbiol. 2009;12:205–214. doi: 10.1016/j.mib.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vafiadi C, Topakas E, Biely P, Christakopoulos P. Purification, characterization and mass spectrometric sequencing of a thermophilic glucuronoyl esterase from Sporotrichum thermophile. FEMS Microbiol Lett. 2009;296:178–184. doi: 10.1111/j.1574-6968.2009.01631.x. [DOI] [PubMed] [Google Scholar]
- 7.Barabote RD, Xie G, Leu DH, Normand P, Necsulea A, Daubin V, Medigue C, Adney WS, Xu XC, Lapidus A, Parales RE, Detter C, Pujic P, Bruce D, Lavire C, Challacombe JF, Brettin TS, Berry AM. Complete genome of the cellulolytic thermophile Acidothermus cellulolyticus 11B provides insights into its ecophysiological and evolutionary adaptations. Genome Res. 2009;19:1033–1043. doi: 10.1101/gr.084848.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asada Y, Endo S, Inoue Y, Mamiya H, Hara A, Kunishima N, Matsunaga T. Biochemical and structural characterization of a short-chain dehydrogenase/reductase of Thermus thermophilus HB8 A hyperthermostable aldose-1-dehydrogenase with broad substrate specificity. Chem Biol Interact. 2009;178:117–126. doi: 10.1016/j.cbi.2008.09.018. [DOI] [PubMed] [Google Scholar]
- 9.Makarova KS, Wolf YI, Koonin EV. Comprehensive comparative-genomic analysis of Type 2 toxin–antitoxin systems and related mobile stress response systems in prokaryotes. Biol Direct. 2009;4:19. doi: 10.1186/1745-6150-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pandey DP, Gerdes K. Toxin–antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005;33:966–976. doi: 10.1093/nar/gki201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blumer-Schuette SE, Kataeva I, Westpheling J, Adams MWW, Kelly RM. Extremely thermophilic microorganisms for biomass conversion: status and prospects. Curr Opin Biotechnol. 2008;19:210–217. doi: 10.1016/j.copbio.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 12.Huber R, Langworthy TA, Konig H, Thomm M, Woese CR, Sleytr UB, Stetter KO. Thermotoga maritima sp nov represents a new genus of unique extremely thermophilic eubacteria growing up to 90°C. Arch Microbiol. 1986;144:324–333. [Google Scholar]
- 13.Chhabra SR, Shockley KR, Conners SB, Scott KL, Wolfinger RD, Kelly RM. Carbohydrate-induced differential gene expression patterns in the hyperthermophilic bacterium Thermotoga maritima. J Biol Chem. 2003;278:7540–7552. doi: 10.1074/jbc.M211748200. [DOI] [PubMed] [Google Scholar]
- 14.Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson LD, Nelson WC, Ketchum KA, McDonald L, Utterback TR, Malek JA, Linher KD, Garrett MM, Stewart AM, Cotton MD, Pratt MS, Phillips CA, Richardson D, Heidelberg J, Sutton GG, Fleischmann RD, Eisen JA, White O, Salzberg SL, Smith HO, Venter JC, Fraser CM. Evidence for lateral gene transfer between Archaea and Bacteria from genome sequence of Thermotoga maritima. Nature. 1999;399:323–329. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- 15.VanFossen AL, Lewis DL, Nichols JD, Kelly RM. Polysaccharide degradation and synthesis by extremely nermophilic anaerobes. Ann NY Acad Sci. 2008;1125:322–337. doi: 10.1196/annals.1419.017. [DOI] [PubMed] [Google Scholar]
- 16.Rainey FA, Donnison AM, Janssen PH, Saul D, Rodrigo A, Bergquist PL, Daniel RM, Stackebrandt E, Morgan HW. Description of Caldicellulosiruptor saccharolyticus gen-nov, spnov—an obligately anaerobic, extremely thermophilic, cellulolytic bacterium. FEMS Microbiol Lett. 1994;120:263–266. doi: 10.1111/j.1574-6968.1994.tb07043.x. [DOI] [PubMed] [Google Scholar]
- 17.VanFossen AL, Verhaart MRA, Kengen SMW, Kelly RM. Carbohydrate utilization patterns for the extremely thermophilic bacterium Caldicellulosiruptor saccharolyticus reveal broad growth substrate preferences. Appl Environ Microb. 2009;75:7718–7724. doi: 10.1128/AEM.01959-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Quantitative mass spectrometry in proteomics: a critical review. Anal Bioanal Chem. 2007;389:1017–1031. doi: 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- 19.Gao J, Friedrichs MS, Dongre AR, Opiteck GJ. Guidelines for the routine application of the peptide hits technique. J Am Soc Mass Spectrom. 2005;16:1231–1238. doi: 10.1016/j.jasms.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Liu HB, Sadygov RG, Yates JR. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 21.Old WM, Meyer-Arendt K, Aveline-Wolf L, Pierce KG, Mendoza A, Sevinsky JR, Resing KA, Ahn NG. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol Cell Proteomics. 2005;4:1487–1502. doi: 10.1074/mcp.M500084-MCP200. [DOI] [PubMed] [Google Scholar]
- 22.Zybailov B, Coleman MK, Florens L, Washburn MP. Correlation of relative abundance ratios derived from peptide ion chromatograms and spectrum counting for quantitative proteomic analysis using stable isotope labeling. Anal Chem. 2005;77:6218–6224. doi: 10.1021/ac050846r. [DOI] [PubMed] [Google Scholar]
- 23.Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J Proteome Res. 2006;5:2339–2347. doi: 10.1021/pr060161n. [DOI] [PubMed] [Google Scholar]
- 24.Gao J, Bauer MW, Shockley KR, Pysz MA, Kelly RM. Growth of hyperthermophilic Archaeon Pyrococcus futiosus on chitin involves two family 18 chitinases. Appl Environ Microbiol. 2003;69:3119–3128. doi: 10.1128/AEM.69.6.3119-3128.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. Ingel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 26.Andrews GL, Shuford CM, Burnett JC, Hawkridge AM, Muddiman DC. Coupling of a vented column with splitless nanoRPLC-ESI-MS for the improved separation and detection of brain natriuretic peptide-32 and its proteolytic peptides. J Chromatogr B. 2009;877:948–954. doi: 10.1016/j.jchromb.2009.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andrews GL, Lewis DL, Notey J, Kelly RM, Muddiman DC. Characterization of the extracellular proteome of the extreme thermophile Caldicellulosiruptor saccharolyticus by GeLC-MS2. J. Proteome Res. 2010 doi: 10.1007/s00216-010-3955-6. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benjamini Y, Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J R Stat Soc Ser B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 29.Weatherly DB, Atwood JA, Minning TA, Cavola C, Tarleton RL, Orlando R. A heuristic method for assigning a false-discovery rate for protein identifications from mascot database search results. Mol Cell Proteomics. 2005;4:762–772. doi: 10.1074/mcp.M400215-MCP200. [DOI] [PubMed] [Google Scholar]
- 30.Gough J, Karplus K, Hughey R, Chothia C. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol. 2001;313:903–919. doi: 10.1006/jmbi.2001.5080. [DOI] [PubMed] [Google Scholar]
- 31.Pfeffer W. Locomotorische Richtungsbewegungen durch chemische Reize. Unters Bot Inst Tub. 1884;1:95–24. [Google Scholar]
- 32.Pfeffer W. Uber chemotaktische Bewegungen von Bakterien, Flagellaten und Volvocineen. Unters Bot Inst Tub. 1888;2:582–663. [Google Scholar]
- 33.Berg HC. Chemotaxis in Bacteria. Annu Rev Biophys Bioeng. 1975;4:119–136. doi: 10.1146/annurev.bb.04.060175.001003. [DOI] [PubMed] [Google Scholar]
- 34.Kolter R, Losick R. Microbiology—one for all and all for one. Science. 1998;280:226–227. doi: 10.1126/science.280.5361.226. [DOI] [PubMed] [Google Scholar]
- 35.Adams MWW. The metabolism of hydrogen by extremely thermophilic sulfur-dependent bacteria. FEMS Microbiol Rev. 1990;75:219–237. [Google Scholar]
- 36.Muralidharan V, Rinker KD, Hirsh IS, Bouwer EJ, Kelly RM. Hydrogen transfer between methanogens and fermentative heterotrophs in hyperthermophilic cocultures. Biotechnol Bioeng. 1997;56:268–278. doi: 10.1002/(SICI)1097-0290(19971105)56:3<268::AID-BIT4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 37.Jansen R, van Embden J, Gasstra W, Schouls L. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol. 2002;43:1565–1575. doi: 10.1046/j.1365-2958.2002.02839.x. [DOI] [PubMed] [Google Scholar]
- 38.Sorek R, Kunin V, Hugenholtz P. CRISPR—a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat Rev Microbiol. 2008;6:181–186. doi: 10.1038/nrmicro1793. [DOI] [PubMed] [Google Scholar]
- 39.Brissette JL, Russel M, Weiner L, Model P. Phage shock protein, a stress protein of Escherichia coli. PNAS. 1990;87:862–866. doi: 10.1073/pnas.87.3.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tjalsma H, Bolhuis A, Jongbloed JDH, Bron S, van Dijl JM. Signal peptide-dependent protein transport in Bacillus subtilis: a genome-based survey of the secretome. Microbiol Mol Biol Rev. 2000;64:515–547. doi: 10.1128/mmbr.64.3.515-547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.