

Abstract
Epidermolysis bullosa acquisita (EBA) is a chronic mucocutaneous autoimmune skin blistering disease. The pathogenic relevance of autoantibodies targeting type VII collagen (COL7) has been well-documented. Therefore, EBA is a prototypical autoimmune disease with a well-characterized pathogenic relevance of autoantibody binding to the target antigen. EBA is a rare disease with an incidence of 0.2 new cases per million and per year. The current treatment of EBA relies on general immunosuppressive therapy, which does not lead to remission in all cases. Therefore, there is a high, so far unmet medical need for the development of novel therapeutic options. During the last 10 years, several novel in vitro and in vivo models of EBA have been established. These models demonstrated a critical role of the genetic background, T cells, and cytokines for mediating the loss of tolerance towards COL7. Neutrophils, complement activation, Fc gamma receptor engagement, cytokines, several molecules involved in cell signaling, release of reactive oxygen species, and matrix metalloproteinases are crucial for autoantibody-induced tissue injury in EBA. Based on this growing understanding of the diseases' pathogenesis, several potential novel therapeutic targets have emerged. In this review, the clinical presentation, pathogenesis, diagnosis, and current treatment options for EBA are discussed in detail.
1. Clinical Presentation of Epidermolysis Bullosa Acquisita
In the beginning of the 20th century, the term “epidermolysis bullosa acquisita” (EBA) was used as a descriptive clinical diagnosis for patients with adult onset and features resembling those of hereditary dystrophic epidermolysis bullosa [1]. Almost 70 years later, EBA was distinguished from other bullous diseases on the basis of distinctive clinical and histological features, implementing the first diagnostic criteria for the disease. Specifically, these included (i) clinical lesions resembling epidermolysis bullosa dystrophica, (ii) adult onset of disease, (iii) a negative family history of epidermolysis bullosa dystrophica, and (iv) exclusion of other bullous diseases [2]. Based on the current understanding of EBA pathogenesis, additional/other criteria define EBA diagnosis today (see Section 13). The cutaneous manifestations in EBA patients are heterogeneous. However, EBA patients can be classified into two major clinical subtypes: noninflammatory (classical or mechanobullous) and inflammatory EBA, which is characterized by cutaneous inflammation resembling bullous pemphigoid, linear IgA disease, mucous membrane pemphigoid, or Brunsting-Perry pemphigoid [3–5]. The clinical presentation of an individual EBA patient may change during the course of the disease, or the same patients may present with two different forms simultaneously.
2. Mechanobullous EBA
The mechanobullous (noninflammatory, classical) variant of EBA is observed in approximately 1/3 of the patients (Table 1) and is characterized by skin fragility, tense blisters, scaring, and milia formation preferably localized to trauma-prone sites and the extensor skin surface. In these patients, nail dystrophy and postinflammatory hyper- and hypopigmentation are also frequently observed. Furthermore, the oral mucosa is often affected in these patients. In mild cases, the clinical presentation is similar to porphyria cutanea tarda, while severe cases are comparable to hereditary recessive dystrophic epidermolysis bullosa [6–9].
Table 1.
Epidemiology and Ig reactivity in EBA.
| Gender distribution | 61% female 39% male |
|
| |
| Age (median, 75 percentile) | 47, 30–66 years* |
|
| |
| Clinical phenotypes | Inflammatory phenotypes: 66% Classical (mechanobullous) phenotype: 33% |
|
| |
| Ig reactivity (DIF positive) | IgG: 39% IgA: 19% IgG and IgA: 35% Not detected: 7% |
3. Inflammatory-Type EBA
The inflammatory variants of EBA, accounting for approximately 2/3 of the cases (Table 1), clinically mimic other autoimmune bullous dermatoses (AIBDs) such as bullous pemphigoid (BP), linear IgA disease (LAD), mucous membrane pemphigoid (MMP), or Brunsting-Perry pemphigoid. In patients with inflammatory EBA, widespread vesiculobullous eruptions are observed, typically involving the trunk, central body, extremities, and skin folds. Usually the patients suffer from pruritus [6–8].
4. Congenital EBA
During pregnancy, IgG is transferred from the mother to the fetus [10]. Therefore, the transfer of anti-COL7 IgG could potentially induce EBA in neonates of mothers with EBA. Interestingly, so far only one case of this congenital EBA has been reported: a mother diagnosed with EBA delivered an otherwise healthy girl, who was noted at birth to have tense blisters and areas of denuded skin. The patient's skin lesions progressed, and the dermatology service was consulted. Direct immunofluorescence of perilesional skin revealed linear deposition of IgG and C3, and ELISA detected anti-COL7 autoantibodies, confirming the diagnosis of EBA. Based on the assumption of disease induction by placental autoantibody transfer, supportive treatment was initiated. This led to cession of new blister formation within 10 days and healing of all erosions within 2 months [11].
5. Extracutaneous EBA Manifestations and Associated Diseases
5.1. Extracutaneous EBA Manifestations
Extracutaneous EBA manifestations include ocular, oral mucosa, esophagus, anal, vaginal, tracheal, and laryngeal lesions [3, 4, 12]. Ocular involvement in EBA predominantly presents with scaring, resembling lesions observed in patients with MMP. It may be the only EBA manifestation in a patient or may be observed in addition to other lesions. In severe cases, ocular involvement may lead to blindness. Interestingly, the predominant Ig class in ocular EBA seems to be IgA; that is, most cases reported noted IgA deposits at the basement membrane. Several EBA patients with ocular involvement have been described. However, the proportion of EBA patients affected by eye lesions has not been systemically evaluated [13–18]. In EBA, erosions and blistering can also affect the oral mucosa. Oral lesions are most commonly observed in patients with noninflammatory EBA and those with MMP-like or LAD-like inflammatory EBA [19, 20]. Esophageal strictures are another extracutaneous manifestation and severe complication in EBA patients. Patients may not be able to swallow foods and thus require endoscopic esophageal dilations, which may have to be repeated several times, if disease activity cannot be controlled [21–23]. Rarely, laryngeal involvement may cause hoarseness, impaired phonation, and loss of voice, and may lead to irreversible respiratory distress. These may be caused by trauma induced by intubation [24, 25].
The expression of COL7 in most of the anatomic regions affected by EBA other than the skin [26–29] may best explain occurrence of these extracutaneous EBA manifestations. Overall, the prevalence of mucosal lesions in EBA is largely unknown. However, one study evaluated the mucosal involvement in 4 EBA patients in a multidisciplinary approach that included dermatologists, ophthalmologists, radiologists, and otolaryngologists. All the 4 patients showed presence of multiple mucosal lesions. Specifically, all had oral lesions ranging between erosions, blisters, tooth loss, and mandibular contraction resulting in impaired ability to open the mouth and alveolar bone loss. Lesions in the nasal mucosa were also noted in all patients, ranging from dryness to extensive fibrosis and synechiae. Symblepharon was also noted in all the patients, as well as pharyngeal and laryngeal strictures. The esophagus dysmotility and/or strictures were observed in 3 of the 4 patients. One patient also suffered from perianal lesions, and another patient presented with erosions on the penile shaft. Hence, these findings have generated awareness to regularity screen EBA patients for (even subclinical) mucosal disease, as the delay of appropriate treatment may lead to severe complications as outlined previously [4, 25].
5.2. Diseases Associated with EBA
On a case report basis, EBA has been noted to be associated with diabetes mellitus cryoglobulinemia, subacute cutaneous/systemic lupus erythematosus, and psoriasis [3, 4, 30]. Interestingly, regarding the association of EBA with psoriasis, in all so far described 4 patients, EBA manifested after psoriasis [31–34]. However, most of these findings are anecdotal reports, and no clear pathogenic interaction of EBA with these diseases has been established. In contrast, accumulating evidence suggests that EBA and inflammatory bowel diseases (IBDs) such as ulcerative colitis (UC) and Crohn's disease (CD) are associated. More specifically, IBD has been reported to be present in approximately 30% of patients with EBA. However, it needs to be taken into account that many of these observations were made before modern diagnostic criteria for EBA were established [35–38]. So far, CD has been noted to be associated with EBA in at least 25 cases [39, 40] as well as four cases of EBA associated with UC [40]. Furthermore, in patients with active IBD, circulating antibodies to COL7 have been noted in frequencies ranging from 6 to 60% [41–43]. Unpublished data from our laboratory indicates that the incidence of anti-COL7 IgG in patients with IBD is at the lower end of the published incidences. Further evidence of a pathogenic link between IBD and EBA has been obtained in EBA mouse models: in both antibody transfer-induced EBA [44] and immunization-induced EBA [45, 46] in addition to skin blistering, blister formation was observed in esophagus, stomach, small intestine, and colon. The prevalence of blister formation in these mouse models paralleled the expression of COL7, which continuously decreased from the one observed in the oral cavity to the distal colon. This anti-COL7-induced gastrointestinal tissue injury is of functional relevance, as a reduced body weight in diseased mice was observed [28].
6. Epidemiology
EBA is a rare disease with an incidence ranging from 0.2 to 0.5 new cases per million and per year [47–49]. As EBA is a chronic disease, the prevalence is presumably higher, but data on EBA prevalence is not available. Three studies have reported epidemiological data from EBA patients [6–8]: this data from a total of 83 patients shows that EBA affects females slightly more often than males. Furthermore, median age of onset is 47 years (75 percentile: 30–66 years). Yet, EBA has also been diagnosed in children [8, 50, 51], and approximately 20% of EBA patients are over 70 years old. Interestingly, black EBA patients of African descent are significantly younger compared to EBA patients in Korea or the Netherlands (Figure 1). Regarding the different clinical phenotypes, the inflammatory variants are observed most commonly (67%), while classical EBA is observed in 33% of the patients. This epidemiological data on EBA is summarized in Table 1.
Figure 1.
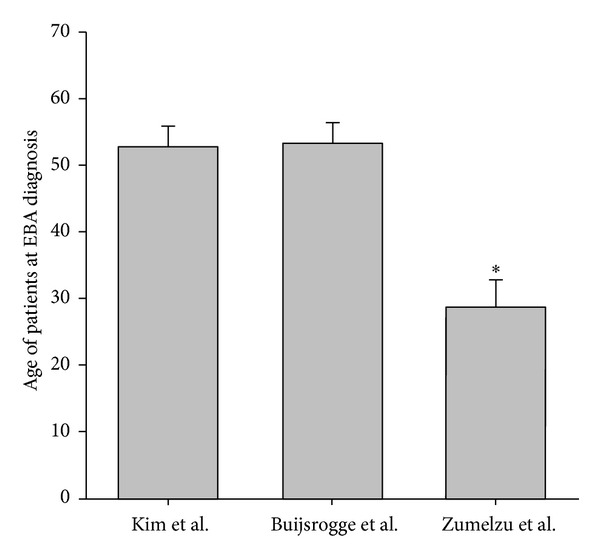
Mean age of EBA onset in Korea, Netherlands, and blacks with African descent. Mean (SEM) patient age (years) at the diagnosis of EBA in 3 large cohorts of patients from Korea, Netherlands, and France (blacks with African descent) [6–8]. ∗ indicates P < 0.05 (ANOVA).
7. Pathogenesis of EBA
Like observed in all other AIBD, in EBA, autoantibodies targeting antigens located within the skin mediate tissue injury [5, 52]. Since the discovery of COL7 as the autoantigen in EBA [53], the development of model systems duplicating several aspects of the human disease has greatly contributed to our current understanding of the diseases' pathogenesis.
8. Identification of Type VII Collagen as the Autoantigen in EBA
COL7 was identified as the autoantigen in EBA approximately 30 years ago. In 1984, Dr. Woodley and colleagues identified a 290 kDa protein located at the basement membrane of human skin, which was distinct from all other known components of the basement membrane [54]. Four years later, the same group identified the carboxyl terminus of COL7 as the autoantigen in EBA [53]. In most EBA patients, the autoantibodies are IgG. In addition to IgG, IgA anti-COL7 autoantibodies are observed either as the only Ig class or in combination with IgG autoantibodies (Table 1). Detailed epitope mapping studies with sera from EBA patients [55–57] demonstrated that most autoantibodies bind to epitopes located within the noncollagenous (NC) 1 domain of COL7 (Figure 2). In only very few patients, antibody reactivity to either the collagenous domain [58] or the NC2 domain [59] can be detected.
Figure 2.
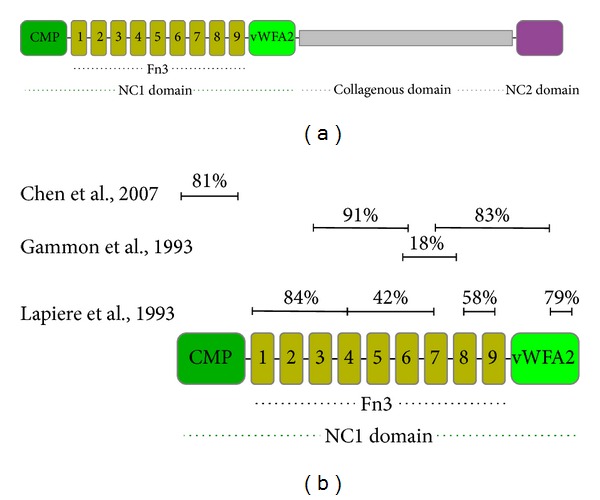
Fine mapping of the autoantibody reactivity in epidermolysis bullosa acquisita. (a) Schematic structure of COL7. In vivo, COL7 chains form a trimer. Single COL7 chain consists of one central collagenous domain, flanked by 2 noncollagenous (NC) domains. The N-terminal NC-1 domain consists of several subdomains with high homologies to adhesion proteins (CMP: cartilage matrix-like, Fn3: fibronectin 3-like, and vWFA2: von Willebrand factor A-like). The C-terminal NC2 domain contains conserved cysteine residues, which accompany an antiparallel assembly of these collagen molecules. The COL7 gene (COL7A1) maps to the locus 3p21 [70–72]. (b) Summary of epitope mapping studies performed within the NC1 domain. The percentages indicated in the figure correspond the percentage of patients with autoantibodies to their regions.
9. Demonstration of the Pathogenic Relevance of Anti-COL7 Antibodies
The pathogenic relevance of anti-COL7 antibodies has been demonstrated in vitro, ex vivo, and in vivo. In detail, in vitro pathogenicity was demonstrated by the incubation of cryosections of human skin with serum, total IgG, or NC1-affinity purified IgG isolated from EBA patients. This led to a linear IgG deposition along the dermal-epidermal junction. Sole antibody binding did not however induce a separation of the epidermis from the dermis. When these sections were subsequently incubated with neutrophils from healthy blood donors, this led to the location of neutrophils to the dermal-epidermal junction, followed by the release of reactive oxygen species (ROS) from the neutrophils and ultimately a separation of the epidermis from the dermis (Table 2, Figure 3) [60]. In addition to the demonstration of the pathogenic relevance of autoantibodies directed against COL7, these experiments also documented the key contribution of neutrophils to blister induction. Based on this insight, neutrophils from healthy blood donors were isolated and incubated with fixed immune complexes of anti-COL7 IgG and recombinant COL7. This led to neutrophil activation, assayed by determination of ROS [61].
Table 2.
Demonstration of pathogenicity of antibodies directed towards type VII collagen.
| Method | Protocol | Readout | Reference |
|---|---|---|---|
| Neutrophil activation in vitro | Incubation of isolated neutrophils with immune complexes of anti-COL7 antibodies and COL7 | (i) ROS release (ii) Elastase release |
[61] |
|
| |||
| Neutrophil activation ex vivo | Incubation of cryosections of human/mouse skin with antibodies against COL7 (serum, IgG, affinity-purified IgG from patients, monoclonal anti-COL7 antibodies, or rabbit anti-COL7 IgG) | (i) Deposition of formazan (ROS detection) (ii) Extent of dermal-epidermal separation |
[60, 62] |
|
| |||
| Anti-COL7 IgG transfer into mice | (a) Repetitive injection of rabbit anti-mouse COL7 IgG into C57Bl/6, BALB/c, or BALB/cnude mice | (i) Body surface area affected by skin lesions (ii) Several secondary endpoint measurements |
[44] |
| (b) Repetitive injection of rabbit anti-human COL7 IgG into SKH1 mice | Same as above | [63] | |
| (c) Repetitive injection of human anti-human COL7 IgG into SKH1 mice | Same as above | [64] | |
| (d) Repetitive injection of human affinity-purified (CMP) anti-human COL7 IgG into SKH1 mice | Same as above | [57] | |
| (e) Repetitive injection of human affinity-purified (Fn3-like domain) anti-human COL7 IgG into SKH1 mice | Same as above | [65] | |
| (f) Repetitive injection of rabbit anti-mouse COL7 IgG (vWFA2-like specific) into several in- or outbred mice | Same as above | [66] | |
| (g) Repetitive injection of rabbit anti-human COL7 IgG into COL7-humanized mice | Same as above | [67] | |
|
| |||
| Immunization of mice with COL7 | (a) Repetitive immunization of SJL/J, BALB/c, and Fc gamma receptor IIB-deficient mice (antigen located within the Fn3-like repeats) | (i) Autoantibody production (ii) Body surface area affected by skin lesions (iii) Several secondary endpoint measurements |
[45] |
| (b) Single immunization of SJL/J, B6.SJL-H2s, C57Bl/10.s, and MRK/MpJ mice (antigen located within the Fn3-like repeats) | Same as above | [68, 69] | |
| (c) Single immunization of SJL/J, B6.SJL-H2s, mice (antigen: vWFA2-like domain) | Same as above | [66] | |
Abbreviations used: COL7: type VII collagen, ROS: reactive oxygen species.
Figure 3.
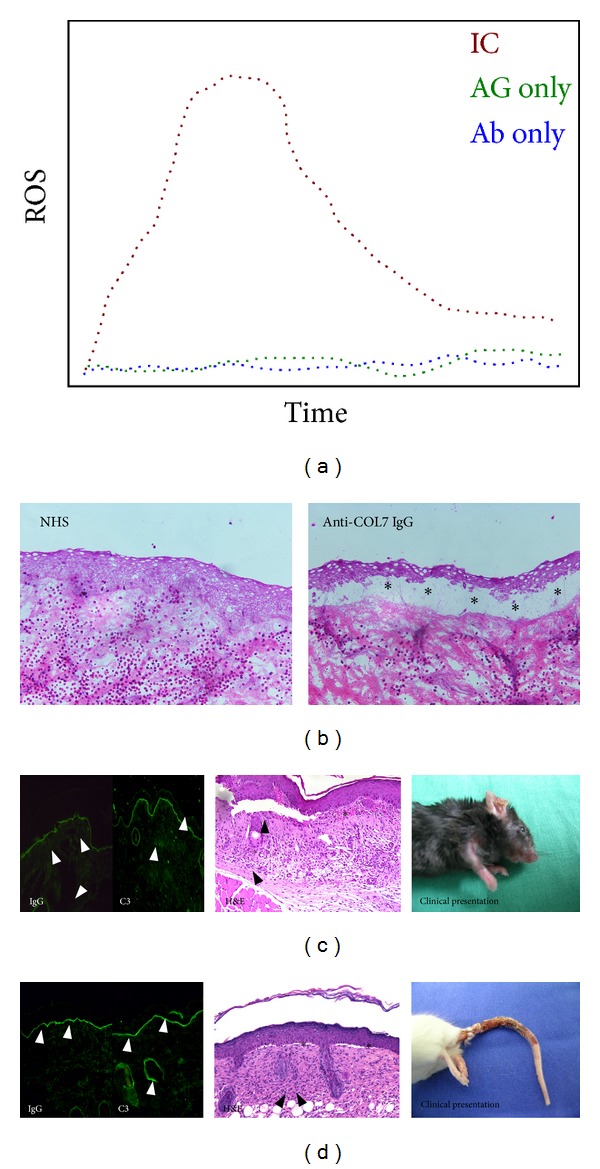
Demonstration of the pathogenicity of anti-type VII collagen antibodies. (a) Immune complexes (IC) of anti-COL7 IgG (Ab) and COL7 (AG) are able to activate neutrophils isolated from either healthy humans or mice. Endpoint measurement is the release of reactive oxygen species (ROS) by chemiluminescence. Detection of elastase by ELISA is an additional measurement for neutrophil activation in this assay. For AIBD, this assay was first described for type XVII collagen and anti-type XVII collagen antibodies obtained from patients with bullous pemphigoid [73]. Later, the same method was adopted for EBA [61]. The figure shows a schematic result (ROS release) from neutrophils activated with either antigen or antibody alone, or with immune complexes. (b) Incubation of cryosections of normal human or mouse skin with antibodies targeting COL7, followed by incubation with neutrophils from healthy donors induces dermal-epidermal separation. This so-called “cryosection assay” also allows to investigate neutrophil activation. This assay was initially developed using sera from BP patients [74, 75] and was later adopted for EBA [60]. Shown are H&E stained sections of cryosections of normal human skin incubated with either normal human serum (NHS) or IgG from an EBA patient. After the washing of antibodies, neutrophils from healthy human donors were added to the sections. While no pathology is observed in the section incubated with NHS, a clear separation of the epidermis from the underlying dermis is present in the section incubated with anti-COL7 IgG. ∗ indicates split formation. (c) Injection of rabbit or human anti-COL7 IgG into mice leads to IgG and complement (C3) deposition along the dermal-epidermal junction (arrow heads). Subsequently, subepidermal blistering (∗) accompanied with a dermal leukocyte infiltration (arrow heads) develops. Clinically, mice present with erythema, crusts, and alopecia. Fresh blisters are rarely observed, most likely due to the thin epidermis in mice. This antibody transfer (passive) EBA mouse model was first described independently in 2005 by two groups [44, 63]. The histology and images are from C57Bl/6 mice injected with rabbit anti-COL7 IgG, 12 days after the first antibody injection. (d) Immunization of susceptible mouse strains with recombinant proteins located within the NC1 domain leads to autoantibody production, which can be detected by direct IF microscopy from skin biopsies. Subsequent to autoantibody deposition at the dermal-epidermal junction, complement activation (evidenced by C3 deposition at the dermal-epidermal junction) and histological and clinical findings duplicating the human disease are observed.
Subsequently, two groups independently documented the pathogenic relevance of autoantibodies targeting COL7 in 2005. Sitaru and colleagues [44] immunized rabbits with recombinant proteins located within the NC1 domain of COL7. Injection of the total immune IgG isolated from these rabbits into C57Bl/6 or BALB/c mice induced subepidermal skin blisters, reproducing human EBA at the clinical, histological, electron microscopical, and immunopathological levels (Table 2, Figure 3). Also in 2005, Woodley and colleagues generated rabbit anti-human COL7 IgG, which also induced experimental EBA when injected into outbred hairless SKH1 mice [63]. This blister-inducing activity of anti-COL7 IgG has been confirmed by similar experiments; that is, experimental EBA can be induced by the injection of (i) affinity-purified anti-COL7 antibodies from EBA patients' sera into adult hairless mice [64], (ii) affinity-purified anti-CMP EBA patient antibodies injected into hairless mice [57], (iii) rabbit anti-human COL7 IgG directed against the Fn3-like repeats of COL7 into hairless mice [65], (iv) rabbit anti-mouse vWFA2 IgG injected into several, but not all, strains of in- and outbred mice [66], and (v) injection of rabbit anti-human COL7 IgG into COL7-humanized mice [67, 76]. Most of these antibody-transfer models of EBA mimic the inflammatory variants of the disease. However, in some models, nail loss is additionally observed [63, 64], which is mostly observed in mechanobullous EBA.
In an attempt to model all aspects of the human disease, several mouse strains were immunized with a protein spanning the amino acid residues 757–967 of murine NC1, which contains a GST-tag for isolation, termed GST-mCOL7C (Figure 3). Mice were immunized with the protein and were subsequently boosted 3 times at 3-week intervals. This induced clinical disease in over 80% of SJL/J and approximately 50% in BALB/c and Fc gamma receptor IIB-deficient mice on a C57Bl/6 genetic background (Table 2, Figure 4). In contrast, SKH1 and C57Bl/6 mice did not develop skin blistering [45]. Experimental EBA can also be induced by immunization in (i) susceptible inbred mouse strains (SJL/J, B6.SJL-H2s, B10.s, and MRL/MpJ) by single immunization with GST-mCOL7C [68], (ii) an autoimmune-prone advanced intercross mouse line (AIL) by single immunization with GST-mCOL7C [77], and (iii) in SJL/J and B6.SJL-H2s mice by single immunization with another immunodominant protein corresponding the vWFA2-like subdomain of COL7s' NC1 domain [66].
Figure 4.

Pathogenesis of autoantibody-induced tissue injury in EBA. (a) In EBA, blister formation is induced by antibody binding to COL7 located at the dermal-epidermal separation. (b) This leads to the activation of the complement system. Cleavage products of complement activation, for example, C5a, mediate CD18-dependent neutrophil extravasation into the skin. (c) Subsequently, cytokines are released from yet to be defined cells. This can either contribute to tissue injury (GM-CSF, IL-1, CXCL1, and CXCL2) or have potent anti-inflammatory effects (IL-6, IL-1ra). (d) Blister formation is mediated by MMP such as elastase and reactive oxygen species (ROS) released from neutrophils after binding to the immune complexes. (e) Recent attention has focused on the resolution of inflammation in EBA. Flightless I (Flii) has potent anti-inflammatory and proresolving effects in experimental EBA.
10. Mechanisms Leading to Loss of Tolerance to COL7
Due to the previous lack of appropriate models [78, 79], investigations on the mechanisms leading to the loss of tolerance have just begun. Clinical observations and MHC genotyping in EBA patients point towards a genetic control of EBA, which was also documented in animal models of the disease. Regarding cellular requirements for the induction of autoantibody production, CD4 T cells have been identified to be crucial. Furthermore, in experimental EBA, the proliferation of CD4 T cells depends on heat-shock protein (HSP) 90. These findings are presented and discussed in detail within the following paragraphs (Table 3).
Table 3.
Mechanisms leading to autoantibody production in EBA.
| Mechanism | Demonstrated by | Reference |
|---|---|---|
| Genes within MHC locus | (i) HLA haplotype in EBA patients (ii) MHC association in experimental EBA |
[8, 68, 80] |
|
| ||
| Genes outside MHC locus | (i) Overrepresentation of black-skinned patients in EBA (ii) Identification of quantitative trait loci (QTL) in experimental EBA |
[8, 77] |
|
| ||
| CD4 T cells | (i) T-cell-deficient nude mice are resistant to induction of immunization-induced EBA (ii) Time-restricted depletion identifies CD4 T cells to be required for induction of immunization-induced EBA |
[66, 83] |
|
| ||
| HSP90 | Blockade of HSP90 prevents induction of and improves already established immunization-induced EBA | [84] |
10.1. EBA Susceptibility Is Genetically Controlled
EBA susceptibility is linked to certain MHC alleles in patients. One study documented an association with HLA-DR2 [80], and in a later study, HLA-DRB1*15:03 was found to be associated with EBA [8]. The observation of cooccurrence of EBA cases in one family further points towards a genetic control of EBA [81]. An MHC restriction is also observed in immunization-induced experimental EBA in mice: after a single immunization with GST-mCOL7C, most inbred mouse strains developed anti-COL7 antibodies. However, only few strains develop subsequently develop clinically manifest skin blistering. Among those, SJL/J and B6.SJL-H2s mice show a high incidence of severe disease, and C57Bl/10.s and MRL/MpJ mice develop mild EBA at a low incidence. NOD/Shilt/J and C57Bl10.q mice were resistant to both anti-COL7 autoantibody production and clinical disease development [68].
Recently, in a cohort of EBA patients diagnosed in a French referral center for autoimmune bullous dermatoses (AIBDs), 54% were black skinned, while only 3% of other AIBD patients were black skinned. Therefore, black-skinned patients have a significantly higher risk to develop EBA, pointing towards a genetic predisposition outside the MHC locus [8]. This assumption is supported by several observations in immunization-induced EBA: in this model, SJL/J, B6.SJL-H2s, and C57Bl/10.s mice are susceptible to disease induction. All strains share the H2s locus but differ genetically regarding genes outside this locus. After immunization, EBA incidence and clinical disease severity are different among these strains [68]. Furthermore, immunization-induced EBA-resistant C57Bl/6 mice become susceptible if they lack the expression of the inhibitory Fc gamma IIB receptor [44]. Overall, these differences indicate that genes outside the MHC locus contribute to EBA susceptibility. In order to define this genetic control of EBA susceptibility, an advanced autoimmune prone intercross mouse line (AIL) was generated. Immunization-induced EBA-resistant parental mouse stains (NZM2410/J, BXD2/TyJ, and Cast) and MRL/MpJ EBA-susceptible mice were intercrossed at an equal strain and sex distribution [82]. Mice of the 4th offspring generation (G4) were then immunized with GST-mCOL7C. Of these genetically diverse mice, 33% developed clinically manifest experimental EBA, while the remaining mice remained clinically healthy. These mice were then genotyped for 1400 single-nucleotide polymorphisms (SNPs) to identify genomic regions controlling EBA susceptibility (quantitative trait locus (QTL)). This led to the identification of several non-MHC QTL controlling EBA susceptibility [77]. Overall, clinical observations and data from experimental models clearly document a contribution of genes in- and outside the MHC locus to EBA susceptibility (Table 3).
10.2. CD4 T Cells Are Required to Induce Autoantibody Production
T-cell-deficient SJLnude mice are completely protected from induction of immunization-induced EBA. In these mice, disease susceptibility could be restored by the transfer of T cells from wild type SJL/J mice that had been immunized with COL7 [83]. Hence, T cells are required for the induction of autoantibody production and possibly also for the maintenance of autoantibody production, as described for other AIBDs such as bullous pemphigoid and pemphigus vulgaris [85–89]. In order to further define T-cell subsets involved in the generation of anti-COL7 antibodies in experimental EBA, CD4- and CD8 T cells were depleted in a time restricted fashion. Depletion of CD4 T cells for two weeks starting one day prior to immunization significantly delayed both autoantibody production and the onset of clinical disease. In contrast, depletion of CD8 T cells for the same time period had no effect [66]. Therefore, CD4 T cells are required for the induction of autoantibody production in experimental EBA (Table 3). It is currently unknown which antigen-presenting cells (APCs) lead to the clonal selection and expansion of COL7 reactive CD4 T cells.
10.3. HSP90 Modulates Autoantibody Production
Challenging the hypothesis that heat-shock protein 90 (Hsp90) inhibition induces cell death of autoreactive plasma cells (as described for malignant plasma cells [90]), mice were treated with HSP90 inhibitors before the induction of immunization-induced EBA (Table 3). In a therapeutic setting, mice with already established immunization-induced EBA were also treated with HSP90 inhibitors. Both HSP90 inhibitors prevented the onset of immunization-induced EBA and also ameliorated clinical disease in already established EBA. Furthermore, HSP90 inhibition led to the suppression of autoantibody production and reduced the dermal leukocyte infiltration. Unexpectedly, total plasma cell numbers, COL7-specific plasma cells, and germinal center B cells were unaffected by HSP90 blockade. Interestingly, T-cell proliferation was significantly inhibited, as evidenced by the reduced response of isolated lymph node cells from immunized mice to in vitro restimulation with anti-CD3/CD28 antibody or autoantigen in the presence of HSP90 inhibitors. These results indicated that HSP90 inhibition has no impact on normal or autoreactive plasma cells in vivo and identified T cells as targets of HSP90 blockade in EBA [84].
11. Autoantibody Production and Maintenance of a High Half-Life of Autoantibodies
11.1. Generation of Anti-COL7 IgG Is Restricted to Peripheral Lymph Nodes in Experimental EBA
In immunization-induced EBA, antigen-specific T cells can be detected in most secondary lymphoid organs (unpublished observation). Interestingly, antigen-specific B cells can only be detected in peripheral lymph nodes [84]. Therefore, COL7 autoantibodies are exclusively produced at this anatomical location. Regarding human EBA patients, the location of autoreactive B cells is unknown. In other AIBD, such as pemphigus vulgaris, antigen-specific B cells can be isolated from blood [91]. Therefore, it is tempting to speculate that anti-COL7 producing B cells are also found in the circulation of EBA patients. This assumption is supported by the detection of COL7-specific T cells in the circulation of EBA patients [92].
11.2. Production of Disease-Inducing Anti-COL7 IgG Is Associated with a Th1 Polarization in Peripheral Lymph Nodes in Experimental EBA
In immunization-induced EBA, most strains develop circulating and tissue-bound anti-COL7 antibodies. However, only few strains develop an overt blistering disease. Comparing the autoantibody response of clinically healthy versus diseased mice after COL7 immunization showed that IgG2 antibodies are associated with clinical EBA manifestation [45, 68, 69]. This antibody response reflects Th1 polarization of the immune response. This assumption is further supported by an increased IFN-γ/IL-4 ratio in the draining lymph nodes of EBA-susceptible mice, compared with EBA-resistant strains [69].
11.3. The Neonatal Fc Receptor Controls the Half-Life of Anti-COL7 Autoantibodies
After the autoantibodies are released into the circulation, their half-life is controlled by the neonatal Fc receptor (FcRn). The FcRn is an MHC class I-like molecule that, among other functions, protects IgG from catabolism [10]. In line, blockade of the FcRn leads to an enhanced clearance of all IgG, including autoantibodies. In animal models of AIBD, including pemphigus, bullous pemphigoid, and EBA, mice are completely blocked from disease induction [93, 94]. This protection can however be overridden by the transfer of excess amounts of antibodies [94].
12. Mechanisms of Autoantibody-Induced Tissue Injury
Experiments employing several model systems have identified several cellular and molecular requirements for autoantibody-induced tissue injury in EBA. This, for example, includes neutrophils, Fc receptors, complement activation, and cytokines. While the contribution of each of these cells and molecules has been well documented, the exact timely and spatial sequence of events leading to blister formation has not been determined in detail. Based on the current understanding of EBA pathogenesis, autoantibody-induced tissue injury in EBA can be divided into the following events: (i) binding of the autoantibodies to their target antigen, (ii) Fab- and Fc-dependent effects on blister formation, including the release of several cytokines, (iii) integrin-dependent extravasation of neutrophils into the skin, (iv) neutrophil activation, including the release of ROS and MMP, and (v) resolving of the autoantibody-induced tissue injury.
12.1. Autoantibody-Induced Tissue Injury Is Genetically Controlled
In other animal models of antibody-induced inflammatory disease, for example, the K/BxN transfer arthritis model, disease susceptibility differs in inbred mouse strains [95]. In line, large differences in clinical disease severity were observed after the transfer of anti-COL7 IgG into different mouse strains. In C57Bl/6 mice, close to 30% of the body surface area was affected by EBA skin lesions 20 days after the first administration of anti-COL7 IgG. In contrast, a lower clinical disease activity was observed in BALB/c mice, while MRL/MpJ and outbred SKH1 mice were completely resistant to EBA induction by the transfer of anti-COL7 IgG. Overall, this points towards a (yet to be determined) genetic control of autoantibody-induced tissue injury in experimental EBA. This assumption is further supported by the observation of a high variation of skin blistering in outbred AIL mice after the transfer of anti-COL7 IgG [66].
12.2. Anti-COL7 Antibodies Rapidly Bind to Their Target Antigen
Morphological investigations on anti-COL7 IgG deposition after transfer into mice by direct immunofluorescence (IF) microscopy showed linear IgG deposits along the dermal-epidermal junction within 24 hours [28]. To further define the kinetics of anti-COL7 IgG deposition to the skin, autoantibodies were fluorescently labeled. These labeled anti-COL7 autoantibodies fully retained their ability to induce blistering in mice. These fluorescently labeled anti-COL7 autoantibodies were i.v. injected into mice, and their binding to the skin was observed using multiphoton in vivo microscopy using an established experimental setup [96]. Within minutes after i.v. injection, anti-COL7 IgG bound to the dermal-epidermal junction (Iwata et al., unpublished). To study clearance rates of circulating and tissue-bound autoantibodies to COL7 in experimental EBA, retention times of diaplacentally transmitted anti-COL7 autoantibodies in serum and in skin were investigated. Immediately after birth, comparable levels of pathogenic antibody concentrations were observed in maternal and neonatal mice. The clearance time of skin-bound autoantibodies was twice as long (8 weeks) as that of circulating autoantibodies (weeks). Interestingly, despite IgG and complement (C3) deposition along the dermal-epidermal junction in the neonatic mice, neither histological, or clinical alterations were observed [46]. In addition to the skin, immunoperoxidase and immunofluorescence staining analyses found COL7 expression to be confined to the basement membranes beneath other stratified squamous epithelia; for example, oesophagus, buccal, anal, and vaginal mucosae, but not in colonic mucosa [26, 27]. On the contrary, others noted scattered COL7 staining in colonic epithelium and in the small intestinal epithelial basement membrane [97, 98]. Furthermore, anchoring fibrils were detected by electron microscopy in the intestine [97], and COL7 expression was detected in oesophagus and colon [41, 99]. In mice, COL7 mRNA and protein can be detected in the skin and in all organs of the gastrointestinal tract. Of note, COL7 expression decreases from proximal to distal parts of the gastrointestinal tract [3].
12.3. F(ab)-Mediated Effects on Blister Formation
As mentioned previously, IgG antibodies from EBA patients bind to a large variety of epitopes located with the NC1 domain of COL7 (Figure 3). F(ab)-mediated effects on blister formation have been described in bullous pemphigoid, for example, a weakening of cell attachment after the binding of anti-BP180 antibodies to keratinocytes [100–102]. In contrast, no data on F(ab)-mediated effects of anti-COL7 antibodies on keratinocyte adhesiveness has been reported. As the NC1 domains multiple binding sites to other proteins located within the dermal-epidermal junction, it seems plausible that autoantibody binding interferes with these interactions [70, 103]. This may explain the pathogenesis of noninflammatory, mechanobullous EBA; however, this assumption needs to be experimentally confirmed.
12.4. Fc-Mediated Effects on Blister Formation
Fc-mediated effects after the binding of anti-COL7 to its autoantigen have been investigated in detail. Several lines of evidence suggest that the Fc portion of anti-COL7 antibodies is a key molecular prerequisite to initiate blister formation. In vitro, only IgG, but not F(ab)2 fragments, directed to COL7 induced dermal-epidermal separation when incubated on cryosections of human skin, followed by the addition of neutrophils from healthy donors [60]. Furthermore, not all isoclasses of anti-COL7 have the potential to induce dermal-epidermal separation. Specifically, only IgG1 and IgG3, but not IgG2 and IgG4, induced ex vivo blister formation [62]. Similar observations are made in mice. While injection of rabbit anti-COL7 IgG induces skin blistering, the administration of corresponding F(ab)2 fragments causes no pathology, despite binding to the dermal-epidermal junction [44]. The absence of skin lesions in mice injected with chicken anti-mouse COL7 IgY, which does not bind murine complement and Fc receptors, further underlines the importance of Fc-Fc receptor interactions for blister formation in experimental EBA [104]. Recent attention has also been attributed to the glycosylation status of IgG, as this has been demonstrated to have a great impact on IgG function [105]. Alterations in the Fc glycosylation patterns have been demonstrated in several chronic inflammatory diseases [106], and modification of IgG glycosylation has been demonstrated to have preventive and/or therapeutic effects in models of inflammation, including EBA [107–114].
The exact timely and special resolution of events occurring after autoantibody binding in EBA has yet to be defined. The current concept of antibody-induced tissue injury is that of a multistep, directed, but not linear, process leading from antibody binding to blister formation. So far, the following cells and molecules have been identified to contribute to blister formation in experimental EBA: (i) cytokines including IL-1, IL-6, GM-CSF, CXCL1, and CXCL2, (ii) complement activation, (iii) adhesion molecules, such as beta-2 integrins, (iv) Fc gamma receptors, (v) neutrophil activation, and (vi) resolving of the inflammatory response. These pathways are discussed later (Figure 4, Table 4).
Table 4.
Mechanisms leading to autoantibody-induced tissue injury in EBA.
| Mechanism | Demonstrated by | Reference |
|---|---|---|
| Anti-COL7 IgG/A binding | Detection of anti-COL7 antibodies in patients, vast in vitro and in vivo (animal models) evidence | [125] |
| Genetic control | Diverse susceptibility of different inbred mouse lines | [66] |
| Neutrophils | Anti-Gr1 treatment completely protects from antibody transfer-induced EBA | [126] |
| Fc-fragment mediated effects | (i) Anti-COL7 IgG, but not corresponding F(ab)2 fragments, induces EBA in vitro and in vivo (ii) Anti-COL7 IgY fails to induce experimental EBA in mice (iii) Enzymatic removal of terminal sugar residues has preventive and therapeutic effects in experimental EBA in mice |
[44, 60, 64, 104, 113] |
|
| ||
| Cytokines | ||
| (a) CXCL1 and CXCL2 | Blockade of the receptors has preventive and therapeutic effects in experimental EBA in mice | [127] |
| (b) GM-CSF | Genetic and pharmacological GM-CSF inhibition has preventive and therapeutic effects in experimental EBA in mice | [128] |
| (c) IL-6 | IL-6 has anti-inflammatory effects in antibody transfer-induced EBA; IL-6 induced IL-1ra, which in turn counteracts proinflammatory events triggered by IL-1 | [123] |
|
| ||
| Complement activation | C5-deficient mice are completely protected from antibody transfer-induced EBA; partial protection is observed in C1q-, Factor B-, or C5aR-deficient mice | [44, 129, 130] |
|
| ||
| Leukocyte extravasation | CD18-deficient mice are completely protected from antibody transfer-induced EBA | [126] |
|
| ||
| Fc gamma RIIB | Fc gamma RIIB-deficient mice show a significantly increased cutaneous blistering in antibody transfer-induced EBA | [124] |
|
| ||
| Fc gamma RIV | Fc gamma RIV-deficient mice are completely protected from antibody transfer-induced EBA | [124] |
|
| ||
| Neutrophil activation | ||
| (a) PI3K beta | PI3K beta-deficient mice are partially protected from antibody transfer-induced EBA | [61] |
| (b) AKT | AKT impairs ROS released from immune omplex activated neutrophils | [131] |
| (c) p38 | Pharmacologic inhibition of p38 phosphorylation partially protected from antibody transfer-induced EBA | [131] |
| (d) ERK1/2 | Pharmacologic inhibition of ERK1/2 phosphorylation partially protected from antibody transfer-induced EBA | [131] |
|
| ||
| ROS | Neutrophil cytosolic factor 1-deficient mice are completely protected from antibody transfer-induced EBA | [126] |
|
| ||
| Elastase/Gelatinase B | Blockade of elastase or gelatinase B completely blocks dermal-epidermal separation | [132] |
|
| ||
| Flii | Overexpression of Flightless I (Flii) increases dermal-epidermal blistering in antibody transfer-induced EBA, and blockade of Flii improves blistering | [133, 134] |
12.5. Differential Effects of Cytokines to Autoantibody-Induced Tissue Injury
There is an ample evidence for an increased expression of several cytokines in AIBD [115]. Despite these findings, and in contrast to other chronic inflammatory diseases [116–120], a cytokine-targeting therapy has not been established in EBA or any other AIBD—with the exception of the relatively well-documented beneficial effects of TNFα inhibition in mucous membrane pemphigoid [121, 122]. Regarding EBA, serum and skin IL-6 expression are increased. Compared to control sera, other cytokines were also increased in EBA but, most likely due to a high degree of variation, were not significantly different [123]. In experimental EBA, induced by the transfer of anti-COL7 IgG into C57Bl/6 or BALB/c mice, increased serum concentrations of several cytokines were noted. Specifically, elevated serum concentrations of TNFα, MIP-1α, GM-CSF, IL-1α, IL-1β, IL-4, IL-6, IL-10, IL-17, RANTES, and KC, but not MIP-1β, G-CSF, IL-2, IL-3, IL-5, IL-9, IL-12, IL-13, eotaxin, IFN-γ, and MCP-1, were observed in experimental EBA (Table 5) [123]. Furthermore, gene expression profiling of skin obtained from mice with experimental EBA additionally identified an increased cutaneous expression of IL-24, CXCL3 (GRO3), and CXCL5 (ENA-78) [124].
Table 5.
Cytokine expression in experimental EBA.
| Cytokine | Serum concentration | Cutaneous expression | Functional relevance | Reference |
|---|---|---|---|---|
| TNF-α | ↑ | ↑ | Proinflammatory (very moderate) | Unpublished |
| MIP-1α | ↑ | ↑ | none | Unpublished |
| GM-CSF | ↑ | ↑ | Proinflammatory (strong) | [128] |
| G-CSF | — | — | nd | |
| IL-1α | ↑ | ↑ | Proinflammatory | [123] |
| IL-1β | ↑ | ↑ | Proinflammatory | [123] |
| IL-2 | Below detection limit | — | nd | |
| IL-3 | — | — | nd | |
| IL-4 | ↑ | nd | nd | |
| IL-5 | — | nd | nd | |
| IL-6 | ↑ | ↑ | Anti-inflammatory | [123] |
| IL-9 | — | nd | nd | |
| IL-10 | ↑ | nd | nd | |
| IL-12 | — | nd | nd | |
| IL-13 | — | nd | nd | |
| IL-17 | ↑ | nd | nd | |
| IL-24 | nd | ↑ | nd | |
| Eotaxin | — | nd | nd | |
| RANTES | ↑ | nd | nd | |
| IFN-γ | — | nd | nd | |
| MCP-1 | — | nd | nd | |
| CXCL1 and CXCL2 | ↑ | ↑ | Proinflammatory | [127] |
| CXCL3 | nd | ↑ | nd | |
| CXCL5 | nd | ↑ | nd |
Functional studies documented little to no contribution of TNFα and MIP-1α in autoantibody-induced tissue injury in experimental EBA (unpublished data). As neutrophils are required to induce skin blistering in mice [126], the contribution of cytokines affecting neutrophil functions such as IL-8 (CXCL1 and CXCL2 in the mouse) and GM-CSF were next evaluated for their contribution in experimental EBA. Regarding CXCL1 and CXCL2, an increased expression was noted in the skin of mice with experimental EBA. In detail, CXCL1 mRNA expression was 50-fold increased comparing lesional to nonlesional skin samples from corresponding anatomical sites from mice with immunization-induced EBA. This was even more pronounced for CXCL2, where an increase over 1,500-fold was noted. Blockade of CXCL1 and CXCL2 functions by oral administration of allosteric CXCR1 and 2 inhibitors (DF2156A) dose dependently, but also strain-dependently, impaired the induction of skin blistering in mice induced by transfer of anti-COL7. In a more therapeutic setting, administration of DF2156A improved clinical EBA manifestation after disease onset in immunization-induced EBA. Compared to high doses of systemic corticosteroids, and inhibition of CXCR1 and 2 was equally effective [127]. Hence, CXCL1 and CXCL2 modulate blister formation in experimental EBA and thus are potential novel therapeutic targets for the treatment of EBA (see Section 14).
Granulocyte macrophage colony-stimulating factor (GM-CSF) stimulates the production, induces migration, and activates many haematopoietic cells. GM-CSF is secreted as a glycosylated protein and consists of a single polypeptide chain. Its receptor consists of a specific GM-CSF binding subunit (CSF2Rα), and a common signal-transduction subunit (CSF2Rβ) which is shared with other cytokines, such as IL-3 and IL-5 in humans [135, 136]. GM-CSF has diverse effects on different inflammatory diseases: application of recombinant GM-CSF improved Crohn's disease in patients [137, 138]. In experimental contact hypersensitivity, characterized by an increased GM-CSF expression [139], blockade of this cytokine had no effect on disease manifestation [140]. In contrast to these observations, blockade of GM-CSF in experimental models of arthritis [141–143], experimental autoimmune encephalitis [144], psoriasis [145], and nephritis [146] protected from disease induction. To test if the observed increased GM-CSF expression in experimental EBA is of functional relevance, anti-COL7 IgG was injected into GM-CSF-deficient mice or mice treated with a function-blocking GM-CSF antibody. Compared to appropriate controls, induction of experimental EBA was impaired if GM-CSF function was blocked. On the molecular level, GM-CSF was required to recruit neutrophils from the bone marrow into the blood and from blood into the skin. In addition, GM-CSF also preactivated the neutrophils, leading to an enhancement of immune complex induced neutrophil activation. Therapeutic blockade of GM-CSF in mice with already established immunization-induced EBA showed beneficial therapeutic effects [128].
IL-6 is predominantly known as a proinflammatory cytokine. This assumption is based on the observation of a correlation of IL-6 serum levels with disease activity in rheumatoid arthritis, Crohn's disease, asthma, and psoriasis [147–150]. Experimentally, the inhibition of IL-6 suppresses the development of inflammatory disease in vivo [151–154] thus highlighting the proinflammatory role of this cytokine. The relevance of IL-6 in contributing to the pathogenesis of inflammatory disease is further supported by the therapeutic effect of tocilizumab on patients with rheumatoid arthritis [155]. Interestingly, most of these proinflammatory effects of IL-6 are mediated by IL-6 transsignaling [156] as (i) protection from experimental arthritis in IL-6-deficient mice could be only restored by the injection of a fusion protein of IL-6 and the sIL-6R (hyper-IL-6), but not by IL-6 alone [157], and (ii) the treatment of experimental arthritis in wild type mice with a fusion protein consisting of the entire extracellular portion of gp130 fused to the Fc region of human IgG1 132 (sgp130) leads to a similar (protected) phenotype as observed in IL-6-deficient mice [157–159]. Contrary, IL-6 may also exert profound anti-inflammatory properties in some experimental models [160–163]. Due to these opposing effects of IL-6 in different inflammatory conditions, the functional relevance of the elevated IL-6 concentrations in experimental EBA was evaluated. In both IL-6-deficient mice and wild type mice treated with anti-IL-6, clinical EBA severity was significantly increased compared to the respective controls. On the contrary, administration of recombinant IL-6 dose-dependently impaired the induction of experimental EBA by the transfer of anti-COL7 IgG. These effects were at least partially due to an induction of IL-1ra by IL-6 and exclusively mediated by classical IL-6 signaling [123]. Therefore, IL-6 has a strong anti-inflammatory role in autoantibody-induced tissue injury in EBA. Despite the fact that the effect of IL-6 on autoantibody production in EBA is not known at the moment, great caution should be taken when considering blocking IL-6 functions in patients with EBA (and possibly other related diseases such as bullous pemphigoid).
Of the other cytokines with noted increased expression in experimental EBA [123, 124], no functional data has been obtained so far (Table 5).
12.6. Complement Activation Is Required for Blister Formation in Experimental EBA
The complement system consists of circulating proteins, which upon activation initiate a highly controlled cascade that is an integral part of the innate humoral immune response [164]. Uncontrolled activation of the complement system has been shown to contribute to the pathogenesis of several chronic inflammatory diseases, such as arthritis [165], asthma [166], and bullous pemphigoid [167]. In line with these observations, C5-deficient mice were completely protected from induction of experimental EBA by antibody transfer [44]. A more detailed investigation of the role of complement in EBA showed that the alternative complement activation is predominantly required for mediation of skin blistering in experimental EBA. Mice deficient for C1q, in which classical complement activation does not take place [168], showed a small and partial but significant protection from EBA induction. In contrast, mice lacking MBL expression, in which most of the lectin activation pathway does not occur [169], did show a similar EBA phenotype compared to wild type controls [129]. Collectively, this data points towards a significant contribution of the complement system in the pathogenesis of the effector phase, that is, autoantibody-induced tissue injury. However, data from asthma [166] suggested that during the initiation of immune responses, complement activation may have immunomodulatory effects. Therefore, the contribution of complement to the early phases of EBA pathogenesis has yet to be defined.
12.7. Integrin-Dependent Extravasation of Leukocytes into the Skin
In order to appropriately locate the limited number of leukocytes to sites of inflammation, leukocytes constantly migrate from the blood to lymphoid organs, as well as to sites of inflammation. This process of leukocyte extravasation is tightly regulated and primarily mediated by adhesion molecules and cytokines. Leukocyte extravasation is initiated by interactions of endothelial adhesion molecules of the selectin family with their leukocytes counterparts. This interaction allows rolling and tethering of leukocytes along the endothelium. During this process, cytokines can lead to the activation of leukocyte integrins, which then mediate firm adhesion. Ultimately, leukocytes leave the vasculature through interactions mediated by junctional adhesion molecules [170–175]. In the effector phase of EBA, that is, autoantibody-induced tissue injury, neutrophils are the main effector cells, as their depletion completely protects from disease induction [126]. Regarding the contribution of adhesion molecules in the pathogenesis of EBA, only one study has been performed. In brief, CD18 deficient mice were completely resistant to autoantibody transfer-induced EBA [126]. However, based on the multistep process of leukocyte extravasation to the skin, it is tempting to speculate that other adhesion molecules also mediate neutrophil extravasation into the skin after the binding of COL7 autoantibodies.
12.8. Fc Receptor Binding Initiates Autoantibody-Induced Tissue Injury in EBA
After their extravasation into the skin, neutrophils bind to the Fc fragments of the tissue-bound anti-COL7 antibodies. The importance of Fc/Fc receptor binding has been demonstrated both in vitro and in vivo: (i) only IgG antibodies but not corresponding F(ab)2 fragments (despite equal binding ability to the skin) induce dermal-epidermal separation in cryosections in the presence of neutrophils [60], (ii) in line, in mice, only anti-COL7 IgG but not the corresponding F(ab)2 fragments induces skin blistering [44, 64], (iii) anti-COL7 IgY, which does not bind to murine Fc receptors, fails to induce skin blistering when transferred into mice [104], and (iv) removal of terminal sugar residues on IgG, which alters their binding acidity to Fc gamma receptors [176] also completely protects mice from EBA induction by antibody transfer [113]. In mice, three different activating Fc gamma receptors (FcgRs) and one inhibitory FcgR have been described: FcgRI, FcgRIII, and FcgRIV are activating FcgR differing with their Fc-binding avidity. FcgRIIB is the only so far described inhibitory FcgR in mice [177]. Data from previously detailed experiments indicated that activating FcgR is required for blister formation in EBA. Furthermore, expression profiling from lesional EBA skin of mice showed an increased expression of FcgRIV [124]. Mice lacking the common, signal-transducing γ-chain of all activating FcgR were completely protected from EBA induction by antibody transfer. Further analysis, using different knock-out mice and FcgR-function-blocking antibodies, identified FcgRIV as the sole mediator of tissue injury in EBA. Blockade of FcgRI, FcgRIII, or even both receptors combined had no effect on EBA manifestation [124]. In humans, albeit the data has been obtained using sera from BP patients, FcgRIIA and FcgRIIIB mediate Fc binding of effector cells of immune complexes [73]. Furthermore, blockade of the FcgRIIB showed that this FcgR protects mice from EBA development [124]. This observation is in line with those obtained in animal models of arthritis and Goodpasture's syndrome [178, 179]. Interestingly, in experimental BP induced by the transfer of rabbit anti-mouse type XVII collagen into neonatal mice, the FcgRIIB had no effect on disease manifestation [180], which may be due to the young age of the mice.
12.9. Complex Interaction of Fc Receptor Ligation and Complement Activation
Interestingly, it had been noted that the complement system can affect the expression and function of FcgR [181, 182]. Hence, complement activation in EBA is thought to increase the threshold for the activation of FcgR expressing cells. It had so far been unclear if IgG-mediated signaling through FcgR is capable to modulate complement-mediated effector functions. Interestingly, injection of IgG1 immune complexes reduced C5a-mediated neutrophil migration into the peritoneum in mice. Detailed analysis of this observation showed galactosylated IgG1 immune complexes through binding to the inhibitory FcgRIIB and dectin-1 block C5aR-mediated proinflammatory events in neutrophils (Figure 5). Thus, the administration of galactosylated IgG1 immune complexes also blocked EBA manifestation in mice [130]. This observation highlights the complexity of autoantibody-induced tissue injury in EBA.
Figure 5.
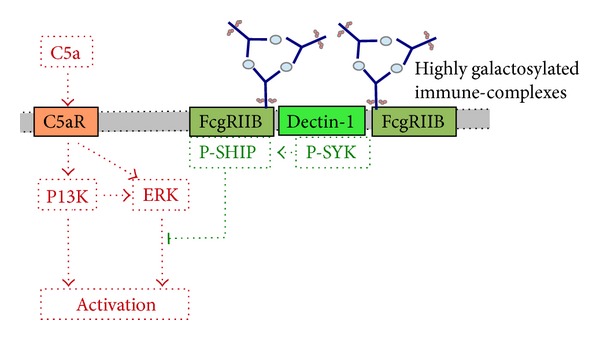
Modulation of complement-driven inflammation through FcgRIIB and dectin-1. Highly galactosylation IgG1 immune complexes bind to FcgRIIB. Galactosylation links FcgRIIB to dectin-1 resulting in tyrosine phosphorylation of the ITAM-like motif downstream of dectin-1 and transient phosphorylation of Syk. This pathway inhibits C5a-mediated ERK1/2 phosphorylation and several cellular effector functions of C5aR.
12.10. Neutrophil Activation
After binding the activating FcgR to the immune complexes located in the skin, this must be signaled in order to provoke a biological response from neutrophils. In macrophages and neutrophils, the Syk protein tyrosine kinase (SYK) is essential for FcgR signaling [183]. Hence, several SYK inhibitors are currently developed and tested for the treatment of several chronic inflammatory diseases [184]. Downstream of SKY, the Akt, ERK1/2, and p38 MAPK phosphorylation have been demonstrated to be involved in the signaling pathways after SYK phosphorylation [185, 186]. In vitro, neutrophil activation can be blocked by inhibition of either Akt, ERK1/2, p38 MAPK phosphorylation. In detail, blockade of either of these pathways dose-dependently inhibited ROS release, as well as dermal-epidermal separation. In contrast, only inhibition of p38 MAPK phosphorylation led to reduced IC-induced degranulation of neutrophils [131]. In vivo, inhibition of ERK1/2 or p38 MAPK phosphorylation impairs induction of skin blistering in antibody transfer-induced EBA [131]. In addition, PI3K beta has recently been identified to be crucial for signal transduction in immune complex activated neutrophils. In detail, blockade of PI3K beta substantially inhibited the production of ROS from neutrophils, and in both antibody transfer-induced EBA and the K/BxN arthritis model, mice were protected from disease induction. In experimental EBA, the effect of PI3K beta could be linked to radiosensitive cells [61].
12.11. Mediators of Blister Formation
Neutrophil activation leads to several biological responses. The release of ROS and proteases is a prominent feature of neutrophil activation [187]. In experimental models of EBA, ROS production has been demonstrated to be crucial for blister formation. In vitro, neutrophils obtained from patients with chronic granulomatous disease, which are not capable to mount an oxidative burst [188], fail to induce dermal-epidermal separation when incubated on cryosections of human skin with deposits of anti-COL7 IgG at the basement membrane [126]. Furthermore, mice lacking cytosolic factor 1 are also completely protected from antibody transfer-induced EBA [126]. In addition to ROS, gelatinase B and elastase have been identified as crucial mediators of dermal-epidermal separation in EBA [132].
12.12. Flii Is a Key Regulator of Resolving Autoantibody-Induced Tissue Injury in EBA
The actin remodeling protein, Flightless I (Flii), has recently been shown to contribute to resolving skin blistering in experimental models of EBA. This work was prompted by previous observations in models of wound healing that demonstrated that reducing Flii expression leads to an improved wound healing. In contrast, Flii over expression resulted in impaired wound healing [189]. In autoantibody transfer-induced EBA, induction of EBA leads to an increased cutaneous Flii expression. This reduced Flii expression in Flii+/− mice significantly impaired blister formation in experimental EBA [133]. Subsequent studies demonstrated that topical treatment with Flii-neutralizing antibodies has therapeutic effects in this model of EBA [134]. These observations point towards significant contribution of pathways that are involved in resolving cutaneous involvement. Therefore, EBA may not only manifest when many proinflammatory stimuli are present, but also (maybe indeed only) when the balance of proinflammatory, anti-inflammatory, and resolving pathways are in a disbalance, which polarizes towards proinflammatory mechanisms.
13. Diagnosis of EBA
The diagnosis of EBA is based on the clinical presentation, the detection of tissue-bound antibodies by direct IF microscopy, and the detection of circulating antibodies directed against COL7 and/or a u-serrated pattern in direct IF microscopy. Additional tests such as transmission electron microscopy or antigen mapping may be performed in unclear cases.
13.1. Clinical Presentation
If the history and clinical presentation of the patient prompts EBA as a potential differential (see Section 1), subsequent laboratory analysis has to be performed to exclude or diagnose EBA. At a minimum, this includes obtaining a lesional biopsy for H&E staining and a perilesional biopsy for direct IF microscopy. In addition, serum analysis for the detection of autoantibody reactivity should be performed. In my personal opinion, other tests should be reserved for special indications.
13.2. Detection of In Vivo Bound Antibodies
The detection of a linear IgG and/or IgA deposition along the basement membrane in a perilesional skin lesion from the patient is observed in almost all EBA cases. In detail, these deposits can be detected in at least 93% of all EBA patients. If the direct IF sections are observed at high magnification, n- and u-serrated patterns can be differentiated. The n-serrated pattern is seen in several subepidermal AIBD, but the u-serrated pattern is unique to EBA (Figure 6). If this criterion is included for EBA diagnosis, the diagnostic sensitivity can be greatly increased [6, 190–192].
Figure 6.
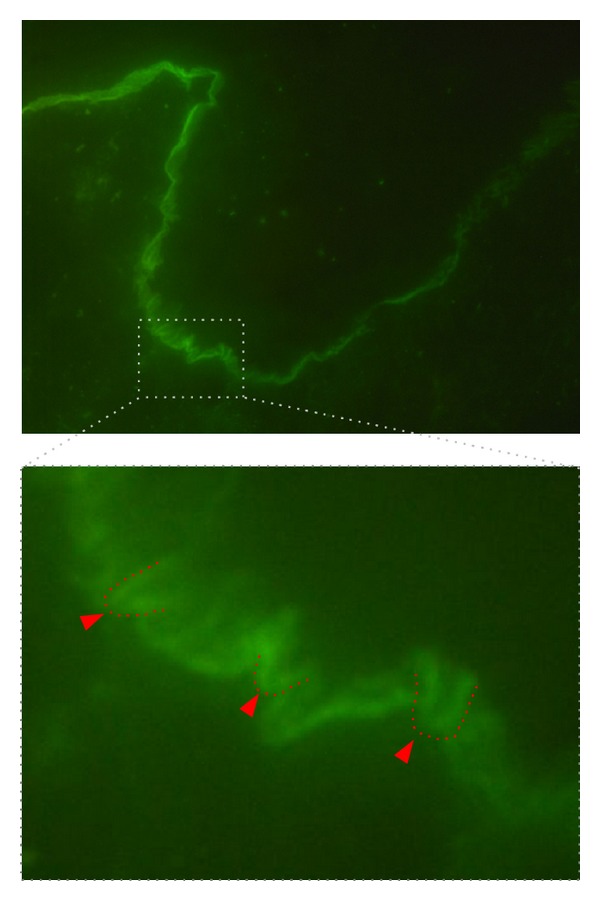
u-serrated pattern in direct IF microscopy in EBA. Direct IF microscopy from perilesional EBA skin (staining for IgG, 400x original magnification). A linear binding along the dermal-epidermal junction is evident. The insert further magnifies the u-serrated binding of the autoantibodies (highlighted in red), which can also be observed in the original direct IF microscopy photograph.
13.3. Detection of Circulating Anti-COL7 Antibodies
Although it can be trained with a web-based program [191], pattern analysis in direct IF microscopy requires an experienced histopathologist. Therefore, detection of circulating anti-COL7 antibodies is a suitable alternative and/or provides additional information to base the diagnosis on. For the detection of anti-COL7 IgG antibodies, three different ELISA systems have been developed, and two of these are commercially available [193–195]. Autoantibodies directed against COL7 can also be specifically detected by IF microscopy using fixed, NC1-expressing HEK293 cells as substrate [195] or by western blotting with NC1 as substrate [56]. In addition, reactivity with a 290-kDa-sized protein of dermal extract and binding to the blister floor in salt-split skin in indirect IF microscopy point towards anti-COL7 antibodies. However, in the later cases, reactivity to laminin gamma-1 and laminin-332 has to be excluded [5].
13.4. Other Diagnostic Tests for the Diagnosis of EBA
13.4.1. Histopathology
For histopathology, the biopsy has to be obtained from lesional skin. In most specimen, subepidermal blister formation is observed. The degree of the dermal leukocyte infiltration, as well as its composition shows great variability. However, to the author's knowledge, no detailed analysis of skin sections from EBA patients has been performed. In summary, the histology in EBA is not pathognomonic but adds to the overall clinical picture, and thus, a skin biopsy should be obtained if EBA is suspected.
13.4.2. Transmission Electron Microscopy
Routine electron microscopy shows blister formation situated in the dermis leaving the basal lamina in the roof of the blister [196]. Transmission electron microscopy is however only used in unclear cases and in specialized centers.
13.4.3. Immunoelectron Microscopy
Direct immunoelectron microscopic examination of skin of EBA patients shows the presence of immune deposits in the anchoring fibril zone, just beneath the lamina densa. This localization is distinct from the deposits in other AIBD [196–198]. Immunoelectron microscopy is considered the gold standard for EBA diagnosis. However, based on practicability and availability, other methods, for example, u-serrated pattern analysis and the combination of direct IF microscopy with the detection of circulating autoantibodies, are more frequently used [199, 200].
13.4.4. Overlay Antigen Mapping
Another elaborate method for the diagnosis of EBA is overlay antigen mapping. Here, perilesional skin specimens from patients are stained for several components of the basement membrane zone. Based on the location of the tissue-deposited autoantibodies (in relation to the other stained structures), this allows to differentiate EBA from other AIBDs.
13.5. Recommended Additional Diagnostic Workup in Patients with EBA
Patients with EBA should be treated by a dermatologist in a multidisciplinary approach. The importance of involving other specialties is based on the findings that EBA is associated with inflammatory bowel disease [201, 202]. Hence, a gastroenterologist on a regular basis should consult EBA patients. Furthermore, as the study by Dr. Luke and colleagues documented a high incidence of (sub)clinical mucosal and eye involvement [25], the respective specialties should also be involved in the treatment and care of EBA patients.
14. Treatment of EBA
Due to the low prevalence, no controlled clinical trials on the treatment of EBA have been performed. Current recommendations for EBA treatment are therefore solely based on the clinical expertise by clinicians specialized in AIBD [203, 204]. In addition, the clinical phenotype and the disease severity have to be taken into account when selecting treatment for EBA patients. However, the increased understanding of the diseases' pathogenesis has identified several potential therapeutic targets. The following paragraphs describe the advantages and disadvantages of the currently applied EBA treatments and discusses the possibility of targeting those of the identified targets, where drugs modulating the respective pathways are already available. However, and of note, the treatment of EBA is challenging. In a cohort of 30 EBA patients, who were initially treated with a combination of methylprednisolone, dapsone, and colchicine, remission was achieved after a median of 9 months on therapy. Long-term followup of these patients showed complete remission in 46% and incomplete remission in another 46% of the patients 6 years after initiation of treatment [7]. Overall, most experts recommend colchicine as first line treatment, as it has fewer adverse events than most of the other medications used for EBA treatment. Below, all drugs used to treat EBA are shortly introduced. The comment at the end indicates the author's personal opinion.
14.1. Currently Applied Treatments for EBA
14.1.1. Corticosteroids
Most EBA patients are treated with systemic corticosteroids. Initial doses range from 0.5 to 1.5 mg/kg per day [3]. In most cases, corticosteroid treatment is combined with other immunosuppressive/modulatory agents to lower the corticosteroid dose. These include almost all of the treatment modalities described below. As steroid treatment often leads to improvement of EBA, it has to be considered as effective. Furthermore, corticosteroid treatment is also effective in antibody transfer-induced EBA in mice [131]. However, due to its known high number of adverse events, other treatment modalities should be taken into consideration before systemic corticosteroid treatment whenever possible. Furthermore, it is tempting to speculate that patients with inflammatory-type EBA respond better to corticosteroids compared to patients with noninflammatory EBA.
14.1.2. Methotrexate and Azathioprine
Both compounds are used as steroid-sparing agents. There are no reports on single use of these compounds in EBA patients. Furthermore, no data is available documenting the effectiveness of either methotrexate or azathioprine as steroid-sparing agents in EBA. Therefore, both compounds should be used only in the treatment of refractory cases.
14.1.3. Cyclosporine
Since its first use in 1987 for EBA [205], cyclosporine has been used in a total of 11 EBA patients [203, 206, 207]. In all patients, the use of cyclosporine was reported to have improved EBA. Of note, in at least 3 patients, cyclosporine was used as monotherapy, leading to remission of EBA. Hence, cyclosporine is most likely an effective treatment of EBA.
14.1.4. Colchicine
Colchicine has been used in well over 40 EBA patients [7, 17, 208, 208–212]. As most reports state beneficial effects of colchicine, it should be used as first line treatment for patients with (inflammatory-type) EBA.
14.1.5. High Dose Intravenous Immunoglobulin (IVIG)
Immunoglobulin G preparations, isolated from human serum, have been used as substitution treatment for patients with antibody deficiencies and severe infections for many decades [213]. Later, intravenous application of high doses of IgG (IVIG) has been established as an effective therapy for many autoimmune diseases, including immune thrombocytopenia (ITP), Guillain-Barre syndrome, multiple sclerosis, myasthenia gravis, pemphigus disease, and Kawasaki disease [214–218]. Until 2010, 12 patients with extensive treatment-resistant EBA had been treated with IVIG, and the reported response was usually favorable [219]. In 2012, results on additional 10 IVIG-treated EBA patients were reported. These patients received IVIG in addition to the previous medication that had failed to control the disease. These earlier drugs were withdrawn over a 5–9-month period, and IVIG was continued as monotherapy for a total duration of 30–52 months. In all 10 patients, a satisfactory clinical response was observed, and during followup (29–123 months), no relapse was observed [220]. Collectively, this data strongly supports the assumption that IVIG is an effective treatment option for EBA.
14.1.6. Dapsone
Several case reports have stated the use of dapsone in EBA [7]. Of note, dapsone monotherapy has been shown to be effective in one EBA patient [221]. Further data, like for all applied treatments in EBA, is needed before final conclusions of the efficacy of dapsone can be drawn.
14.1.7. Cyclophosphamide
Cyclophosphamide has been rarely used in EBA. Therefore, no interpretation on its efficacy for controlling EBA can be drawn [222].
14.1.8. Rituximab
Anti-CD20 treatment with rituximab has been increasingly used for the treatment of AIBD [91, 223, 224], and currently, a large phase III clinical trial compares rituximab treatment to general corticosteroid therapy treatment in patients with pemphigus (NCT00784589). Regarding EBA, 12 cases have been reported with rituximab treatment. In most cases, a favorable outcome was reported. Most patients treated with rituximab, several previous treatments had failed. In most cases, anti-CD20 was administered in an adjuvant setting. One patient, unresponsive to previous treatments, was treated with immunoadsorption to lower antibody titers rapidly, followed by rituximab monotherapy. This led to rapid and lasting clinical remission. Another patient was treated with additional rituximab as azathioprine monotherapy had not improved EBA. Approximately 2 weeks later, the patient died of bacterial pneumonia [225–235]. Hence, anti-CD20 treatment should be taken into account when selecting treatments for patients with either severe and/or relapsing EBA.
14.1.9. Plasmapheresis and Immunoadsorption
Although several reports and case report series have documented a beneficial effect of plasmapheresis and immunoadsorption in AIBD—especially in pemphigus [236], only very limited experience with this treatment modality has been described in EBA. Taken together, 3 cases of either plasmapheresis or immunoadsorption in EBA patients have been published so far, and in all cases, plasmapheresis and immunoadsorption have been used in addition to other treatments [228, 233, 237]. Despite the improvement that was reported in all 3 cases, the currently available data does not allow drawing a final conclusion on the effectiveness of these methods for EBA patients.
14.1.10. Extracorporeal Photochemotherapy
In principle, similar conclusions like for plasmapheresis and immunoadsorbtion can be drawn for extracorporeal photochemotherapy. Currently, data on 8 EBP patients successfully treated with extracorporeal photochemotherapy has been reported [16, 238–241]. Hence, this method seems promising for the management of treatment-refractory patients.
14.2. Therapeutic Targets for Future EBA Therapy
Based on the increasing, but so far from complete, understanding of EBA pathogenesis, several potential therapeutic targets have been identified. This paragraph briefly describes those potential therapeutic targets, where compounds are already in use for other indications.
14.2.1. Targeting the Generation of Antibodies
Several HSP90 inhibitors are in phase I-II clinical trials for oncology indications [242]. As the blockade of HSP90 prevented the onset of immunization-induced EBA and improved already established disease [84], blockade of HSP90 has also emerged as a potential therapeutic target for EBA. However, as HSP90 inhibition is associated with several adverse events, including treatment-related death [243], the dosing has to be adjusted in EBA patients, or less toxic HSP inhibitors such as TCLB-145 [84] must be used. To achieve this, detailed preclinical pharmacodynamics studies and dose-finding studies have to be performed. With the documented relevance of T cells for the generation of autoantibodies in experimental EBA [83] and the detection of COL7-specific T cells in (albeit few) EBA patients [92], T-cell depleting antibodies may be an option for severe and treatment refractory cases. Currently, several anti-CD3 and anti-CD4 antibodies are tested in inflammatory and malignant diseases [244–248].
14.2.2. Removal of Autoantibodies
As an adjuvant treatment of AIBD (mainly pemphigus), immunoadsorption is performed [249]. Currently, a large phase III trial evaluates the efficacy and safety of immunoadsorption in pemphigus patients [250]. So far, experience with this technique is limited to few EBA patients (see Section 14), but overall immunoadsorption seems well suited to quickly reduce the levels of circulating autoantibodies, resulting in relatively rapid therapeutic response. Ultimately, as the autoantigen and the pathogenic relevance of autoantibodies are well documented, an antigen-specific immunoadsorption would be the ideal method to induce rapid clinical response in EBA patients. Antigen-specific removal of autoantibodies is well documented in dilative cardiomyopathy [251], and hence the principle seems applicable to other diseases, including EBA.
14.2.3. Modulation of Autoantibody-Induced Tissue Injury
The binding of activating FcgR to the immune complexes located in the skin is a key feature for autoantibody-mediated blister formation in EBA [124]. Hence, blockade of this interaction seems an attractive therapeutic approach. Currently, SM101, a soluble FcgRIIB which competes with FcgR expressed on immune cells for pathogenic immune complexes [252], is in phase II studies for primary immune thrombocytopenia (ITP) and systemic lupus erythematosus (SLE) [253]. In experimental models of lupus and arthritis SM101 prevented disease onset and/or improved already established disease [254, 255]. Hence, application of SM101 seems a promising treatment option for patients with treatment refractory EBA. As the lack of CD18 expression prevented neutrophil extravasation into the skin and subsequently clinical disease manifestation in experimental EBA [126], modulation of leukocyte extravasation could be targeted in EBA patients. Potential drug candidate is anti-alpha4 integrin (natalizumab), licensed for the treatment of multiple sclerosis [256]. Due to the observed progressive multifocal leukoencephalopathy under natalizumab treatment [257], risks and benefits have to be carefully considered, and patients must be monitored regularly. Other treatments targeting leukocyte extravasation, for example, anti-CD11a (efalizumab), have been withdrawn [258], have not been successful in sufficiently controlling skin inflammation [259, 260], or have not been tested in inflammatory skin conditions, such as KW-0761, which is used in the treatment of relapsed adult T-cell leukemia-lymphoma [261]. In principle, anti-CCR4 could be a therapeutic target in EBA, as CCR4 is involved in leukocyte recruitment into the skin [262]. An alternative to the targeted disruption of leukocyte extravasation by use of recombinant antibodies could be modified heparins. Besides its potent anti-coagulant activity, heparin has potent anti-inflammatory activities [263, 264]. Structural modifications of structurally defined glucan sulfates obtained by chemical modifications of neutral homoglycans produced by algae, bacteria, or fungi led to the production of compounds with reduced anticoagulant activity but stronger anti-inflammatory and/or antimetastatic activity compared to heparin [265]. One of these modified homoglycans, termed PS3, inhibits cutaneous inflammation [266].
In addition, selected cytokines such as CXCL1 and CXCL2 have been identified as novel therapeutic targets (see Section 7). Several compounds blocking CXCL1 and/or CXCL2 have been developed, for example, reparixin that enhanced pancreatic islet survival after transplantation in a phase 2 randomized, open-label pilot study [267]. In experimental models, a similar allosteric CXCR1/2 inhibitor prevented the onset of antibody-transfer-induced EBA and improved already established immunization-induced EBA [127]. Therefore, inhibition of CXCL1/2 by receptor-blockade seems a valid therapeutic approach for patients with treatment refractory EBA. GM-CSF also potently affects neutrophils functions. As detailed previously, blockade of this cytokine has beneficial effects both in antibody transfer- and immunization-induced EBA. GM-CSF has emerged as a target for rheumatoid arthritis, and three different anti-GM-CSF antibodies are currently evaluated in phase I-II clinical trials for this indication (MOR103, MorphoSys AG, NCT01023256; KB003, KaloBios Pharmaceuticals, NCT00995449; MT203, Takeda Pharma A/S, NCT01317797). Furthermore, the identification of the anti-inflammatory effects of IL-6, which is at least partially mediated by regulation of IL-1ra, makes use of recombinant IL-1ra (Anakinra). Anakinra is very effective in patients with Schnitzler's syndrome [268]. To the author's knowledge, Anakinra has not been used to treat AIBD or EBA patients. However, based on the efficacy of the compound in other inflammatory conditions and the preclinical data in experimental EBA, the use of compounds targeting IL-1 would be expected to have beneficial effects on EBA patients.
Acknowledgments
The author thanks all of his colleagues who contributed to several of the studies reviewed in this paper. This study has been made possible by the financial support of the Deutsche Forschungsgemeinschaft (EXC 306/1 and 2, GRK 1727/1, GRK 1734/1, and LU877/5-1), the University of Lübeck (Focus Program Autoimmunity), the Dr. Robert Pfleger Foundation, and project research grants from Biotest AG, Dompé s.p.a., and Euroimmun AG.
References
- 1.Elliott GT. Two cases of epidermolysis bullosa. Journal of Cutaneous and Genito-Urinary Diseases. 1895;13(article 10) [Google Scholar]
- 2.Roenigk HH, Jr., Ryan JG, Bergfeld WF. Epidermolysis bullosa acquisita. Report of three cases and review of all published cases. Archives of Dermatology. 1971;103(1):1–10. doi: 10.1001/archderm.103.1.1. [DOI] [PubMed] [Google Scholar]
- 3.Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? Journal of Dermatology. 2010;37(3):220–230. doi: 10.1111/j.1346-8138.2009.00799.x. [DOI] [PubMed] [Google Scholar]
- 4.Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clinics in Dermatology. 2012;30(1):60–69. doi: 10.1016/j.clindermatol.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmidt E, Zillikens D. Pemphigoid diseases. The Lancet. 2013;381:320–332. doi: 10.1016/S0140-6736(12)61140-4. [DOI] [PubMed] [Google Scholar]
- 6.Buijsrogge JJA, Diercks GFH, Pas HH, Jonkman MF. The many faces of epidermolysis bullosa acquisita after serration pattern analysis by direct immunofluorescence microscopy. British Journal of Dermatology. 2011;165(1):92–98. doi: 10.1111/j.1365-2133.2011.10346.x. [DOI] [PubMed] [Google Scholar]
- 7.Kim JH, Kim YH, Kim S-C. Epidermolysis bullosa acquisita: a retrospective clinical analysis of 30 cases. Acta Dermato-Venereologica. 2011;91(3):307–312. doi: 10.2340/00015555-1065. [DOI] [PubMed] [Google Scholar]
- 8.Zumelzu C, Le Roux-Villet C, Loiseau P, et al. Black patients of african descent and HLA-DRB115:03 frequency overrepresented in epidermolysis bullosa acquisita. Journal of Investigative Dermatology. 2011;131(12):2386–2393. doi: 10.1038/jid.2011.231. [DOI] [PubMed] [Google Scholar]
- 9.Lam C, Vleugels RA. Images in clinical medicine. Epidermolysis bullosa acquisita. The New England Journal of Medicine. 2013;368(article e17) doi: 10.1056/NEJMicm1204895. [DOI] [PubMed] [Google Scholar]
- 10.Kuo TT, Baker K, Yoshida M, et al. Neonatal Fc receptor: from immunity to therapeutics. Journal of Clinical Immunology. 2010;30(6):777–789. doi: 10.1007/s10875-010-9468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abrams ML, Smidt A, Benjamin L, Chen M, Woodley D, Mancini AJ. Congenital epidermolysis bullosa acquisita: vertical transfer of maternal autoantibody from mother to infant. Archives of Dermatology. 2011;147(3):337–341. doi: 10.1001/archdermatol.2010.317. [DOI] [PubMed] [Google Scholar]
- 12.Chen M, Kim GH, Prakash L, Woodley DT. Epidermolysis bullosa acquisita: autoimmunity to anchoring fibril collagen. Autoimmunity. 2012;45(1):91–101. doi: 10.3109/08916934.2011.606450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lang PG, Jr., Tapert MJ. Severe ocular involvement in a patient with epidermolysis bullosa acquisita. Journal of the American Academy of Dermatology. 1987;16(2):439–443. doi: 10.1016/s0190-9622(87)70057-7. [DOI] [PubMed] [Google Scholar]
- 14.Zierhut M, Thiel H-J, Weidle EG, Steuhl K-P, Sonnichsen K, Schaumburg-Lever G. Ocular involvement in epidermolysis bullosa acquisita. Archives of Ophthalmology. 1989;107(3):398–401. doi: 10.1001/archopht.1989.01070010408035. [DOI] [PubMed] [Google Scholar]
- 15.Caux F, Kirtschig G, Lemarchand-Venencie F, et al. IgA-epidermolysis bullosa acquisita in a child resulting in blindness. British Journal of Dermatology. 1997;137(2):270–275. doi: 10.1046/j.1365-2133.1997.18191915.x. [DOI] [PubMed] [Google Scholar]
- 16.Camara A, Bécherel P-A, Bussel A, et al. Resistant epidermolysis bullous acquisita with severe ocular involvement: successful extracorporeal photochemotherapy. Annales de Dermatologie et de Venereologie. 1999;126(8-9):612–615. [PubMed] [Google Scholar]
- 17.Bauer JW, Schaeppi H, Metze D, et al. Ocular involvement in IgA-epidermolysis bullosa acquisita. British Journal of Dermatology. 1999;141(5):887–892. doi: 10.1046/j.1365-2133.1999.03163.x. [DOI] [PubMed] [Google Scholar]
- 18.Vodegel RM, de Jong MCJM, Pas HH, Jonkman MF. IgA-mediated epidermolysis bullosa acquisita: two cases and review of the literature. Journal of the American Academy of Dermatology. 2002;47(6):919–925. doi: 10.1067/mjd.2002.125079. [DOI] [PubMed] [Google Scholar]
- 19.Kurzhals G, Stolz W, Meurer M, Kunze J, Braun-Falco O, Krieg T. Acquired epidermolysis bullosa with the clinical feature of Brunsting-Perry cicatricial bullous pemphigoid. Archives of Dermatology. 1991;127(3):391–395. [PubMed] [Google Scholar]
- 20.Wieme N, Lambert J, Moerman M, Geerts ML, Temmerman L, Naeyaert JM. Epidermolysis bullosa acquisita with combined features of bullous pemphigoid and cicatricial pemphigoid. Dermatology. 1999;198(3):310–313. doi: 10.1159/000018139. [DOI] [PubMed] [Google Scholar]
- 21.Taniuchi K, Inaoki M, Nishimura Y, Mori T, Takehara K. Nonscarring inflammatory epidermolysis bullosa acquisita with esophageal involvement and linear IgG deposits. Journal of the American Academy of Dermatology. 1997;36(2):320–322. doi: 10.1016/s0190-9622(97)80408-2. [DOI] [PubMed] [Google Scholar]
- 22.Harman KE, Whittam LR, Wakelin SH, Black MM. Severe, refractory epidermolysis bullosa acquisita complicated by an oesophageal stricture responding to intravenous immune globulin. British Journal of Dermatology. 1998;139(6):1126–1127. doi: 10.1046/j.1365-2133.1998.2576m.x. [DOI] [PubMed] [Google Scholar]
- 23.Shipman AR, Agero AL, Cook I, et al. Epidermolysis bullosa acquisita requiring multiple oesophageal dilatations. Clinical and Experimental Dermatology. 2008;33(6):787–789. doi: 10.1111/j.1365-2230.2008.02875.x. [DOI] [PubMed] [Google Scholar]
- 24.Hester JE, Arnstein DP, Woodley D. Laryngeal manifestations of epidermolysis bullosa acquisita. Archives of Otolaryngology—Head and Neck Surgery. 1995;121(9):1042–1044. doi: 10.1001/archotol.1995.01890090078015. [DOI] [PubMed] [Google Scholar]
- 25.Luke MC, Darling TN, Hsu R, et al. Mucosal morbidity in patients with epidermolysis bullosa acquisita. Archives of Dermatology. 1999;135(8):954–959. doi: 10.1001/archderm.135.8.954. [DOI] [PubMed] [Google Scholar]
- 26.Paller AS, Queen LL, Woodley DT. Organ-specific, phylogenetic, and ontogenetic distribution of the epidermolysis bullosa acquisita antigen. Journal of Investigative Dermatology. 1986;86(4):376–379. doi: 10.1111/1523-1747.ep12285623. [DOI] [PubMed] [Google Scholar]
- 27.Visser R, Arends JW, Leigh IM, Bosman FT. Paterns and composition of basement membranes in colon adenomas and adenocarcinomas. Journal of Pathology. 1993;170(3):285–290. doi: 10.1002/path.1711700311. [DOI] [PubMed] [Google Scholar]
- 28.Ishii N, Recke A, Mihai S, et al. Autoantibody-induced intestinal inflammation and weight loss in experimental epidermolysis bullosa acquisita. Journal of Pathology. 2011;224(2):234–244. doi: 10.1002/path.2857. [DOI] [PubMed] [Google Scholar]
- 29.Furue M, Iwata M, Tamaki K, Ishibashi Y. Anatomical distribution and immunological characteristics of epidermolysis bullosa acquisita antigen and bullous pemphigoid antigen. British Journal of Dermatology. 1986;114(6):651–659. doi: 10.1111/j.1365-2133.1986.tb04872.x. [DOI] [PubMed] [Google Scholar]
- 30.Lee CW. Prevalences of subacute cutaneous lupus erythematosus and epidermolysis bullosa acquisita among Korean/Oriental populations. Dermatology. 1998;197(2, article 187) doi: 10.1159/000017997. [DOI] [PubMed] [Google Scholar]
- 31.Endo Y, Tamura A, Ishikawa O, Miyachi Y, Hashimoto T. Psoriasis vulgaris coexistent with epidermolysis bullosa acquisita. British Journal of Dermatology. 1997;137(5):783–786. [PubMed] [Google Scholar]
- 32.Morris SD, Mallipeddi R, Oyama N, et al. Psoriasis bullosa acquisita. Clinical and Experimental Dermatology. 2002;27(8):665–669. doi: 10.1046/j.1365-2230.2002.01100.x. [DOI] [PubMed] [Google Scholar]
- 33.Hoshina D, Sawamura D, Nomura T, et al. Epidermolysis bullosa acquisita associated with psoriasis vulgaris. Clinical and Experimental Dermatology. 2007;32(5):516–518. doi: 10.1111/j.1365-2230.2007.02430.x. [DOI] [PubMed] [Google Scholar]
- 34.Kabashima R, Hino R, Bito T, et al. Epidermolysis bullosa acquisita associated with psoriasis. Acta Dermato-Venereologica. 2010;90(3):314–316. doi: 10.2340/00015555-0832. [DOI] [PubMed] [Google Scholar]
- 35.Sherry-Dottridge F. Case for diagnosis: acquired epidermatolysis bullosa? Proceedings of the Royal Society of Medicine. 1962;55, article 409 [PMC free article] [PubMed] [Google Scholar]
- 36.Labeille B, Gineston J-L, Denoeux J-P, Capron J-P. Epidermolysis bullosa acquisita and Crohn’s disease. A case report with immunological and electron microscopic studies. Archives of Internal Medicine. 1988;148(6):1457–1459. [PubMed] [Google Scholar]
- 37.Raab B, Fretzin DF, Bronson DM. Epidermolysis bullosa acquisita and inflammatory bowel disease. Journal of the American Medical Association. 1983;250(13):1746–1748. [PubMed] [Google Scholar]
- 38.Schattenkirchner S, Lémann M, Prost C, et al. Localized epidermolysis bullosa acquisita of the esophagus in a patient with Crohn’s disease. American Journal of Gastroenterology. 1996;91(8):1657–1659. [PubMed] [Google Scholar]
- 39.Livden JK, Nilsen R, Thunold S, Schjonsby H. Epidermolysis bullosa acquisita and Crohn’s disease. Acta Dermato-Venereologica. 1978;58(3):241–244. [PubMed] [Google Scholar]
- 40.Hughes BR, Horne J. Epidermolysis bullosa acquisita and total ulcerative colitis. Journal of the Royal Society of Medicine. 1988;81(8):473–475. doi: 10.1177/014107688808100821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have autoantibodies to type VII collagen. Journal of Investigative Dermatology. 2002;118(6):1059–1064. doi: 10.1046/j.1523-1747.2002.01772.x. [DOI] [PubMed] [Google Scholar]
- 42.Oostingh GJ, Sitaru C, Zillikens D, Kromminga A, Lührs H. Subclass distribution of type VII collagen-specific autoantibodies in patients with inflammatory bowel disease. Journal of Dermatological Science. 2005;37(3):182–184. doi: 10.1016/j.jdermsci.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 43.Licarete E, Ganz S, Recknagel M, et al. Prevalence of collagen VII-specific autoantibodies in patients with autoimmune and inflammatory diseases. BMC Immunology. 2012;13(article 16) doi: 10.1186/1471-2172-13-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sitaru C, Mihai S, Otto C, et al. Induction of dermal-epidermal separation in mice by passive transfer of antibodies specific to type VII collagen. Journal of Clinical Investigation. 2005;115(4):870–878. doi: 10.1172/JCI21386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sitaru C, Chiriac MT, Mihai S, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. Journal of Immunology. 2006;177(5):3461–3468. doi: 10.4049/jimmunol.177.5.3461. [DOI] [PubMed] [Google Scholar]
- 46.Kasperkiewicz M, Hirose M, Recke A, Schmidt E, Zillikens D, Ludwig RJ. Clearance rates of circulating and tissue-bound autoantibodies to type VII collagen in experimental epidermolysis bullosa acquisita. British Journal of Dermatology. 2010;162(5):1064–1070. doi: 10.1111/j.1365-2133.2010.09680.x. [DOI] [PubMed] [Google Scholar]
- 47.Bernard P, Vaillant L, Labeille B, et al. Incidence and distribution of subepidermal autoimmune bullous skin diseases in three French regions. Bullous Diseases French Study Group. Archives of Dermatology. 1995;131(1):48–52. [PubMed] [Google Scholar]
- 48.Wong SN, Chua SH. Spectrum of subepidermal immunobullous disorders seen at the National Skin Centre, Singapore: a 2-year review. British Journal of Dermatology. 2002;147(3):476–480. doi: 10.1046/j.1365-2133.2002.04919.x. [DOI] [PubMed] [Google Scholar]
- 49.Bertram F, Bröcker E-B, Zillikens D, Schmidt E. Prospective analysis of the incidence of autoimmune bullous disorders in Lower Franconia, Germany. Journal of the German Society of Dermatology. 2009;7(5):434–440. doi: 10.1111/j.1610-0387.2008.06976.x. [DOI] [PubMed] [Google Scholar]
- 50.Yang B, Wang C, Wang N, et al. Childhood epidermolysis bullosa acquisita: report of a Chinese case. Pediatric Dermatology. 2012;29:614–617. doi: 10.1111/j.1525-1470.2011.01509.x. [DOI] [PubMed] [Google Scholar]
- 51.Bordier-Lamy F, Eschard C, Coste M, et al. Epidermolysis bullosa acquisita of childhood. Annales de Dermatologie et de Venereologie. 2009;136(6-7):513–517. doi: 10.1016/j.annder.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 52.Stanley JR, Amagai M. Pemphigus, bullous impetigo, and the staphylococcal scalded-skin syndrome. New England Journal of Medicine. 2006;355(17):1800–1810. doi: 10.1056/NEJMra061111. [DOI] [PubMed] [Google Scholar]
- 53.Woodley DT, Burgeson RE, Lunstrum G, Bruckner-Tuderman L, Reese MJ, Briggaman RA. Epidermolysis bullosa acquisita antigen is the globular carboxyl terminus of type VII procollagen. Journal of Clinical Investigation. 1988;81(3):683–687. doi: 10.1172/JCI113373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Woodley DT, Briggaman RA, O’Keefe EJ. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. New England Journal of Medicine. 1984;310(16):1007–1013. doi: 10.1056/NEJM198404193101602. [DOI] [PubMed] [Google Scholar]
- 55.Lapiere J-C, Woodley DT, Parente MG, et al. Epitope mapping of type VII collagen. Identification of discrete peptide sequences recognized by sera from patients with acquired epidermolysis bullosa. Journal of Clinical Investigation. 1993;92(4):1831–1839. doi: 10.1172/JCI116774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gammon WR, Murrell DF, Jenison MW, et al. Autoantibodies to type VII collagen recognize epitopes in a fibronectin-like region of the noncollagenous (NC1) domain. Journal of Investigative Dermatology. 1993;100(5):618–622. doi: 10.1111/1523-1747.ep12472291. [DOI] [PubMed] [Google Scholar]
- 57.Chen M, Doostan A, Bandyopadhyay P, et al. The cartilage matrix protein subdomain of type VII collagen is pathogenic for epidermolysis bullosa acquisita. American Journal of Pathology. 2007;170(6):2009–2018. doi: 10.2353/ajpath.2007.061212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ishii N, Yoshida M, Ishida-Yamamoto A, et al. Some epidermolysis bullosa acquisita sera react with epitopes within the triple-helical collagenous domain as indicated by immunoelectron microscopy. British Journal of Dermatology. 2009;160(5):1090–1093. doi: 10.1111/j.1365-2133.2008.08952.x. [DOI] [PubMed] [Google Scholar]
- 59.Ishii N, Yoshida M, Hisamatsu Y, et al. Epidermolysis bullosa acquisita sera react with distinct epitopes on the NC1 and NC2 domains of type VII collagen: study using immunoblotting of domain-specific recombinant proteins and postembedding immunoelectron microscopy. British Journal of Dermatology. 2004;150(5):843–851. doi: 10.1111/j.1365-2133.2004.05933.x. [DOI] [PubMed] [Google Scholar]
- 60.Sitaru C, Kromminga A, Hashimoto T, Bröcker EB, Zillikens D. Autoantibodies to type VII collagen mediate Fcγ-dependent neutrophil activation and induce dermal-epidermal separation in cryosections of human skin. American Journal of Pathology. 2002;161(1):301–311. doi: 10.1016/s0002-9440(10)64182-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kulkarni S, Sitaru C, Jakus Z, et al. PI3Kβ plays a critical role in neutrophil activation by immune complexes. Science Signaling. 2011;4(168, article ra23) doi: 10.1126/scisignal.2001617. [DOI] [PubMed] [Google Scholar]
- 62.Recke A, Sitaru C, Vidarsson G, et al. Pathogenicity of IgG subclass autoantibodies to type VII collagen: induction of dermal-epidermal separation. Journal of Autoimmunity. 2010;34(4):435–444. doi: 10.1016/j.jaut.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 63.Woodley DT, Chang C, Saadat P, Ram R, Liu Z, Chen M. Evidence that anti-type VII collagen antibodies are pathogenic and responsible for the clinical, histological, and immunological features of epidermolysis bullosa acquisita. Journal of Investigative Dermatology. 2005;124(5):958–964. doi: 10.1111/j.0022-202X.2005.23702.x. [DOI] [PubMed] [Google Scholar]
- 64.Woodley DT, Ram R, Doostan A, et al. Induction of epidermolysis bullosa acquisita in mice by passive transfer of autoantibodies from patients. Journal of Investigative Dermatology. 2006;126(6):1323–1330. doi: 10.1038/sj.jid.5700254. [DOI] [PubMed] [Google Scholar]
- 65.Wang X, Gupta R, Garlapati A, Cogan J, Woodley D, Chen M. Type IV collagen binding-site within type VII collagen is a pathogenic epitope for EBA autoantibodies. Journal of Investigative Dermatology. 2011;131:p. S7. [Google Scholar]
- 66.Iwata H, Leinweber S, Samavedam U, et al. Antibodies to the von Willebrand Factor A domain of type VII collagen induce strain-dependent subepidermal blistering in mice. Experimental Dermatology. 2012;21(article e31) [Google Scholar]
- 67.Vorobyev A, Recke A, Buijsrogge JJ, et al. Human type VII collagen harbors multiple pathogenically relevant epitopes. Journal of Investigative Dermatology. 2011;131:p. S19. [Google Scholar]
- 68.Ludwig RJ, Recke A, Bieber K, et al. Generation of antibodies of distinct subclasses and specificity is linked to H2s in an active mouse model of epidermolysis bullosa acquisita. Journal of Investigative Dermatology. 2011;131(1):167–176. doi: 10.1038/jid.2010.248. [DOI] [PubMed] [Google Scholar]
- 69.Hammers CM, Bieber K, Kalies K, et al. Complement-fixing anti-type VII collagen antibodies are induced in Th1-polarized lymph nodes of epidermolysis bullosa acquisita-susceptible mice. Journal of Immunology. 2011;187(10):5043–5050. doi: 10.4049/jimmunol.1100796. [DOI] [PubMed] [Google Scholar]
- 70.Leineweber S, Schönig S, Seeger K. Insight into interactions of the von-Willebrand-factor-A-like domain 2 with the FNIII-like domain 9 of collagen VII by NMR and SPR. FEBS Letters. 2011;585(12):1748–1752. doi: 10.1016/j.febslet.2011.04.071. [DOI] [PubMed] [Google Scholar]
- 71.Parente MG, Chung LC, Ryynanen J, et al. Human type VII collagen: cDNA cloning and chromosomal mapping of the gene. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(16):6931–6935. doi: 10.1073/pnas.88.16.6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wegener H, Leineweber S, Seeger K. The vWFA2 domain of type VII collagen is responsible for collagen binding. Biochemical and Biophysical Research Communications. 2013;430:449–453. doi: 10.1016/j.bbrc.2012.11.119. [DOI] [PubMed] [Google Scholar]
- 73.Yu X, Holdorf K, Kasper B, Zillikens D, Ludwig RJ, Petersen F. FcγRIIA and FcγRIIIB are required for autoantibody-induced tissue damage in experimental human models of bullous pemphigoid. Journal of Investigative Dermatology. 2010;130(12):2841–2844. doi: 10.1038/jid.2010.230. [DOI] [PubMed] [Google Scholar]
- 74.Gammon WR, Merritt CC, Lewis DM. An in vitro model of immune complex-mediated basement membrane zone separation caused by pemphigoid antibodies, leukocytes, and complement. Journal of Investigative Dermatology. 1982;78(4):285–290. doi: 10.1111/1523-1747.ep12507222. [DOI] [PubMed] [Google Scholar]
- 75.Sitaru C, Schmidt E, Petermann S, Munteanu LS, Bröcker E-B, Zillikens D. Autoantibodies to bullous pemphigoid antigen 180 induce dermal-epidermal separation in cryosections of human skin. Journal of Investigative Dermatology. 2002;118(4):664–671. doi: 10.1046/j.1523-1747.2002.01720.x. [DOI] [PubMed] [Google Scholar]
- 76.Umemoto H, Akiyama M, Domon T, et al. Type VII collagen deficiency causes defective tooth enamel formation due to poor differentiation of ameloblasts. The American Journal of Pathology. 2012;181:1659–1671. doi: 10.1016/j.ajpath.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 77.Ludwig RJ, Müller S, Marques ADC, et al. Identification of quantitative trait loci in experimental epidermolysis bullosa acquisita. Journal of Investigative Dermatology. 2012;132(5):1409–1415. doi: 10.1038/jid.2011.466. [DOI] [PubMed] [Google Scholar]
- 78.Bieber K, Sun S, Ishii N, et al. Animal models for autoimmune bullous dermatoses. Experimental Dermatology. 2010;19(1):2–11. doi: 10.1111/j.1600-0625.2009.00948.x. [DOI] [PubMed] [Google Scholar]
- 79.Ludwig RJ. Model systems duplicating epidermolysis bullosa acquisita: a methodological review. Autoimmunity. 2012;45(1):102–110. doi: 10.3109/08916934.2011.606451. [DOI] [PubMed] [Google Scholar]
- 80.Gammon WR, Heise ER, Burke WA, Fine J-D, Woodley DT, Briggaman RA. Increased frequency of HLA-DR2 in patients with autoantibodies to epidermolysis bullosa acquisita antigen: evidence that the expression of autoimmunity to type VII collagen is HLA class II allele associated. Journal of Investigative Dermatology. 1988;91(3):228–232. doi: 10.1111/1523-1747.ep12470317. [DOI] [PubMed] [Google Scholar]
- 81.Noe MH, Chen M, Woodley DT, Fairley JA. Familial epidermolysis bullosa acquisita. Dermatology Online Journal. 2008;14(12, article 2) [PubMed] [Google Scholar]
- 82.Asghari F, Fitzner B, Holzhüter S-A, et al. Identification of quantitative trait loci for murine autoimmune pancreatitis. Journal of Medical Genetics. 2011;48(8):557–562. doi: 10.1136/jmg.2011.089730. [DOI] [PubMed] [Google Scholar]
- 83.Sitaru AG, Sesarman A, Mihai S, et al. T cells are required for the production of blister-inducing autoantibodies in experimental epidermolysis bullosa acquisita. Journal of Immunology. 2010;184(3):1596–1603. doi: 10.4049/jimmunol.0901412. [DOI] [PubMed] [Google Scholar]
- 84.Kasperkiewicz M, Müller R, Manz R, et al. Heat-shock protein 90 inhibition in autoimmunity to type VII collagen: evidence that nonmalignant plasma cells are not therapeutic targets. Blood. 2011;117(23):6135–6142. doi: 10.1182/blood-2010-10-314609. [DOI] [PubMed] [Google Scholar]
- 85.Amagai M, Tsunoda K, Suzuki H, Nishifuji K, Koyasu S, Nishikawa T. Use of autoantigen-knockout mice in developing an active autoimmune disease model for pemphigus. Journal of Clinical Investigation. 2000;105(5):625–631. doi: 10.1172/JCI8748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tsunoda K, Ota T, Suzuki H, et al. Pathogenic autoantibody production requires loss of tolerance against desmoglein 3 in both T and B cells in experimental pemphigus vulgaris. European Journal of Immunology. 2002;32:627–633. doi: 10.1002/1521-4141(200203)32:3<627::AID-IMMU627>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 87.Ujiie H, Shibaki A, Nishie W, et al. Noncollagenous 16A domain of type XVII collagen-reactive CD4+ T cells play a pivotal role in the development of active disease in experimental bullous pemphigoid model. Clinical Immunology. 2012;142(2):167–175. doi: 10.1016/j.clim.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 88.Ujiie H, Shibaki A, Nishie W, et al. A novel active mouse model for bullous pemphigoid targeting humanized pathogenic antigen. Journal of Immunology. 2010;184(4):2166–2174. doi: 10.4049/jimmunol.0903101. [DOI] [PubMed] [Google Scholar]
- 89.Ujiie H, Shimizu H. Evidence for pathogenicity of autoreactive T cells in autoimmune bullous diseases shown by animal disease models. Experimental Dermatology. 2012;21:901–905. doi: 10.1111/exd.12011. [DOI] [PubMed] [Google Scholar]
- 90.Giménez Ortiz A, Montalar Salcedo J. Heat shock proteins as targets in oncology. Clinical and Translational Oncology. 2010;12(3):166–173. doi: 10.1007/s12094-010-0486-8. [DOI] [PubMed] [Google Scholar]
- 91.Colliou N, Picard D, Caillot F, et al. Long-term remissions of severe pemphigus after rituximab therapy are associated with prolonged failure of desmoglein B cell response. Science Translational Medicine. 2013;5(article 175ra30) doi: 10.1126/scitranslmed.3005166. [DOI] [PubMed] [Google Scholar]
- 92.Müller R, Dahler C, Möbs C, et al. T and B cells target identical regions of the non-collagenous domain 1 of type VII collagen in epidermolysis bullosa acquisita. Clinical Immunology. 2010;135(1):99–107. doi: 10.1016/j.clim.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 93.Li N, Zhao M, Hilario-Vargas J, et al. Complete FcRn dependence for intravenous Ig therapy in autoimmune skin blistering diseases. Journal of Clinical Investigation. 2005;115(12):3440–3450. doi: 10.1172/JCI24394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sesarman A, Sitaru AG, Olaru F, Zillikens D, Sitaru C. Neonatal Fc receptor deficiency protects from tissue injury in experimental epidermolysis bullosa acquisita. Journal of Molecular Medicine. 2008;86(8):951–959. doi: 10.1007/s00109-008-0366-7. [DOI] [PubMed] [Google Scholar]
- 95.Ji H, Gauguier D, Ohmura K, et al. Genetic influences on the end-stage effector phase of arthritis. Journal of Experimental Medicine. 2001;194(3):321–330. doi: 10.1084/jem.194.3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Textor J, Peixoto A, Henrickson SE, Sinn M, von Andrian UH, Westermann J. Defining the quantitative limits of intravital two-photon lymphocyte tracking. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(30):12401–12406. doi: 10.1073/pnas.1102288108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lohi J, Leivo I, Tani T, et al. Laminins, tenascin and type VII collagen in colorectal mucosa. Histochemical Journal. 1996;28(6):431–440. doi: 10.1007/BF02331434. [DOI] [PubMed] [Google Scholar]
- 98.Leivo I, Tani T, Laitinen L, et al. Anchoring complex components laminin-5 and type VII collagen in intestine: association with migrating and differentiating enterocytes. Journal of Histochemistry and Cytochemistry. 1996;44(11):1267–1277. doi: 10.1177/44.11.8918902. [DOI] [PubMed] [Google Scholar]
- 99.Wetzels RHW, Robben HCM, Leigh IM, Schaafsma HE, Vooijs GP, Ramaekers FCS. Distribution patterns of type VII collagen in normal and malignant human tissues. American Journal of Pathology. 1991;139(2):451–459. [PMC free article] [PubMed] [Google Scholar]
- 100.Iwata H, Kamio N, Aoyama Y, et al. IgG from patients with bullous pemphigoid depletes cultured keratinocytes of the 180-kDa bullous pemphigoid antigen (type XVII collagen) and weakens cell attachment. Journal of Investigative Dermatology. 2009;129(4):919–926. doi: 10.1038/jid.2008.305. [DOI] [PubMed] [Google Scholar]
- 101.Natsuga K, Nishie W, Shinkuma S, et al. Antibodies to pathogenic epitopes on type XVII collagen cause skin Ffragility in a complement-dependent and -independent manner. The Journal of Immunology. 2012;188(11):5792–5799. doi: 10.4049/jimmunol.1003402. [DOI] [PubMed] [Google Scholar]
- 102.Iwata H, Kitajima Y. Bullous pemphigoid: role of complement and mechanisms for blister formation within the lamina lucida. Experimental Dermatology. 2013;22(6):381–385. doi: 10.1111/exd.12146. [DOI] [PubMed] [Google Scholar]
- 103.Villone D, Fritsch A, Koch M, Bruckner-Tuderman L, Hansen U, Bruckner P. Supramolecular interactions in the dermo-epidermal junction zone: anchoring fibril-collagen VII tightly binds to banded collagen fibrils. Journal of Biological Chemistry. 2008;283(36):24506–24513. doi: 10.1074/jbc.M802415200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sesarman A, Mihai S, Chiriac MT, et al. Binding of avian IgY to type VII collagen does not activate complement and leucocytes and fails to induce subepidermal blistering in mice. British Journal of Dermatology. 2008;158(3):463–471. doi: 10.1111/j.1365-2133.2007.08388.x. [DOI] [PubMed] [Google Scholar]
- 105.Lauc G, Huffman JE, Pucic M, et al. Loci associated with N-glycosylation of human immunoglobulin G show pleiotropy with autoimmune diseases and haematological cancers. PLOS Genetics. 2013;9 doi: 10.1371/journal.pgen.1003225.e1003225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ercan A, Barnes MG, Hazen M, et al. Multiple juvenile idiopathic arthritis subtypes demonstrate proinflammatory IgG glycosylation. Arthritis & Rheumatism. 2012;64:3025–3033. doi: 10.1002/art.34507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Collin M, Olsén A. EndoS, a novel secreted protein from Streptococcus pyogenes with endoglycosidase activity on human IgG. The EMBO Journal. 2001;20(12):3046–3055. doi: 10.1093/emboj/20.12.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nandakumar KS, Collin M, Olsén A, et al. Endoglycosidase treatment abrogates IgG arthritogenicity: importance of IgG glycosylation in arthritis. European Journal of Immunology. 2007;37(10):2973–2982. doi: 10.1002/eji.200737581. [DOI] [PubMed] [Google Scholar]
- 109.Albert H, Collin M, Dudziak D, Ravetch JV, Nimmerjahn F. In vivo enzymatic modulation of IgG glycosylation inhibits autoimmune disease in an IgG subclass-dependent manner. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(39):15005–15009. doi: 10.1073/pnas.0808248105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.van Timmeren MM, van der Veen BS, Stegeman CA, et al. IgG glycan hydrolysis attenuates ANCA-mediated glomerulonephritis. Journal of the American Society of Nephrology. 2010;21(7):1103–1114. doi: 10.1681/ASN.2009090984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yang R, Otten MA, Hellmark T, et al. Successful treatment of experimental glomerulonephritis with IdeS and EndoS, IgG-degrading streptococcal enzymes. Nephrology Dialysis Transplantation. 2010;25(8):2479–2486. doi: 10.1093/ndt/gfq115. [DOI] [PubMed] [Google Scholar]
- 112.Allhorn M, Briceño JG, Baudino L, et al. The IgG-specific endoglycosidase EndoS inhibits both cellular and complement-mediated autoimmune hemolysis. Blood. 2010;115(24):5080–5088. doi: 10.1182/blood-2009-08-239020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hirose M, Vafia K, Kalies K, et al. Enzymatic autoantibody glycan hydrolysis alleviates autoimmunity against type VII collagen. Journal of Autoimmunity. 2012;39(4):304–314. doi: 10.1016/j.jaut.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 114.Benkhoucha M, Molnarfi N, Santiago-Raber ML, et al. IgG glycan hydrolysis by EndoS inhibits experimental autoimmune encephalomyelitis. J Neuroinflammation. 2012;9(article 209) doi: 10.1186/1742-2094-9-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ludwig RJ, Schmidt E. Cytokines in autoimmune bullous skin diseases. Epiphenomena or contribution to pathogenesis? Giornale Italiano di Dermatologia e Venereologia. 2009;144(4):339–349. [PubMed] [Google Scholar]
- 116.Kuemmerle-Deschner JB, Tyrrell PN, Koetter I, et al. Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle-Wells syndrome. Arthritis & Rheumatism. 2011;63(3):840–849. doi: 10.1002/art.30149. [DOI] [PubMed] [Google Scholar]
- 117.Lovell DJ, Ruperto N, Goodman S, et al. Adalimumab with or without methotrexate in juvenile rheumatoid arthritis. New England Journal of Medicine. 2008;359(8):810–820. doi: 10.1056/NEJMoa0706290. [DOI] [PubMed] [Google Scholar]
- 118.Elliott MJ, Maini RN, Feldmann M, et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor α (cA2) versus placebo in rheumatoid arthritis. The Lancet. 1994;344(8930):1105–1110. doi: 10.1016/s0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- 119.Mease PJ, Goffe BS, Metz J, Vanderstoep A, Finck B, Bürge DJ. Etanercept in the treatment of psoriatic arthritis and psoriasis: a randomised trial. The Lancet. 2000;356(9227):385–390. doi: 10.1016/S0140-6736(00)02530-7. [DOI] [PubMed] [Google Scholar]
- 120.Sandborn WJ, Rutgeerts P, Enns R, et al. Adalimumab induction therapy for Crohn disease previously treated with infliximab: a randomized trial. Annals of Internal Medicine. 2007;146(12):829–838. doi: 10.7326/0003-4819-146-12-200706190-00159. [DOI] [PubMed] [Google Scholar]
- 121.John H, Whallett A, Quinlan M. Successful biologic treatment of ocular mucous membrane pemphigoid with anti-TNF-α . Eye. 2007;21(11):1434–1435. doi: 10.1038/sj.eye.6702948. [DOI] [PubMed] [Google Scholar]
- 122.Kennedy JS, Devillez RL, Henning JS. Recalcitrant cicatricial pemphigoid treated with the anti-TNF-alpha agent etanercept. Journal of Drugs in Dermatology. 2010;9(1):68–70. [PubMed] [Google Scholar]
- 123.Samavedam UK, Scheller J, Gupta Y, et al. Recombinant IL-6 treatment protects mice from organ specific autoimmune disease by IL-6 classical signalling-dependent IL-1ra induction. Journal of Autoimmunity. 2013;40:74–85. doi: 10.1016/j.jaut.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 124.Kasperkiewicz M, Nimmerjahn F, Wende S, et al. Genetic identification and functional validation of FcγRIV as key molecule in autoantibody-induced tissue injury. Journal of Pathology. 2012;228(1):8–19. doi: 10.1002/path.4023. [DOI] [PubMed] [Google Scholar]
- 125.Ludwig RJ, Zillikens D. Pathogenesis of epidermolysis bullosa acquisita. Dermatologic Clinics. 2011;29(3):493–501. doi: 10.1016/j.det.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 126.Chiriac MT, Roesler J, Sindrilaru A, Scharffetter-Kochanek K, Zillikens D, Sitaru C. NADPH oxidase is required for neutrophil-dependent autoantibody-induced tissue damage. Journal of Pathology. 2007;212(1):56–65. doi: 10.1002/path.2157. [DOI] [PubMed] [Google Scholar]
- 127.Hirose M, Brandolini L, Zimmer D, et al. The allosteric CXCR1/2 inhibitor DF2156A improves experimental epidermolysis bullosa acquisita. Journal of Genetic Syndromes & Gene Therapy. 2013 [Google Scholar]
- 128.Samavedam U, Mueller S, Recke A, Schmidt E, Zillikens D, Ludwig RJ. A crucial role of granulocyte-macrophage colony-stimulating factor in the pathogenesis of experimental epidermolysis bullosa acquisita. Experimental Dermatology. 2012;21(article e11) [Google Scholar]
- 129.Mihai S, Chiriac MT, Takahashi K, et al. The alternative pathway of complement activation is critical for blister induction in experimental epidermolysis bullosa acquisita. Journal of Immunology. 2007;178(10):6514–6521. doi: 10.4049/jimmunol.178.10.6514. [DOI] [PubMed] [Google Scholar]
- 130.Karsten CM, Pandey MK, Figge J, et al. Galactosylated IgG1 links FcγRIIB and Dectin-1 to blockcomplement-mediated inflammation. Nature Medicine. 2012;18(9):1401–1406. doi: 10.1038/nm.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hellberg L, Holdorf K, Hänsel M, et al. Methylprednisolone blocks autoantibody-induced tissue damage through inhibition of neutrophil activation. Journal of Investigative Dermatology. 2013 doi: 10.1038/jid.2013.91. [DOI] [PubMed] [Google Scholar]
- 132.Shimanovich I, Mihai S, Oostingh GJ, et al. Granulocyte-derived elastase and gelatinase B are required for dermal-epidermal separation induced by autoantibodies from patients with epidermolysis bullosa acquisita and bullous pemphigoid. Journal of Pathology. 2004;204(5):519–527. doi: 10.1002/path.1674. [DOI] [PubMed] [Google Scholar]
- 133.Kopecki Z, Arkell RM, Strudwick XL, et al. Overexpression of the Flii gene increases dermal-epidermal blistering in an autoimmune ColVII mouse model of epidermolysis bullosa acquisita. Journal of Pathology. 2011;225(3):401–413. doi: 10.1002/path.2973. [DOI] [PubMed] [Google Scholar]
- 134.Kopecki Z, Ruzehaji N, Turner C, et al. Topically applied Flightless I neutralising antibodies improve healing of blistered skin in a murine model of epidermolysis bullosa acquisita. Journal of Investigative Dermatology. 2013;133(4):1008–1016. doi: 10.1038/jid.2012.457. [DOI] [PubMed] [Google Scholar]
- 135.Whetton AD, Dexter TM. Myeloid haemopoietic growth factors. Biochimica et Biophysica Acta. 1989;989(2):111–132. doi: 10.1016/0304-419x(89)90038-3. [DOI] [PubMed] [Google Scholar]
- 136.Griffin JD, Cannistra SA, Sullivan R, Demetri GD, Ernst TJ, Kanakura Y. The biology of GM-CSF: regulation of production and interaction with its receptor. International Journal of Cell Cloning. 1990;8(1):35–45. doi: 10.1002/stem.5530080705. [DOI] [PubMed] [Google Scholar]
- 137.Korzenik JR, Dieckgraefe BK, Valentine JF, Hausman DF, Gilbert MJ. Sargramostim for active Crohn’s disease. New England Journal of Medicine. 2005;352(21):2193–2201. doi: 10.1056/NEJMoa041109. [DOI] [PubMed] [Google Scholar]
- 138.Sainathan SK, Hanna EM, Gong Q, et al. Granulocyte macrophage colony-stimulating factor ameliorates DSS-induced experimental colitis. Inflammatory Bowel Diseases. 2008;14(1):88–99. doi: 10.1002/ibd.20279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kondo S, Pastore S, Shivji GM, McKenzie RC, Sauder DN. Characterization of epidermal cytokine profiles in sensitization and elicitation phases of allergic contact dermatitis as well as irritant contact dermatitis in mouse skin. Lymphokine and Cytokine Research. 1994;13(6):367–375. [PubMed] [Google Scholar]
- 140.Gillessen S, Mach N, Small C, Mihm M, Dranoff G. Overlapping roles for granulocyte-macrophage colony-stimulating factor and interleukin-3 in eosinophil homeostasis and contact hypersensitivity. Blood. 2001;97(4):922–928. doi: 10.1182/blood.v97.4.922. [DOI] [PubMed] [Google Scholar]
- 141.Campbell IK, Bendele A, Smith DA, Hamilton JA. Granulocyte-macrophage colony stimulating factor exacerbates collagen induced arthritis in mice. Annals of the Rheumatic Diseases. 1997;56(6):364–368. doi: 10.1136/ard.56.6.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cook AD, Braine EL, Campbell IK, Rich MJ, Hamilton JA. Blockade of collagen-induced arthritis post-onset by antibody to granulocyte-macrophage colony-stimulating factor (GM-CSF): requirement for GM-CSF in the effector phase of disease. Arthritis Research. 2001;3(5):293–298. doi: 10.1186/ar318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Plater-Zyberk C, Joosten LAB, Helsen MMA, Hepp J, Baeuerle PA, van den Berg WB. GM-CSF neutralisation suppresses inflammation and protects cartilage in acute streptococcal cell wall arthritis of mice. Annals of the Rheumatic Diseases. 2007;66(4):452–457. doi: 10.1136/ard.2006.057182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Codarri L, Gyülvészii G, Tosevski V, et al. RORγ3t drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nature Immunology. 2011;12(6):560–567. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- 145.Schön M, Denzer D, Kubitza RC, Ruzicka T, Schön MP. Critical role of neutrophils for the generation of psoriasiform skin lesions in flaky skin mice. Journal of Investigative Dermatology. 2000;114(5):976–983. doi: 10.1046/j.1523-1747.2000.00953.x. [DOI] [PubMed] [Google Scholar]
- 146.Kitching AR, Huang XR, Turner AL, Tipping PG, Dunn AR, Holdsworth SR. The requirement for granulocyte-macrophage colony-stimulating factor and granulocyte colony-stimulating factor in leukocyte-mediated immune glomerular injury. Journal of the American Society of Nephrology. 2002;13(2):350–358. doi: 10.1681/ASN.V132350. [DOI] [PubMed] [Google Scholar]
- 147.Houssiau FA, Devogelaer J-P, van Damme J, Nagant de Deuxchaisnes C, van Snick J. Interleukin-6 in synovial fluid and serum of patients with rheumatoid arthritis and other inflammatory arthritides. Arthritis & Rheumatism. 1988;31(6):784–788. doi: 10.1002/art.1780310614. [DOI] [PubMed] [Google Scholar]
- 148.Mahida YR, Kurlac L, Gallagher A, Hawkey CJ. High circulating concentrations of interleukin-6 in active Crohn’s disease but not ulcerative colitis. Gut. 1991;32(12):1531–1534. doi: 10.1136/gut.32.12.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yokoyama A, Kohno N, Fujino S, et al. Circulating interleukin-6 levels in patients with bronchial asthma. American Journal of Respiratory and Critical Care Medicine. 1995;151(5):1354–1358. doi: 10.1164/ajrccm.151.5.7735584. [DOI] [PubMed] [Google Scholar]
- 150.Grossman RM, Krueger J, Yourish D, et al. Interleukin 6 is expressed in high levels of psoriatic skin and stimulates proliferation of cultured human keratinocytes. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(16):6367–6371. doi: 10.1073/pnas.86.16.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Eriksson U, Kurrer MO, Schmitz N, et al. Interleukin-6-deficient mice resist development of autoimmune myocarditis associated with impaired upregulation of complement C3. Circulation. 2003;107(2):320–325. doi: 10.1161/01.cir.0000043802.38699.66. [DOI] [PubMed] [Google Scholar]
- 152.Ohshima S, Saeki Y, Mima T, et al. Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(14):8222–8226. doi: 10.1073/pnas.95.14.8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Alonzi T, Fattori E, Lazzaro D, et al. Interleukin 6 is required for the development of collagen-induced arthritis. Journal of Experimental Medicine. 1998;187(4):461–468. doi: 10.1084/jem.187.4.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Samoilova EB, Horton JL, Hilliard B, Liu T-ST, Chen Y. IL-6-deficient mice are resistant to experimental autoimmune encephalomyelitis: roles of IL-6 in the activation and differentiation of autoreactive T cells. Journal of Immunology. 1998;161(12):6480–6486. [PubMed] [Google Scholar]
- 155.Maini RN, Taylor PC, Szechinski J, et al. Double-blind randomized controlled clinical trial of the interleukin-6 receptor antagonist, tocilizumab, in European patients with rheumatoid arthritis who had an incomplete response to methotrexate. Arthritis & Rheumatism. 2006;54(9):2817–2829. doi: 10.1002/art.22033. [DOI] [PubMed] [Google Scholar]
- 156.Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochimica et Biophysica Acta. 2011;1813(5):878–888. doi: 10.1016/j.bbamcr.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 157.Nowell MA, Richards PJ, Horiuchi S, et al. Soluble IL-6 receptor governs IL-6 activity in experimental arthritis: blockade of arthritis severity by soluble Glycoprotein 130. Journal of Immunology. 2003;171(6):3202–3209. doi: 10.4049/jimmunol.171.6.3202. [DOI] [PubMed] [Google Scholar]
- 158.Nowell MA, Williams AS, Carty SA, et al. Therapeutic targeting of IL-6 trans signaling counteracts STAT3 control of experimental inflammatory arthritis. Journal of Immunology. 2009;182(1):613–622. doi: 10.4049/jimmunol.182.1.613. [DOI] [PubMed] [Google Scholar]
- 159.Waetzig GH, Rose-John S. Hitting a complex target: an update on interleukin-6 trans-signalling. Expert Opinion on Therapeutic Targets. 2012;16(2):225–236. doi: 10.1517/14728222.2012.660307. [DOI] [PubMed] [Google Scholar]
- 160.Poffenberger MC, Straka N, El Warry N, Fang D, Shanina I, Horwitz MS. Lack of IL-6 during coxsackievirus infection heightens the early immune response resulting in increased severity of chronic autoimmune myocarditis. PLoS ONE. 2009;4(7, article e6207) doi: 10.1371/journal.pone.0006207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Xing Z, Gauldie J, Cox G, et al. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. Journal of Clinical Investigation. 1998;101(2):311–320. doi: 10.1172/JCI1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Grivennikov S, Karin E, Terzic J, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15(2):103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jin JO, Han X, Yu Q. Interleukin-6 induces the generation of IL-10-producing Tr1 cells and suppresses autoimmune tissue inflammation. Journal of Autoimmunity. 2013;40:28–44. doi: 10.1016/j.jaut.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Klos A, Tenner AJ, Johswich K-O, Ager RR, Reis ES, Köhl J. The role of the anaphylatoxins in health and disease. Molecular Immunology. 2009;46(14):2753–2766. doi: 10.1016/j.molimm.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wang Y, Kristan J, Hao L, Lenkoski CS, Shen Y, Matis LA. A role for complement in antibody-mediated inflammation: C5-deficient DBA/1 mice are resistant to collagen-induced arthritis. Journal of Immunology. 2000;164(8):4340–4347. doi: 10.4049/jimmunol.164.8.4340. [DOI] [PubMed] [Google Scholar]
- 166.Zhang X, Kohl J. A complex role for complement in allergic asthma. Expert Review of Clinical Immunology. 2010;6(2):269–277. doi: 10.1586/eci.09.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Liu Z, Giudice GJ, Swartz SJ, et al. The role of complement in experimental bullous pemphigoid. Journal of Clinical Investigation. 1995;95(4):1539–1544. doi: 10.1172/JCI117826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Botto M, Dell’Agnola C, Bygrave AE, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nature Genetics. 1998;19(1):56–59. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- 169.Takahashi K, Gordon J, Liu H, et al. Lack of mannose-binding lectin-A enhances survival in a mouse model of acute septic peritonitis. Microbes and Infection. 2002;4(8):773–784. doi: 10.1016/s1286-4579(02)01597-6. [DOI] [PubMed] [Google Scholar]
- 170.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nature Reviews Immunology. 2013;13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 171.Mora JR, von Andrian UH. T-cell homing specificity and plasticity: new concepts and future challenges. Trends in Immunology. 2006;27(5):235–243. doi: 10.1016/j.it.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 172.Radeke HH, Ludwig RJ, Boehnche W-H. Experimental approaches to lymphocyte migration in dermatology in vitro and in vivo . Experimental Dermatology. 2005;14(9):641–666. doi: 10.1111/j.0906-6705.2005.00350.x. [DOI] [PubMed] [Google Scholar]
- 173.Schön MP, Ludwig RJ. Lymphocyte trafficking to inflamed skin—molecular mechanisms and implications for therapeutic target molecules. Expert Opinion on Therapeutic Targets. 2005;9(2):225–243. doi: 10.1517/14728222.9.2.225. [DOI] [PubMed] [Google Scholar]
- 174.Ludwig RJ, Zollner TM, Santoso S, et al. Junctional adhesion molecules (JAM)-B and -C contribute to leukocyte extravasation to the skin and mediate cutaneous inflammation. Journal of Investigative Dermatology. 2005;125(5):969–976. doi: 10.1111/j.0022-202X.2005.23912.x. [DOI] [PubMed] [Google Scholar]
- 175.Ludwig RJ, Hardt K, Hatting M, et al. Junctional adhesion molecule (JAM)-B supports lymphocyte rolling and adhesion through interaction with α4β1 integrin. Immunology. 2009;128(2):196–205. doi: 10.1111/j.1365-2567.2009.03100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Allhorn M, Collin M. Sugar-free antibodies—the bacterial solution to autoimmunity? Annals of the New York Academy of Sciences. 2009;1173:664–669. doi: 10.1111/j.1749-6632.2009.04739.x. [DOI] [PubMed] [Google Scholar]
- 177.Nimmerjahn F, Ravetch JV. Fcγ receptors as regulators of immune responses. Nature Reviews Immunology. 2008;8(1):34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 178.Yuasa T, Kubo S, Yoshino T, et al. Deletion of Fcγ receptor IIB renders H-2b mice susceptible to collagen-induced arthritis. Journal of Experimental Medicine. 1999;189(1):187–194. doi: 10.1084/jem.189.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Nakamura A, Yuasa T, Ujike A, et al. Fcγ receptor IIB-deficient mice develop goodpasture’s syndrome upon immunization with type IV collagen: a novel murine model for autoimmune glomerular basement membrane disease. Journal of Experimental Medicine. 2000;191(5):899–905. doi: 10.1084/jem.191.5.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhao M, Trimbeger ME, Li N, Diaz LA, Shapiro SD, Liu Z. Role of FcRs in animal model of autoimmune bullous pemphigoid. Journal of Immunology. 2006;177(5):3398–3405. doi: 10.4049/jimmunol.177.5.3398. [DOI] [PubMed] [Google Scholar]
- 181.Shushakova N, Skokowa J, Schulman J, et al. C5a anaphylatoxin is a major regulator of activating versus inhibitory FcγRs in immune complex-induced lung disease. Journal of Clinical Investigation. 2002;110(12):1823–1830. doi: 10.1172/JCI200216577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Godau J, Heller T, Hawlisch H, et al. C5a initiates the inflammatory cascade in immune complex peritonitis. Journal of Immunology. 2004;173(5):3437–3445. doi: 10.4049/jimmunol.173.5.3437. [DOI] [PubMed] [Google Scholar]
- 183.Kiefer F, Brumell J, Al-Alawi N, et al. The Syk protein tyrosine kinase is essential for Fcγ/receptor signaling in macrophages and neutrophils. Molecular and Cellular Biology. 1998;18(7):4209–4220. doi: 10.1128/mcb.18.7.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tan SL, Liao C, Lucas MC, Stevenson C, Demartino JA. Targeting the SYK-BTK axis for the treatment of immunological and hematological disorders: recent progress and therapeutic perspectives. Pharmacology & Therapeutics. 2013;138:294–309. doi: 10.1016/j.pharmthera.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 185.Rider LG, Hirasawa N, Santini F, Beaven MA. Activation of the mitogen-activated protein kinase cascade is suppressed by low concentrations of dexamethasone in mast cells. Journal of Immunology. 1996;157(6):2374–2380. [PubMed] [Google Scholar]
- 186.Chen J, Tang H, Hay N, Xu J, Ye RD. Akt isoforms differentially regulate neutrophil functions. Blood. 2010;115(21):4237–4246. doi: 10.1182/blood-2009-11-255323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes and Infection. 2003;5(14):1317–1327. doi: 10.1016/j.micinf.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 188.Leiding JW, Holland SM. Chronic Granulomatous Disease. GeneReviews, 1993. [PubMed]
- 189.Cowin AJ, Adams DH, Strudwick XL, et al. Flightless I deficiency enhances wound repair by increasing cell migration and proliferation. Journal of Pathology. 2007;211(5):572–581. doi: 10.1002/path.2143. [DOI] [PubMed] [Google Scholar]
- 190.Vodegel RM, Jonkman MF, Pas HH, De Jong MCJM. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. British Journal of Dermatology. 2004;151(1):112–118. doi: 10.1111/j.1365-2133.2004.06006.x. [DOI] [PubMed] [Google Scholar]
- 191.Terra JB, Meijer JM, Jonkman MF, Diercks GF. The n- versus u-serration is a learnable criterion to differentiate pemphigoid from epidermolysis bullosa acquisita in direct immunofluorescence serration pattern analysis. British Journal of Dermatology. 2013 doi: 10.1111/bjd.12308. [DOI] [PubMed] [Google Scholar]
- 192.Terra JB, Pas HH, Hertl M, Dikkers FG, Kamminga N, Jonkman MF. Immunofluorescence serration pattern analysis as a diagnostic criterion in antilaminin-332 mucous membrane pemphigoid: immunopathological findings and clinical experience in 10 Dutch patients. British Journal of Dermatology. 2011;165(4):815–822. doi: 10.1111/j.1365-2133.2011.10474.x. [DOI] [PubMed] [Google Scholar]
- 193.Chen M, Chan LS, Cai X, O’Toole EA, Sample JC, Woodley DT. Development of an ELISA for rapid detection of anti-type VII collagen autoantibodies in epidermolysis bullosa acquisita. Journal of Investigative Dermatology. 1997;108(1):68–72. doi: 10.1111/1523-1747.ep12285634. [DOI] [PubMed] [Google Scholar]
- 194.Saleh MA, Ishii K, Kim Y-J, et al. Development of NC1 and NC2 domains of Type VII collagen ELISA for the diagnosis and analysis of the time course of epidermolysis bullosa acquisita patients. Journal of Dermatological Science. 2011;62(3):169–175. doi: 10.1016/j.jdermsci.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 195.Komorowski L, Müller R, Vorobyev A, et al. Sensitive and specific assays for routine serological diagnosis of epidermolysis bullosa acquisita. Journal of the American Academy of Dermatology. 2012;68:3, e89–e95. doi: 10.1016/j.jaad.2011.12.032. [DOI] [PubMed] [Google Scholar]
- 196.Nieboer C, Boorsma DM, Woerdeman MJ, Kalsbeek GL. Epidermolysis bullosa acquisita. Immunofluorescence, electron microscopic and immuno-electron microscopic studies in four patients. British Journal of Dermatology. 1980;102(4):383–392. doi: 10.1111/j.1365-2133.1980.tb06550.x. [DOI] [PubMed] [Google Scholar]
- 197.Yaoita H, Briggaman RA, Lawley TJ. Epidermolysis bullosa acquisita: ultrastructural and immunological studies. Journal of Investigative Dermatology. 1981;76(4):288–292. doi: 10.1111/1523-1747.ep12526124. [DOI] [PubMed] [Google Scholar]
- 198.Caux F. Epidermolysis bullosa acquisita. Presse Medicale. 2010;39(10):1081–1088. doi: 10.1016/j.lpm.2010.04.021. [DOI] [PubMed] [Google Scholar]
- 199.de Jong MCJM, Bruins S, Heeres K, et al. Bullous pemphigoid and epidermolysis bullosa acquisita: differentiation by fluorescence overlay antigen mapping. Archives of Dermatology. 1996;132(2):151–157. doi: 10.1001/archderm.132.2.151. [DOI] [PubMed] [Google Scholar]
- 200.Kazama T, Yamamoto Y, Hashimoto T, Komai A, Ito M. Application of confocal laser scanning microscopy to differential diagnosis of bullous pemphigoid and epidermolysis bullosa acquisita. British Journal of Dermatology. 1998;138(4):593–601. doi: 10.1046/j.1365-2133.1998.02167.x. [DOI] [PubMed] [Google Scholar]
- 201.Hundorfean G, Neurath MF, Sitaru C. Autoimmunity against type VII collagen in inflammatory bowel disease. Journal of Cellular and Molecular Medicine. 2010;14(10):2393–2403. doi: 10.1111/j.1582-4934.2009.00959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clinical and Experimental Dermatology. 2013;38:225–230. doi: 10.1111/ced.12114. [DOI] [PubMed] [Google Scholar]
- 203.Engineer L, Ahmed AR. Emerging treatment for epidermolysis bullosa acquisita. Journal of the American Academy of Dermatology. 2001;44(5):818–828. doi: 10.1067/mjd.2001.113693. [DOI] [PubMed] [Google Scholar]
- 204.Kim JH, Kim SC. Epidermolysis bullosa acquisita. Journal of the European Academy of Dermatology and Venereology. 2013 doi: 10.1111/jdv.12096. [DOI] [PubMed] [Google Scholar]
- 205.Connolly SM, Sander HM. Treatment of epidermolysis bullosa acquisita with cyclosporine. Journal of the American Academy of Dermatology. 1987;16(4):p. 890. doi: 10.1016/s0190-9622(87)80239-6. [DOI] [PubMed] [Google Scholar]
- 206.Khatri ML, Benghazeil M, Shafi M. Epidermolysis bullosa acquisita responsive to cyclosporin therapy. Journal of the European Academy of Dermatology and Venereology. 2001;15(2):182–184. doi: 10.1046/j.1468-3083.2001.00174-2.x. [DOI] [PubMed] [Google Scholar]
- 207.Maize JC, Jr., Cohen JB. Cyclosporine controls epidermolysis bullosa acquisita co-occuring with acquired factor VIII deficiency. International Journal of Dermatology. 2005;44(8):692–694. doi: 10.1111/j.1365-4632.2004.02245.x. [DOI] [PubMed] [Google Scholar]
- 208.Megahed M, Scharfletter-Kochanek K. Epidermolysis bullosa acquisita—successful treatment with colchicine. Archives of Dermatological Research. 1994;286(1):35–40. doi: 10.1007/BF00375841. [DOI] [PubMed] [Google Scholar]
- 209.Cunningham BB, Kirchmann TTT, Woodley D. Colchicine for epidermolysis bullosa acquisita. Journal of the American Academy of Dermatology. 1996;34(5):781–784. doi: 10.1016/s0190-9622(96)90013-4. [DOI] [PubMed] [Google Scholar]
- 210.Arora KP, Sachdeva B, Singh N, Bhattacharya SN. Remission of recalcitrant epidermolysis bullosa acquisita (EBA) with colchicine monotherapy. Journal of Dermatology. 2005;32(2):114–119. doi: 10.1111/j.1346-8138.2005.tb00727.x. [DOI] [PubMed] [Google Scholar]
- 211.Tanaka N, Dainichi T, Ohyama B, et al. A case of epidermolysis bullosa acquisita with clinical features of Brunsting-Perry pemphigoid showing an excellent response to colchicine. Journal of the American Academy of Dermatology. 2009;61(4):715–719. doi: 10.1016/j.jaad.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 212.Kiniwa Y, Ashida A, Ohashi A, et al. A case of epidermolysis bullosa acquisita associated with laryngeal stenosis. Acta Dermato-Venereologica. 2012;92(1):93–94. doi: 10.2340/00015555-1163. [DOI] [PubMed] [Google Scholar]
- 213.Driessen G, van der Burg M. Educational paper: primary antibody deficiencies. European Journal of Pediatrics. 2011;170(6):693–702. doi: 10.1007/s00431-011-1474-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Cines DB, Blanchette VS. Medical progress: immune thrombocytopenic purpura. New England Journal of Medicine. 2002;346(13):995–1008. doi: 10.1056/NEJMra010501. [DOI] [PubMed] [Google Scholar]
- 215.Yuki N. Infectious origins of, and molecular mimicry in, Guillain-Barré and Fisher syndromes. Lancet Infectious Diseases. 2001;1(1):29–37. doi: 10.1016/S1473-3099(01)00019-6. [DOI] [PubMed] [Google Scholar]
- 216.Fergusson D, Hutton B, Sharma M, et al. Use of intravenous immunoglobulin for treatment of neurologic conditions: a systematic review. Transfusion. 2005;45(10):1640–1657. doi: 10.1111/j.1537-2995.2005.00581.x. [DOI] [PubMed] [Google Scholar]
- 217.Kobayashi T, Saji T, Otani T, et al. Efficacy of immunoglobulin plus prednisolone for prevention of coronary artery abnormalities in severe Kawasaki disease (RAISE study): a randomised, open-label, blinded-endpoints trial. The Lancet. 2012;379(9826):1613–1620. doi: 10.1016/S0140-6736(11)61930-2. [DOI] [PubMed] [Google Scholar]
- 218.Amagai M, Ikeda S, Shimizu H, et al. A randomized double-blind trial of intravenous immunoglobulin for pemphigus. Journal of the American Academy of Dermatology. 2009;60(4):595–603. doi: 10.1016/j.jaad.2008.09.052. [DOI] [PubMed] [Google Scholar]
- 219.Ishii N, Hashimoto T, Zillikens D, Ludwig RJ. High-dose intravenous immunoglobulin (IVIG) therapy in autoimmune skin blistering diseases. Clinical Reviews in Allergy and Immunology. 2010;38(2-3):186–195. doi: 10.1007/s12016-009-8153-y. [DOI] [PubMed] [Google Scholar]
- 220.Ahmed AR, Gürcan HM. Treatment of epidermolysis bullosa acquisita with intravenous immunoglobulin in patients non-responsive to conventional therapy: clinical outcome and post-treatment long-term follow-up. Journal of the European Academy of Dermatology and Venereology. 2012;26:1074–1083. doi: 10.1111/j.1468-3083.2011.04205.x. [DOI] [PubMed] [Google Scholar]
- 221.Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. Journal of Cutaneous Medicine and Surgery. 2001;5(5):397–399. doi: 10.1007/s10227-001-0008-y. [DOI] [PubMed] [Google Scholar]
- 222.Kirtschig G, Murrell D, Wojnarowska F, Khumalo N. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database of Systematic Reviews. 2003;(1) doi: 10.1002/14651858.CD004056.CD004056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Joly P, Mouquet H, Roujeau J-C, et al. A single cycle of rituximab for the treatment of severe pemphigus. New England Journal of Medicine. 2007;357(6):545–552. doi: 10.1056/NEJMoa067752. [DOI] [PubMed] [Google Scholar]
- 224.Kasperkiewicz M, Shimanovich I, Ludwig RJ, Rose C, Zillikens D, Schmidt E. Rituximab for treatment-refractory pemphigus and pemphigoid: a case series of 17 patients. Journal of the American Academy of Dermatology. 2011;65(3):552–558. doi: 10.1016/j.jaad.2010.07.032. [DOI] [PubMed] [Google Scholar]
- 225.McKinley SK, Huang JT, Tan J, Kroshinsky D, Gellis S. A case of recalcitrant epidermolysis bullosa acquisita responsive to rituximab therapy. Pediatric Dermatology. 2012 doi: 10.1111/pde.12006. [DOI] [PubMed] [Google Scholar]
- 226.Kim JH, Lee SE, Kim S-C. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. Journal of Dermatology. 2012;39(5):477–479. doi: 10.1111/j.1346-8138.2011.01360.x. [DOI] [PubMed] [Google Scholar]
- 227.Meissner C, Hoefeld-Fegeler M, Vetter R, et al. Severe acral contractures and nail loss in a patient with mechano-bullous Epidermolysis bullosa acquisita. European Journal of Dermatology. 2010;20(4):543–544. doi: 10.1684/ejd.2010.1002. [DOI] [PubMed] [Google Scholar]
- 228.Kubisch I, Diessenbacher P, Schmidt E, Gollnick H, Leverkus M. Premonitory epidermolysis bullosa acquisita mimicking eyelid dermatitis: successful treatment with rituximab and protein a immunoapheresis. American Journal of Clinical Dermatology. 2010;11(4):289–293. doi: 10.2165/11533210-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 229.Saha M, Cutler T, Bhogal B, Black MM, Groves RW. Correspondence: refractory epidermolysis bullosa acquisita: successful treatment with rituximab. Clinical and Experimental Dermatology. 2009;34(8):e979–e980. doi: 10.1111/j.1365-2230.2009.03608.x. [DOI] [PubMed] [Google Scholar]
- 230.Cavailhes A, Balme B, Gilbert D, Skowron F. Successful use of combined corticosteroids and rituximab in the treatment of recalcitrant epidermolysis bullosa acquisita. Annales de Dermatologie et de Venereologie. 2009;136(11):795–799. doi: 10.1016/j.annder.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 231.Mercader P, Rodenas JM, Peña A, Mascaro JM., Jr. Fatal Pseudomona pneumonia following rituximab therapy in a patient with epidermolysis bullosa acquisita. Journal of the European Academy of Dermatology and Venereology. 2007;21(8):1141–1142. doi: 10.1111/j.1468-3083.2006.02127.x. [DOI] [PubMed] [Google Scholar]
- 232.Sadler E, Schafleitner B, Lanschuetzer C, et al. Treatment-resistant classical epidermolysis bullosa acquisita responding to rituximab. British Journal of Dermatology. 2007;157(2):417–419. doi: 10.1111/j.1365-2133.2007.08048.x. [DOI] [PubMed] [Google Scholar]
- 233.Niedermeier A, Eming R, Pfütze M, et al. Clinical response of severe mechanobullous epidermolysis bullosa acquisita to combined treatment with immunoadsorption and rituximab (anti-CD20 monoclonal antibodies) Archives of Dermatology. 2007;143(2):192–198. doi: 10.1001/archderm.143.2.192. [DOI] [PubMed] [Google Scholar]
- 234.Crichlow SM, Mortimer NJ, Harman KE. A successful therapeutic trial of rituximab in the treatment of a patient with recalcitrant, high-titre epidermolysis bullosa acquisita. British Journal of Dermatology. 2007;156(1):194–196. doi: 10.1111/j.1365-2133.2006.07596.x. [DOI] [PubMed] [Google Scholar]
- 235.Schmidt E, Benoit S, Bröcker E-B, Zillikens D, Goebeler M. Successful adjuvant treatment of recalcitrant epidermolysis bullosa acquisita with anti-CD20 antibody rituximab. Archives of Dermatology. 2006;142(2):147–150. doi: 10.1001/archderm.142.2.147. [DOI] [PubMed] [Google Scholar]
- 236.Schmidt E, Zillikens D. Immunoadsorption in dermatology. Archives of Dermatological Research. 2010;302(4):241–253. doi: 10.1007/s00403-009-1024-9. [DOI] [PubMed] [Google Scholar]
- 237.Furue M, Iwata M, Yoon H-I. Epidermolysis bullosa acquisita: clinical response to plasma exchange therapy and circulating anti-basement membrane zone antibody titer. Journal of the American Academy of Dermatology. 1986;14(5):873–878. doi: 10.1016/s0190-9622(86)70103-5. [DOI] [PubMed] [Google Scholar]
- 238.Miller JL, Stricklin GP, Fine JD, King LE, del Carmen Arzubiaga M, Ellis DL. Remission of severe epidermolysis bullosa acquisita induced by extracorporeal photochemotherapy. British Journal of Dermatology. 1995;133(3):467–471. doi: 10.1111/j.1365-2133.1995.tb02680.x. [DOI] [PubMed] [Google Scholar]
- 239.Gordon KB, Chan LS, Woodley DT. Treatment of refractory epidermolysis bullosa acquisita with extracorporeal photochemotherapy. British Journal of Dermatology. 1997;136(3):415–420. [PubMed] [Google Scholar]
- 240.Sanli H, Akay BN, Ayyildiz E, Anadolu R, Ilhan O. Remission of severe autoimmune bullous disorders induced by long-term extracorporeal photochemotherapy. Transfusion and Apheresis Science. 2010;43(3):353–359. doi: 10.1016/j.transci.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 241.Baroudjian B, Le Roux-Villet C, Brechignac S, et al. Long-term efficacy of extracorporeal photochemotherapy in a patient with refractory epidermolysis bullosa acquisita. European Journal of Dermatology. 2012;22(6):795–797. doi: 10.1684/ejd.2012.1840. [DOI] [PubMed] [Google Scholar]
- 242.Usmani SZ, Chiosis G. HSP90 inhibitors as therapy for multiple myeloma. Clinical Lymphoma, Myeloma and Leukemia. 2011;11(supplement 1):S77–S81. doi: 10.1016/j.clml.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 243.Pacey S, Wilson RH, Walton M, et al. A phase I study of the heat shock protein 90 inhibitor alvespimycin (17-DMAG) given intravenously to patients with advanced solid tumors. Clinical Cancer Research. 2011;17(6):1561–1570. doi: 10.1158/1078-0432.CCR-10-1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.ClinicalTrials.gov. Clinical studies using teplizumab. 2012.
- 245.ClinicalTrials.gov. Clinical studies using otelixizumab. 2012.
- 246.Kim YH, Duvic M, Obitz E, et al. Clinical efficacy of zanolimumab (HuMax-CD4): two phase 2 studies in refractory cutaneous T-cell lymphoma. Blood. 2007;109(11):4655–4662. doi: 10.1182/blood-2006-12-062877. [DOI] [PubMed] [Google Scholar]
- 247.van Oosten BW, Lai M, Hodgkinson S, et al. Treatment of multiple sclerosis with the monoclonal anti-CD4 antibody cM-T412: results of a randomized, double-blind, placebo-controlled, MR- monitored phase II trial. Neurology. 1997;49(2):351–357. doi: 10.1212/wnl.49.2.351. [DOI] [PubMed] [Google Scholar]
- 248.Stronkhorst A, Radema S, Yong S-L, et al. CD4 antibody treatment in patients with active Crohn’s disease: a phase 1 dose finding study. Gut. 1997;40(3):320–327. doi: 10.1136/gut.40.3.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Meyersburg D, Schmidt E, Kasperkiewicz M, Zillikens D. Immunoadsorption in dermatology. Therapeutic Apheresis and Dialysis. 2012;16:311–320. doi: 10.1111/j.1744-9987.2012.01075.x. [DOI] [PubMed] [Google Scholar]
- 250. IA-PEM., 2013, https://drks-neu.uniklinik-freiburg.de/drks_web/
- 251.Nagatomo Y, Baba A, Ito H, et al. Specific immunoadsorption therapy using a tryptophan column in patients with refractory heart failure due to dilated cardiomyopathy. Journal of Clinical Apheresis. 2011;26(1):1–8. doi: 10.1002/jca.20268. [DOI] [PubMed] [Google Scholar]
- 252.Sondermann P, Jacob U. Human Fcγ receptor IIb expressed in Escherichia coli reveals IgG binding capability. Biological Chemistry. 1999;380(6):717–721. doi: 10.1515/BC.1999.090. [DOI] [PubMed] [Google Scholar]
- 253.Clinical trials using SM101. 2012, http://clinicaltrials.gov/
- 254.Werwitzke S, Trick D, Sondermann P, et al. Treatment of lupus-prone NZB/NZW F1 mice with recombinant soluble Fcγ receptor II (CD32) Annals of the Rheumatic Diseases. 2008;67(2):154–161. doi: 10.1136/ard.2006.068981. [DOI] [PubMed] [Google Scholar]
- 255.Magnusson SE, Andrén M, Nilsson KE, Sondermann P, Jacob U, Kleinau S. Amelioration of collagen-induced arthritis by human recombinant soluble FcγRIIb. Clinical Immunology. 2008;127(2):225–233. doi: 10.1016/j.clim.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 256.Tubridy N, Behan PO, Capildeo R, et al. The effect of anti-α4 integrin antibody on brain lesion activity in MS. Neurology. 1999;53(3):466–472. doi: 10.1212/wnl.53.3.466. [DOI] [PubMed] [Google Scholar]
- 257.Langer-Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. New England Journal of Medicine. 2005;353(4):375–381. doi: 10.1056/NEJMoa051847. [DOI] [PubMed] [Google Scholar]
- 258.Reichert JM. Marketed therapeutic antibodies compendium. MAbs. 2012;4:413–415. doi: 10.4161/mabs.19931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Friedrich M, Bock D, Philipp S, et al. Pan-selectin antagonism improves psoriasis manifestation in mice and man. Archives of Dermatological Research. 2006;297(8):345–351. doi: 10.1007/s00403-005-0626-0. [DOI] [PubMed] [Google Scholar]
- 260.Bhushan M, Bleiker TO, Ballsdon AE, et al. Anti-e-selectin is ineffective in the treatment of psoriasis: a randomized trial. British Journal of Dermatology. 2002;146(5):824–831. doi: 10.1046/j.1365-2133.2002.04743.x. [DOI] [PubMed] [Google Scholar]
- 261.Ishida T, Joh T, Uike N, et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study. Journal of Clinical Oncology. 2012;30(8):837–842. doi: 10.1200/JCO.2011.37.3472. [DOI] [PubMed] [Google Scholar]
- 262.Mirshahpanah P, Li Y-YY, Burkhardt N, Asadullah K, Zollner TM. CCR4 and CCR10 ligands play additive roles in mouse contact hypersensitivity. Experimental Dermatology. 2008;17(1):30–34. doi: 10.1111/j.1600-0625.2007.00630.x. [DOI] [PubMed] [Google Scholar]
- 263.Ludwig RJ, Alban S, Boehncke W-H. Structural requirements of heparin and related molecules to exert a multitude of anti-inflammatory activities. Mini-Reviews in Medicinal Chemistry. 2006;6(9):1009–1023. doi: 10.2174/138955706778195180. [DOI] [PubMed] [Google Scholar]
- 264.Fritzsche J, Alban S, Ludwig RJ, et al. The influence of various structural parameters of semisynthetic sulfated polysaccharides on the P-selectin inhibitory capacity. Biochemical Pharmacology. 2006;72(4):474–485. doi: 10.1016/j.bcp.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 265.Becker M, Franz G, Alban S. Inhibition of PMN-elastase activity by semisynthetic glucan sulfates. Thrombosis and Haemostasis. 2003;89(5):915–925. [PubMed] [Google Scholar]
- 266.Alban S, Ludwig RJ, Bendas G, et al. PS3, A semisynthetic Β-1,3-glucan sulfate, diminishes contact hypersensitivity responses through inhibition of L- and P-selectin functions. Journal of Investigative Dermatology. 2009;129(5):1192–1202. doi: 10.1038/jid.2008.358. [DOI] [PubMed] [Google Scholar]
- 267.Citro A, Cantarelli E, Maffi P, et al. CXCR1/2 inhibition enhances pancreatic islet survival after transplantation. The Journal of Clinical Investigation. 2012;122:3647–3651. doi: 10.1172/JCI63089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Schneider SW, Gaubitz M, Luger TA, Bonsmann G. Prompt response of refractory Schnitzler syndrome to treatment with anakinra. Journal of the American Academy of Dermatology. 2007;56(5):S120–S122. doi: 10.1016/j.jaad.2006.05.057. [DOI] [PubMed] [Google Scholar]