

Abstract
The commonest pediatric brain tumors are low-grade gliomas (LGGs). We utilized whole genome sequencing to discover multiple novel genetic alterations involving BRAF, RAF1, FGFR1, MYB, MYBL1 and genes with histone-related functions, including H3F3A and ATRX, in 39 LGGs and low-grade glioneuronal tumors (LGGNTs). Only a single non-silent somatic alteration was detected in 24/39 (62%) tumors. Intragenic duplications of the FGFR1 tyrosine kinase domain (TKD) and rearrangements of MYB were recurrent and mutually exclusive in 53% of grade II diffuse LGGs. Transplantation of Trp53-null neonatal astrocytes containing TKD-duplicated FGFR1 into brains of nude mice generated high-grade astrocytomas with short latency and 100% penetrance. TKD-duplicated FGFR1 induced FGFR1 autophosphorylation and upregulation of the MAPK/ERK and PI3K pathways, which could be blocked by specific inhibitors. Focusing on the therapeutically challenging diffuse LGGs, our study of 151 tumors has discovered genetic alterations and potential therapeutic targets across the entire range of pediatric LGGs/LGGNTs.
Low-grade gliomas (LGGs) arise most frequently in children and young adults and are the commonest pediatric central nervous system (CNS) neoplasms 1-3. Although LGGs grow slowly, those that cannot be surgically resected cause considerable morbidity and premature death. Current adjuvant therapies with irradiation and pharmaceuticals extend survival but contribute to morbidity; thus, there is an urgent need for targeted therapeutics in patients with inoperable disease 1,3-10.
Studies of pediatric LGGs and related low-grade glioneuronal tumors (LGGNTs) have implicated abnormalities of the MAPK/ERK pathway in their oncogenesis 11-14, but detailed knowledge of driver mutations in these diverse tumors is lacking. Up to 15% of children with the hereditary tumor syndrome neurofibromatosis type 1 (NF-1) develop a pilocytic astrocytoma (PA), the commonest type of LGG 15,16. Neurofibromin 1, protein product of the NF1 tumor-suppressor gene, is a negative regulator of RAS in the MAPK/ERK pathway 17. NF-1-related LGGs account for less than 15% of pediatric LGGs; however, almost all sporadic cerebellar PAs demonstrate MAPK/ERK pathway activation secondary to a KIAA1549-BRAF fusion gene, in which BRAF lacks its auto-inhibitory domain and becomes constitutively active 11,12. Other mechanisms activating the MAPK/ERK pathway in LGGs are comparatively rare and include RAF1 fusion genes and BRAF:p.V600E or KRAS mutations, though the BRAF:p.V600E mutation is present in a proportion of LGGNTs 11,12,18-21. While nearly all World Health Organization (WHO) grade I LGGs from the intracranial posterior fossa will harbor one of the above mutations, they occur less frequently in supratentorial grade I LGGs and rarely in diffuse grade II tumors 21,22. Importantly, the genetics of inoperable disease that causes significant morbidity and mortality in children, particularly midline supratentorial and diffusely infiltrating LGGs, remain poorly characterized.
In this study, we sequenced the whole genomes of 39 pediatric LGGs and LGGNTs, along with their matching normal DNAs, identifying multiple novel genetic abnormalities. The principal novel findings, duplication of the tyrosine kinase domain (TKD) of fibroblast growth factor receptor-1 (FGFR1) and rearrangements of MYB or MYBL1, occur most frequently in diffuse cerebral LGGs.
Results
The genomic landscape of LGGs and LGGNTs
The study cohort (Supplementary Fig. 1) consisted of 151 tumors from 149 patients in three series: (i) tumors analyzed by whole genome sequencing (WGS; n=39) and/or transcriptome sequencing (mRNA-seq; n=46), (ii) diverse LGGs/LGGNTs for evaluating the frequency and clinicopathological associations of all mutated genes discovered through WGS or mRNA-seq (n=84), and (iii) non-cerebellar LGGs/LGGNTs without matching germline samples included to increase representation among those tumors for which genetic abnormalities are largely unknown (n=22).
Tumor series 1 included a ‘discovery set’ of 39 paired tumor / germline samples analyzed by WGS at an average of 45x haploid coverage (Supplementary Fig. 2; Supplementary Table 1). All somatic structural variations (SVs) and sequence mutations (single nucleotide variations (SNVs) and small insertions or deletions (indels)) in RefSeq exons were validated by orthogonal sequencing methods. Exhaustive validation of all somatic SVs and all somatic sequence mutations in non-repetitive regions of the reference human genome was undertaken for 16 tumors using custom capture arrays. The background mutation rate ascertained from validated SNVs in these tumors ranged from 5.7×10-9 to 8.7×10-8 (Supplementary Note; Supplementary Table 2). Seven tumors from the WGS series were also analyzed by high-coverage exome sequencing (245x average coverage), which showed that WGS was able to detect >85% of somatic coding variants, including subclonal mutations in these tumors (Supplementary Note; Supplementary Table 3).
Remarkably, the median number of non-silent somatic sequence mutations and SVs per tumor in the WGS (discovery) series was one, suggesting that few genetic alterations are required for oncogenesis (Fig. 1; Supplementary Fig. 3). Despite this low lesion burden, we found multiple recurrent abnormalities among different histopathological subtypes, including KIAA1549-BRAF fusions in PAs, frequent BRAF:p.V600E mutations in pleomorphic xanthoastrocytomas (PXAs), rearrangements and amplification of MYB in diffuse gliomas, and intragenic TKD duplications of FGFR1, all of which recurred at a frequency of more than 6% when sought across the cohort of 151 tumors (Figs. 1-4; Supplementary Table 4). Other validated WGS coding alterations, SVs and sequence mutations (SNVs and small indels), occurred at a frequency of less than 4% across the study cohort (Figs. 2-4; Supplementary Tables 4-8). However, among these were NF1 and FGFR1 sequence mutations, episome-associated FGFR1-TACC1 and FGFR3-TACC3 gene fusions, a rearrangement of MYBL1, an H3F3A:p.K27M mutation in three supratentorial diffuse astrocytomas, and three novel gene fusions involving BRAF or RAF1;FXR1-BRAF, BRAF-MACF1, and QKI-RAF1. When considering sequence mutations alone, the only genes with a mutation rate significantly higher than the background rate were BRAF, NF1, H3F3A and FGFR1 (Supplementary Table 9).
Figure 1. Clinicopathological characteristics and genetic alterations in tumors from series 1 examined by WGS or mRNA-seq.
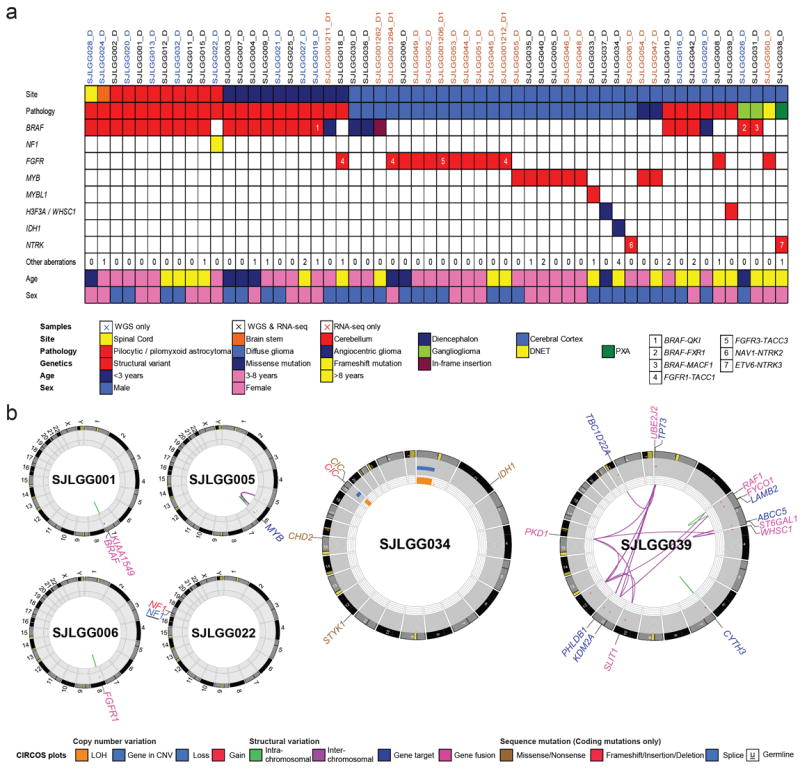
a Summary of clinicopathological characteristics (Tumor site, Pathology, Age at surgery, Sex) and key genetic alterations identified in 56 LGGs/LGGNTs by WGS (n=38) or mRNA-seq (n=44). Very few SVs or SNVs are found across the WGS tumor series. Numbers of aberrations for tumor samples highlighted in beige were based on analysis of mRNA-seq alone. Numbers in colored cells refer to gene fusions listed in box at right below table. b Selected CIRCOS plots summarizing aspects of WGS data, including a solitary KIAA1549-BRAF fusion (SJLGG001), MYB rearrangement (SJLGG005), FGFR1 TKD-duplication (SJLGG006), and NF1 mutation (SJLGG022). The genomic profiles of two oligodendroglial tumors, SJLGG006 and SJLGG034, illustrate some key differences between this type of tumor in children and in adults, the latter demonstrating IDH1 and CIC mutations alongside 1p/19q co-deletion. SJLGG039 exemplifies five LGGs with complex rearrangements.
Figure 4. Genetic alterations in LGGNTs.
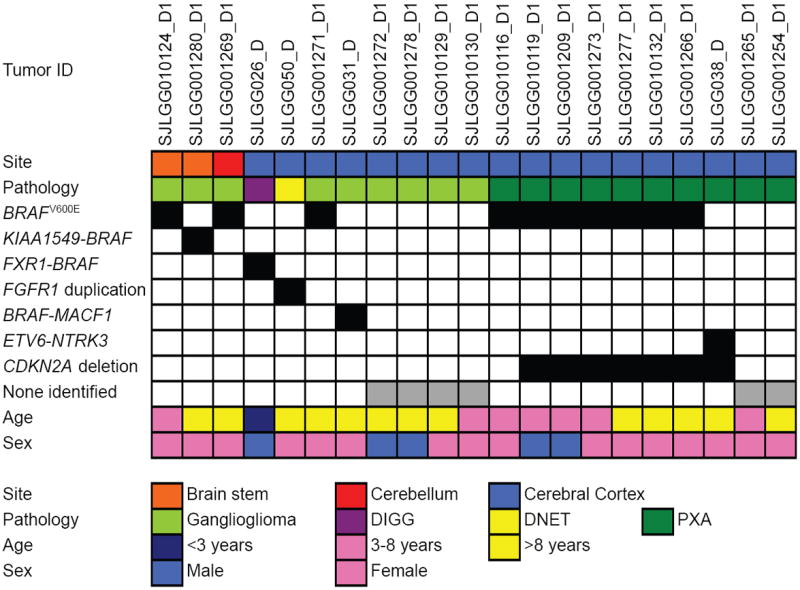
Associations between genetic alterations and clinicopathological characteristics for LGGNTs across the tumor cohort.
Abbreviations: DIGG – desmoplastic infantile ganglioglioma, DNET – dysembryoplastic neuroepithelial tumor, PXA – pleomorphic xanthoastrocytoma
Figure 2. Genetic alterations in supratentorial LGGs.
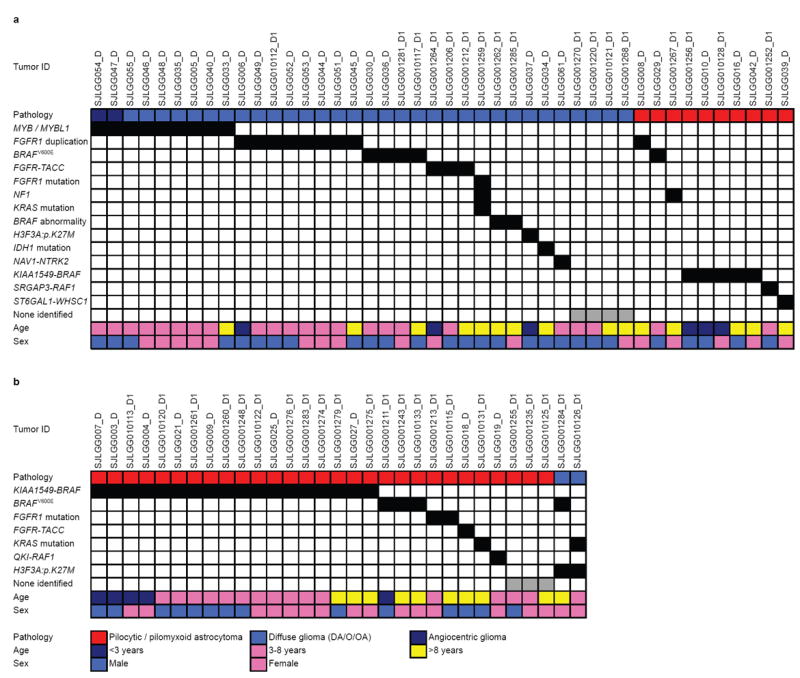
Associations between genetic alterations and clinicopathological characteristics for LGGs in the (a) cerebral hemispheres or (b) diencephalon.
Only four of 39 tumors (10%) in the WGS series lacked a MYB/MYBL1 rearrangement, FGFR1 alteration, or aberration of a gene in the NF1/RAS/RAF pathway. One of these, SJLGG034, was an oligodendroglioma from a patient aged 15 years that demonstrated genetic aberrations characteristic of adult-type disease; an IDH1 mutation and co-deletion of chromosomes 1p and 19q (Fig. 1). This tumor also had the highest number of sequence mutations in the WGS series, with six non-silent mutations in five genes: IDH1:p.R132H, CIC:p.V676fs and p.S726R, CHD2:p.D1722V, STYK1:p.P101L, BAI3:p.I869 splice site. No other tumor in the entire study cohort harbored an IDH1/2 mutation.
Key abnormalities in the other three tumors from the WGS series were an ETV6-NTRK3 fusion associated with CDKN2A deletion, an H3F3A mutation, and a rearrangement of WHSC1 (Supplementary Table 4). H3F3A, WHSC1 and three other genes found to have mutations in ‘discovery series’ tumors, ATRX, EP300, and CHD2, have histone-related functions.
Despite the paucity of somatic lesions in most tumors, multiple SVs probably resulting from a single complex rearrangement event were detected in five cerebral tumors: SJLGG039, SJLGG038, SJLGG033, SJLGG035 and SJLGG005, with 19, 13, 5, 4 and 3 SVs respectively. For SJLGG039, 18 of 19 SVs are interconnected interchromosomal SVs (Fig. 1b). Each SV breakpoint corresponds to the end of a (low-level) 0.12 copy number gain (1.8Mb-3.3Mb) that was scattered across chromosomes 1, 3, 4, 10, 11, 12, 16 and 22. This alteration suggests a copy number gain of 1 in approximately 25% of cells in the tissue sample, while the pattern of SVs and copy number variations (CNVs) suggests that focal amplicons are remnants of low-level chromosomal gains followed by loss of DNA in a complex rearrangement termed “chromothripsis” 23. WGS identified fusion of ST6GAL1 (exon 2) to the first coding exon (exon 4) of WHSC1, and this was validated by mRNA-seq, Sanger sequencing and iFISH (Supplementary Fig. 4). More details of the SVs in SJLGG038, SJLGG033, SJLGG035 and SJLGG005 are provided in Supplementary Information on-line (Supplementary Note; Supplementary Figs. 5-8).
Across the tumor cohort, we identified two tumors with a germline NF1 mutation (Supplementary Table 8). The first, a series 1 tumor (SJLGG022), had a germline splice mutation affecting R2214 at exon 43, as well as a somatic 4-bp frameshift mutation at T2263. No additional somatic mutations were detected in this cerebellar PA. The second, SJLGG001225, showed a germline nonsense mutation, W571*. Loss of wild-type NF1 was due to somatically acquired LOH at this locus. This was first suggested in the sequence chromatogram by an increase in the mutant allele fraction from 50% in germline to 80% in tumor (Supplementary Fig. 9). LOH was apparently caused by somatic copy number loss of chromosome 17 (Supplementary Fig. 10b). Both tumors with an NF1 germline mutation lost the second wild-type allele, one by acquiring a frameshift mutation, the other by LOH.
WGS and SNP array data demonstrated the presence of aneuploidy in a subset (11%) of LGGs/LGGNTs, but most tumors showed very few somatic copy number alterations (Supplementary Fig. 10). Paired copy number analysis using WGS identified subclonal gain or deletion across multiple chromosomes in three tumors: SJLGG008, SJLGG039, and SJLGG042 (Supplementary Fig. 11).
Across the study cohort of 151 tumors (Figs. 2-4; Supplementary Table 4), KIAA1549-BRAF fusions were detected in PAs, two pilomyxoid astrocytomas (PMAs), and a single brainstem ganglioglioma, and were present in 59%, 90%, and 80% of PAs/PMAs in the supratentorial, posterior fossa, and spinal anatomic compartments, respectively. BRAF:p.V600E mutations were detected in a high proportion of pleomorphic xanthoastrocytomas (70%) and at lower frequencies in diffuse astrocytomas (23%), gangliogliomas (33%) and PAs (6%). Abnormalities of genes encoding proteins influencing the MAPK/ERK pathway were detected in almost all (95%) PAs/PMAs and 82% of all LGGs/LGGNTs in the study. Among LGGs characterized by diffuse infiltration of adjacent brain, grade II gliomas and angiocentric gliomas, aberrations of MYB, MYBL1, or FGFR1/3 (TKD duplication, missense mutation, or TACC1/3 fusion) were detected in 68%. Of the rest, one oligodendroglioma was characterized by alterations typical of adult-type disease (SJLGG034), three contained an H3F3A:p.K27M mutation, four others harbored a BRAF:p.V600E mutation, and one had a FAM131B-BRAF fusion gene. Only 9.9% of LGGs/LGGNTs, all non-series 1 tumors and mostly cerebral in location, had no detectable genetic alteration.
FGFR1 alterations in LGGs/LGGNTs
WGS identified an intragenic duplication of the entire TKD of FGFR1 in two of 39 tumors. This encompassed exons 10-18 to produce an in-frame fusion gene separated by a ‘linker’ element of variable length (Fig. 5). Altogether across the study cohort, there were 13 tumors with this SV, four of which represented two pairs of primary and recurrent tumors. First and second surgeries were 17 or 19 months apart, and no anaplastic progression was found in either recurrent tumor. Genomic profiles of the paired primary and relapsed tumor samples analyzed by WGS (SJLGG006_D/R) were identical; alongside the FGFR1 duplication, there were no tier 1 SNVs, one tier 2 SNV, and 6 tier 3 SNVs.
Figure 5. FGFR1 aberrations in LGGs/LGGNTs.
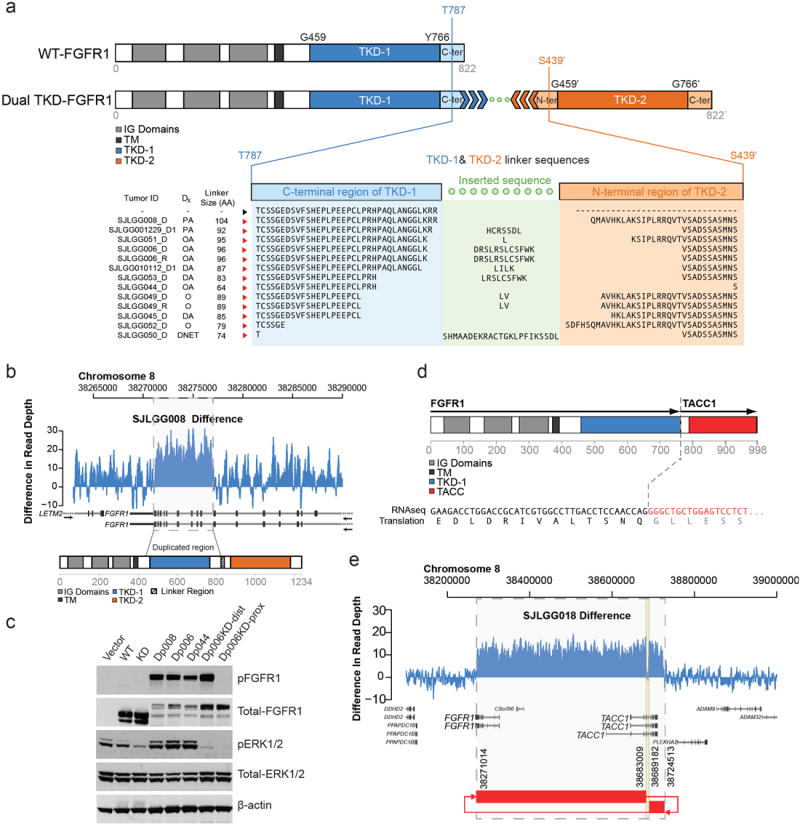
a FGFR1 – WT and with TKD duplication. With TKD duplication, two full length TKDs (TKD-1 & TKD-2) are separated by a linker sequence between the end (TKD-1) and beginning (TKD-2) of the auto-inhibited, activation-competent kinase domain spanning amino acids 459-766. Linker length varies between 74 and 104 amino acids. b Normalized (tumor minus germline) WGS read-coverage of FGFR1 in sample SJLGG008, with a 5kb duplication encompassing exons 10-18. c Western blot showing autophosphorylation of TKD-duplicated FGFR1 (Dp006, Dp008, Dp044) in 293T cells. FGFR1 autophosphorylation is associated with activation of the MAPK pathway, as indicated by raised p-ERK1/2 levels. Introducing a kinase-dead (KD) mutation (D623A) into either TKD-1 (Dp006KD-prox) or TKD-2 (Dp006KD-dist) of Dp006 abrogates MAPK pathway upregulation, but an active proximal TKD will still autophosphorylate FGFR1, while one with an active distal TKD will not. d FGFR1-TACC1 fusion protein detected in SJLGG018 preserving the TKD. e Normalized (tumor minus germline) WGS read coverage plot and SV connection plot for FGFR1/TACC1 locus suggesting formation of an episome.
Abbreviations: WT – wild-type, TKD – tyrosine kinase domain, IG – Ig-like, TM – transmembrane domain, Dx – diagnosis, PA – pilocytic astrocytoma, OA – oligoastrocytoma, DA – diffuse astrocytoma, O – oligodendroglioma, DNET – dysembryoplastic neuroepithelial tumor, KD – kinase-dead, TACC – TACC domain.
All but three of the 11 primary tumors with FGFR1 TKD duplication were diffuse (grade II) gliomas, and all but one were located in the cerebral cortex (Figs. 2-5). Although relatively infrequent across the entire cohort of LGGs/LGGNTs (7.4%), FGFR1 TKD duplication was present in 24% of grade II diffuse cerebral gliomas. The entire tumor cohort was screened by iFISH for FGFR1 amplification or rearrangement, and none was found. However, FGFR1 TKD missense mutations were detected in three LGGs (Supplementary Table 4).
Two SVs identified in WGS data from sample SJLGG018 were predicted to form an episome that connects the 3’ UTR of FGFR1 to the intron 6 sense strand of TACC1 (Supplementary Fig. 12). The region consists of two segments with copy number gain of one and an unamplified 6kb segment caused by loss of DNA during episome formation. Using both the split reads from mRNA-seq data and RT-PCR, we confirmed an in-frame FGFR1-TACC1 fusion transcript that joins exon 18 of FGFR1 with exon 7 of TACC1. Using copy number data from SNP array analysis or mRNA-seq, we were able to identify and then to validate by RT-PCR two additional tumors (SJLGG01212, SJLGG01264) with amplification segments on FGFR1 and TACC1 and fusion of these genes. These methods also disclosed a single FGFR3-TACC3 fusion (SJLGG01206). Substantial activation of both MAPK/ERK and PI3K pathways (pERK1/2 – 166x level in normal brain; pAKT – 58x level in normal brain) was demonstrated by multiplex immunoassay for SJLGG018.
Pediatric high-grade gliomas (n=33) were screened for FGFR1 TKD duplication, and only one example was detected, in an anaplastic oligoastrocytoma (grade III) that had progressed from a grade II tumor. No FGFR1 TKD duplication was detected in 11 adult-type oligodendrogliomas, all having IDH1 mutation and 1p/19q co-deletion.
MYB / MYBL1 rearrangements in diffuse LGGs
Four tumors analyzed by WGS harbored a novel rearrangement of MYB or MYBL1, all of which were cerebral grade II diffuse astrocytomas. Subsequent analysis of the entire study cohort using iFISH with MYB, MYBL1, and MYBL2 probes revealed a potential MYB rearrangement or copy number abnormality in a further five tumors: two diffuse astrocytomas, two angiocentric gliomas, and one oligodendroglioma (Figs.2, 6; SupplementaryFigs. 6-8, 13). No other tumor showed a potential MYBL1/2 rearrangement. MYB amplification, manifesting as episome formation, was detected in two tumors by WGS, mRNA-seq and iFISH. All non-WGS LGGs with MYB rearrangement were analyzed by mRNA-seq, which detected several partner genes (Supplementary Fig. 13). All SVs were associated with deletion of the MYB 3’ regulatory region. Of two tumors with MYB amplification, one also showed a 3’ deletion, but the amplicon in the other (SJLGG035) extended beyond the 3’UTR and miRNA binding sites. In all tumors with MYB rearrangement or amplification, MYB expression was elevated at the protein level (Fig. 6).
Figure 6. MYB and MYBL1 aberrations in diffusely infiltrating LGGs.
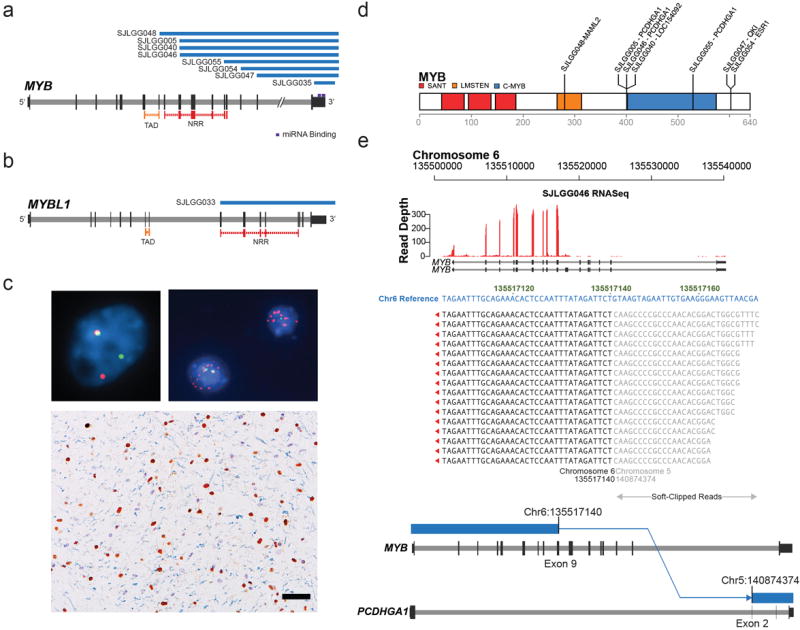
a, b Deletion through re-arrangement (blue bars) of MYB in 8 diffuse cerebral gliomas (a) or of MYBL1 in 1 diffuse cerebral glioma (b). c FISH - ‘break-apart’ probes were used to screen the study cohort for MYB, MYBL1, and MYBL2 rearrangements: (top left) probes targeting MYB are contiguous (yellow) at the normal locus, but split (green/red) where MYB is rearranged, (top right) MYB amplification (red probe), (bottom) cerebral diffuse astrocytoma with overexpression (brown reaction product) of MYB protein in the nuclei of neoplastic cells, but not normal glial cells (blue/gray hematoxylin counterstain). Scale bar = 50μm. d MYB protein showing breakpoints for rearrangements with other genes. The MYB transactivation domain is intact in all fused proteins, except in SJLGG048, where fusion with MAML2 may cause hyper-activation. In other fusions, the negative regulatory region or regulatory miRNA binding site is disrupted, or there is episome formation. e mRNA-seq and WGS data for SJLGG046 demonstrating MYB and PCDHGA1 fusion and over-expression of fused MYB.
Abbreviations: TAD – transactivating domain, NRR – negative regulatory region.
MYB alterations occurred only in cerebral gliomas with an infiltrative behavior; diffuse astrocytomas, oligodendrogliomas, and angiocentric gliomas. Although relatively infrequent (6%) across the entire cohort of LGGs/LGGNTs, MYB or MYBL1 aberrations were present in 25% of diffuse cerebral gliomas (Fig. 2; Supplementary Table 4). No MYB and MYBL1/2 alterations were identified by FISH in 33 pediatric high-grade gliomas or 79 ependymomas.
Expression profiling and activation of signaling pathways in LGGs/LGGNTs
Gene expression profiling of LGGs/LGGNTs using both Affymetrix™ U133plus2 arrays and mRNA-seq clearly showed clustering according to genetic abnormality. Tumors also clustered according to anatomic site and pathology, because of the strong associations between these variables and specific genetic abnormalities (Supplementary Note; Supplementary Fig. 14). No pattern was noted for gender.
Multiplex immunoassays and western blotting demonstrated activation of the MAPK/ERK and PI3K pathways in groups of LGGs/LGGNTs characterized by KIAA1549-BRAF fusion, FGFR1 TKD duplication, or MYB alteration (Fig. 7). Components of other signaling pathways tested by the multiplex immunoassay, such as the JAK-STAT pathway, showed no consistent alterations.
Figure 7. Activation of MAPK and PI3K pathways in LGGs/LGGNTs with FGFR1, MYB and BRAF abnormalities.
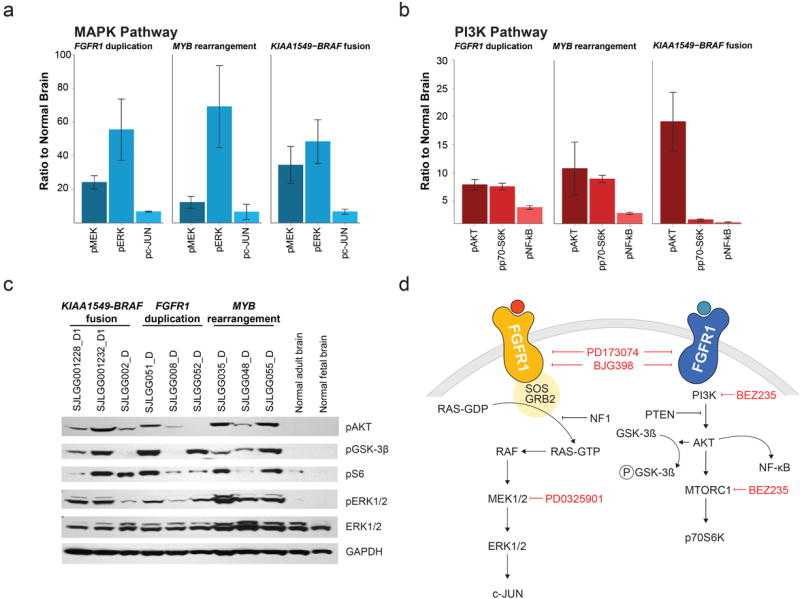
a, b Multiplex immunoassay levels for three MAPK (a) and three PI3K (b) pathway components are given relative to levels in normal brain for tumors with either FGFR1 duplication (n=11), or MYB rearrangement (n=6), or KIAA1549-BRAF fusion (n=18); bars represent the standard error of the mean. Gene expression profiling showed no significant elevation in the levels of corresponding mRNAs, with the exception of c-JUN. c Western blot data showing upregulation of the MAPK pathway (pERK1/2) and PI3K pathway (pAKT/pS6) in three groups of LGGs characterized by KIAA1549-BRAF fusion, FGFR1 duplication, or MYB rearrangement (AKT inhibits GSK-3β by phosophorylation). d Diagrams of MAPK (left) and PI3K (right) pathway components downstream of FGFR1, showing targets of pharmaceuticals used to inhibit the actions of TKD-duplicated FGFR1 (see Fig. 8).
TKD-duplicated FGFR1 is transforming and activates MAPK/ERK and PI3K signaling pathways
Neonatal (P2) Tp53-null astrocytes transfected with a TKD-duplicated FGFR1 construct (Dp006 / Dp008) and transplanted into the brains of nude mice generated high-grade astrocytic tumors with a short latency and complete penetrance (Fig. 8). Transplanted cells containing empty vector or wild-type FGFR1 constructs have failed to generate tumors in mice imaged at 60 days post-transplant. Tumors generated with both Dp006 and Dp008 were characterized by activation of the MAPK/ERK and PI3K pathways (Fig. 8).
Figure 8. TKD-duplicated FGFR1 in neonatal astrocytes generates gliomas in vivo.
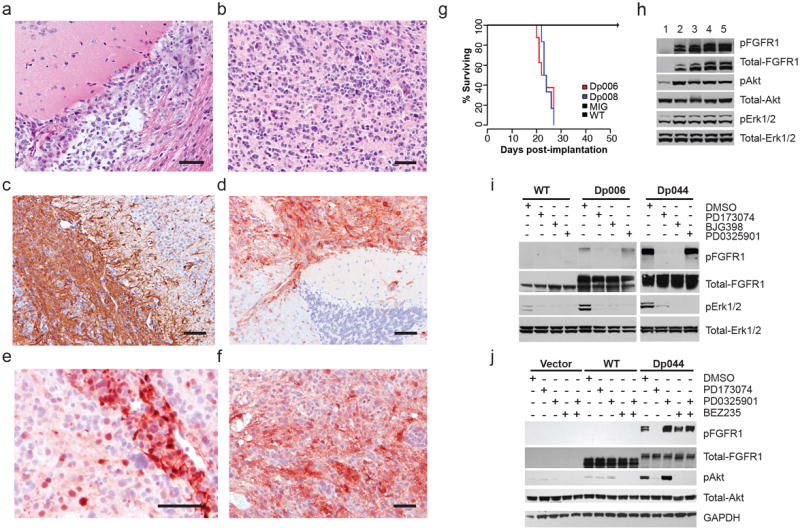
a-f Histopathology of TKD-duplicated FGFR1 tumors in mouse brain. Diffuse gliomas could demonstrate either an astrocytic (a) or oligodendroglial (b) phenotype, and were focally immunopositive for GFAP (c). Tumor cells, but not normal brain, showed overexpression of FGFR1 (d), pMAPK (e), or pAKT (f). Scale bar = 100μm. g Survival curves for mice transplanted with neonatal astrocytes containing empty vector (MIG), wild-type FGFR1 (WT), or two variants of TKD-duplicated FGFR1 (Dp006, Dp008). All Dp006/8 mice died from the effects of a glioma with a median survival of 23 days. h Western blot of proteins from normal brain (1) or tumor cell lysates (separate mice: 2,3 – Dp006; 4,5 – Dp008). TKD-duplicated FGFR1 is associated with increased levels of pFGFR1 (Y463/464), pAKT (S473), and pMAPK (T202/204). i Inhibitor studies showing that FGFR1 inhibitors PD173074 and BGJ398 block both autophosphorylation of duplicated FGFR1 (Dp044 & Dp006) and downstream activation of the MAPK pathway. The MEK1 inhibitor PD0325901 does not block FGFR1 autophosphorylation, but inhibits MAPK pathway activation. j Inhibitor studies showing that the FGFR1 inhibitor PD173074 blocks autophophorylation of duplicated FGFR1 and downstream activation of the PI3K pathway. The PI3K/mTOR inhibitor BEZ235 does not block FGFR1 autophosphorylation, but inhibits PI3K pathway activation.
When transfected into 293T cells, TKD-duplicated FGFR1 constructs with ‘linker’ elements of different lengths demonstrated receptor autophosphorylation and activation of the MAPK/ERK pathway (Figs. 5,8; Supplementary Fig. 15). FGFR1 inhibitors blocked autophosphorylation and downstream activation of the MAPK/ERK pathway, and MEK1 inhibitors abrogated MAPK/ERK activity (Fig. 8). When transfected into MCF7 cells, TKD-duplicated FGFR1 constructs produced receptor autophosphorylation and activation of the PI3K pathway, which were both blocked by a specific FGFR1 inhibitor. A PI3K/mTOR inhibitor, but not a MEK inhibitor, also switched off PI3K activation.
Discussion
LGGs encompass the WHO grade I PA, the less prevalent infiltrative WHO grade II diffuse gliomas, which have astrocytic, oligodendroglial, or mixed oligoastrocytic cytological features, and rare entities, such as the PXA and angiocentric glioma 24. In addition, low-grade glioneuronal tumors (LGGNTs), such as the ganglioglioma, contain a glial component with LGG histopathology and are usually grouped with LGGs in therapeutic classifications (Supplementary Table 10).
Most pediatric LGGs are cerebellar PAs. These are circumscribed tumors, which are usually amenable to surgical resection and have a low recurrence rate 25,26. However, PAs at other sites, such as the brainstem or optic pathways, are less well delineated from vital structures in adjacent brain, and complete excision is usually impossible. Inoperable tumors, including the diffuse grade II LGGs, grow slowly and may respond to adjuvant therapies, but over time cause significant morbidity and premature death 1,3-10. The molecular genetics of pediatric diffuse grade II LGGs have not been well characterized, unlike those of their adult counterparts 21,27-29, yet it is among these tumors that we have found the principal novel recurrent abnormalities reported in this study.
Using WGS, we have mapped the genomic landscape of 39 pediatric LGGs/LGGNTs and demonstrated that most pediatric LGGs/LGGNTs have only one somatic genetic event that affects protein coding. Furthermore, only one of 151 tumors (SJLGG001259_D1), with an NF1 frameshift mutation, an activating FGFR1 mutation, and a KRAS mutation, harbored genetic abnormalities with potentially overlapping effects on the MAPK/ERK pathway. In other tumors, NF1/RAF/RAS, FGFR1, and MYB/MYBL1 abnormalities were mutually exclusive. WGS also revealed several novel RAF abnormalities; FXR1-BRAF fusion, BRAF-MACF1 fusion, and QKI-RAF1 fusion, augmenting previous accounts of other rare defects in MAPK/ERK pathway genes, such as SRGAP3-RAF1 and FAM131B-BRAF fusions 11,30,31.
In the present study, 24% of diffuse WHO grade II cerebral gliomas showed a previously unreported duplication of the FGFR1 TKD, which produces FGFR1 autophosphorylation and activation of both MAPK/ERK and PI3K pathways. Autophosphorylation of FGFR1 may arise from ligand-independent homodimerization facilitated by an extended intracytoplasmic peptide tail and apposition of duplicated TKDs. A dual-TKD construct with truncated linker (Dp-NLK) did not stimulate the MAPK/ERK pathway, suggesting that downstream signaling is dependent on linker length as well as flexibility (Supplementary Fig. 14). Duplication of the EGFR TKD domain has been reported in a single glioblastoma 32, but FGFR1 TKD duplication has not been previously described in other CNS tumors.
Our dataset includes other infrequent FGFR aberrations in LGGs/LGGNTs; missense FGFR1 mutations and FGFR1-TACC1 and FGFR3-TACC3 fusions that are all predicted to result in constitutive FGFR signaling. Missense FGFR1 mutations have been reported as a rare event in glioblastoma and malignant melanoma 33,34, and fusions with TACC genes have recently been reported at low frequency (3%) in glioblastomas 35. This study also reported micro-amplification at the FGFR3-TACC3 locus suggesting that the FGFR3-TACC3 fusion is likely to arise from tandem duplication, because both FGFR3 and TACC3 are transcribed in the same orientation on the reference genome and are less than 50kb apart. By contrast, FGFR1 and TACC1 are transcribed in opposite orientations and separated by 400kb. An episome structure, which was supported by the presence of two SVs and two amplification segments in this region, makes it possible to alter the relative orientation of the two genes to generate a fusion protein between the FGFR1 TKD domain and the TACC domain of TACC1. FGFR1 amplifications and other FGFR1 fusions, which are important oncogenetic mechanisms in several neoplasms 36-38, were not detected in our series of LGGs/LGGNTs.
The region containing FGFR1 and TACC1 on chromosome 8 encompasses two other genes: WHSC1L1 and LETM2. Its paralogous region on chromosome 4 contains, in the following order: TACC3, FGFR3, LETM1 and WHSC1. The gene order synteny was maintained in both paralogous duplications. Aside from these fusions, recurrent somatic mutations and SVs in FGFR1, LETM1 (S580R) and WHSC1 (ST6GAL1-WHSC1 fusion) were found in tumors from our study cohort, suggesting in the setting of minimal somatic lesions that disruption of this highly conserved multi-gene group may be important in gliomagenesis.
MYB or MYBL1 abnormalities were evident in 25% of cerebral gliomas with a diffusely infiltrative architecture, including two angiocentric gliomas. Angiocentric gliomas share some histological features with ependymoma 24,39, but we found no MYB or MYBL1/2 alterations in a large series of ependymomas from across the neuraxis. We previously identified two structural alterations that generate MYB overexpression in pediatric diffuse cerebral gliomas: episome-associated amplification encompassing MYB’s transcription activating domain and focal deletion of its negative regulatory domain plus an inhibitory miRNA-binding regulatory domain in its 3’UTR 22. In the present study, WGS and mRNA-seq revealed novel MYB and MYBL1 rearrangements that involved fusion with several different genes. While some MYB fusion partners have reported roles in oncogenesis, all detected aberrations can produce MYB overexpression by one of the two mechanisms described above. Overall, mutually exclusive FGFR1 or MYB/MYBL1 aberrations were present in 56% of diffuse gliomas.
Our comprehensive analysis of NF1/RAF/RAS, FGFR1, and MYB abnormalities across a series of LGG/LGGNTs representative of the disease demonstrated that nearly all LGGs/LGGNTs in the spinal and posterior fossa compartments, which are dominated by PAs, are characterized by KIAA1549-BRAF fusion genes, while cerebral tumors, including most diffuse gliomas, are more heterogeneous. A subset of 7 LGGs (4.7%), most with a concurrent BRAF abnormality, demonstrated H3F3A mutations or abnormalities in other genes linked to histone function; ATRX, EP300, WHSC1, and CHD2. In our series, the genetics of only 9.9% LGGs/LGGNTs remained completely uncharacterized.
H3F3A mutations have recently been demonstrated in up to one third of pediatric glioblastomas, the most aggressive of high-grade gliomas (HGGs). Midline tumors are associated with an H3F3A:p.K27M mutation and are particularly prevalent in diffuse pontine gliomas 40-42. Although we detected an H3F3A:p.K27M mutation in only three (1.9%) of tumors in our series, this finding does indicate some overlap between the genetics of pediatric LGGs and HGGs, and it is notable that two of three diffuse grade II astrocytomas in which we found this mutation were thalamic; the other was from the cerebral cortex. One child with a thalamic tumor has died within two years of diagnosis, but the others have a progression-free survival beyond 10 years. None of these three tumors also contained a TP53 or ATRX mutation; instead one contained a KRAS:p.Q61H mutation and another a BRAF:p.V600E mutation. Only one of 33 tested HGGs (3%), an anaplastic oligoastrocytoma that had progressed from a grade II tumor, contained an FGFR1 TKD duplication, and no MYB abnormalities were found. Any overlap between the genetics of adult high-grade gliomas (HGGs) and pediatric LGGs seems to be confined to rare FGFR1 missense mutations and FGFR-TACC fusions.
The histopathologic features of WHO grade II diffuse gliomas occurring in children or adults appear very similar, yet their clinical behaviors and genetics are distinct. Over a period of 10-15 years post-surgery and despite adjuvant therapies, up to two thirds of adult grade II gliomas will progress to high-grade disease (WHO grade III/IV), heralding a poor prognosis 43-45. In contrast, childhood grade II gliomas can show relentless slow growth, but pathologic progression occurs much less frequently 7,46.
Our data support the hypothesis that distinct sets of genetic aberrations underlie clinicopathologic differences between adult and pediatric disease. Most adult grade II gliomas (>80%) display an IDH1 or IDH2 mutation, usually IDH1:p.R132H, which is considered to be an early transforming event. About two thirds of adult diffuse gliomas with an astrocytic phenotype have a concurrent TP53 mutation, and >80% of grade II oligodendrogliomas show co-deletion of chromosomes 1p and 19q 28,29,47-51. Progression to high-grade pathology is accompanied by the acquisition of additional genetic abnormalities, such that the range of adult diffuse gliomas from grade II to grade IV (glioblastoma) is characterized by stepwise accumulation of specific genetic abnormalities 52,53. Rarely, adult-type grade II disease can present in childhood 40, and our series contained one such example, an oligodendroglioma with an IDH1:p.R132H mutation, 1p/19q co-deletion, and CIC mutations. There is a high concordance between IDH1 and CIC mutations in adult oligodendrogliomas, suggesting cooperation between these genes 54,55. In contrast, our data suggest that a separate set of genetic aberrations characterizes pediatric diffuse gliomas and a single genetic aberration can be transforming in the majority of cases.
LGGs with duplication of the FGFR1 TKD or MYB overexpression show activation of the MAPK/ERK and PI3K pathways, demonstrating immunoassay profiles that are similar to PAs with KIAA1549-BRAF fusions and suggesting potential targets for therapeutic intervention. Combined activation of these pathways was also demonstrated in our functional studies of TKD-duplicated FGFR1. Against a facilitative Tp53-null background in transplanted neonatal astrocytes, TKD-duplicated FGFR1 was transforming, rapidly generating high-grade astrocytic tumors that demonstrated combined activation of these signaling pathways. In vitro studies utilizing two cell lines transfected with TKD-duplicated FGFR1 constructs showed that the specific FGFR1 inhibitors PD173074 and BGJ398 and MEK1 inhibitor PD0325901 could block FGFR1 autophosphorylation and constitutive activation of the MAPK/ERK pathway respectively and that upregulation of the PI3K pathway could be blocked by the specific inhibitor BEZ235. Such findings present a potential opportunity for the use of targeted therapies in the care of patients with LGGs that are unresectable and cause significant morbidity, as they do for patients with other cancers in which FGFRs play a critical role 56-58.
WGS of pediatric LGGs/LGGNTs has demonstrated multiple previously unreported oncogenetic mechanisms and facilitated discovery of a greatly extended genetic profile for pediatric diffuse (WHO grade II) gliomas. Our comprehensive analysis has also emphasized the potential therapeutic benefit of targeting upregulation of the MAPK/ERK and PI3K pathways in a disease that causes considerable morbidity and early mortality.
On-line Methods
Patient cohorts and sample details
The study cohort consisted of 151 tumors from 149 patients (Supplementary Fig. 1). Tissue was available at the time of diagnosis and relapse for 2 tumors (SJLGG006_D / R, SJLGG049_D / R). Archived series of 33 pediatric high-grade gliomas (WHO grade III/IV), 79 ependymomas and 11 adult anaplastic oligodendrogliomas (WHO grade III) were screened for relevant alterations.
Tissue samples had been snap-frozen at the time of first resection, which in all cases predated adjuvant therapy. DNA and RNA were extracted from frozen tissue and peripheral blood leukocytes 11. Archived formalin-fixed paraffin-embedded (FFPE) blocks and slides were retrieved for pathology review and specific analyses.
Whole genome and transcriptome sequencing and analysis
WGS, mRNA-seq, exome sequencing and SNP or gene expression profiling by array were performed as previously described 59,60. For both WGS and mRNA-seq, paired-end sequencing was performed using the Illumina GAIIx or HighSeq platform with 100bp read length.
WGS mapping, coverage and quality assessment, SNV / indel detection, tier annotation for sequence mutations, prediction of deleterious effects of missense mutations, and identification of loss of heterozygosity have been described previously 60. SVs were analyzed using CREST and annotated as before 60,61. The reference human genome assembly NCBI Build 37 was used for mapping all samples. CNVs were identified by evaluating the difference in read depth for each tumour and matched normal tissue using a novel algorithm, CONSERTING (COpy Number SEgmentation by Regression Tree In Next-Gen sequencing).
SNVs were classified into the following three tiers, as previously described 60:
-
Tier 1
coding synonymous, nonsynonymous, splice-site, and non-coding RNA variants
-
Tier 2
conserved variants (cutoff: conservation score ≥ 500, based on either the phastConsElements28way table or the phastConsElements17way table from the UCSC genome browser, and variants in regulatory regions annotated by UCSC annotation (Regulatory annotations included are targetScanS, ORegAnno, tfbsConsSites, vistaEnhancers, eponine, firstEF, L1 TAF1 Valid, Poly(A), switchDbTss, encodeUViennaRnaz, laminB1, cpgIslandExt)
-
Tier 3
variants in non-repeat masked regions
Paired-end reads from mRNA-seq were aligned to the following 4 databases using BWA (0.5.5) aligner 62: (i) human NCBI Build 37 reference sequence, (ii) RefSeq, (iii) a sequence file that represents all possible combinations of non-sequential pairs in RefSeq exons, and (iv) AceView flat file (UCSC), representing transcripts constructed from human ESTs. Final BAM files were constructed by selecting the best alignment in the four databases. SV detection was carried out using CREST and deFuse 61,63.
High-throughput sequencing of 32 candidate genes in tumors from series 2
All coding exons of the following genes were screened in 84 LGGs/LGGNTs: ADAMTS9, ATRX, BAI3, BRAF, CDK13, CHD2, CIC, DCTN1, DSG1, EP300, FGFR1, FGFR2, FGFR3, FLT1, FXR1, KRAS, LETM1, MAML2, MYB, MYBL1, NEURL4, NF1, NSMAF, PRIC285, PTEN, QKI, RAF1, SPHK1, STYK1, TFDP1, TMPRSS11D, and TP53. This list includes every gene with a validated non-silent mutation present in the dominant clone (i.e. mutant allele fraction >0.25 by WGS) of tumors from series 1, genes with SVs (e.g. FGFR1), genes with a related biology to that of mutated genes (e.g. FGFR2), and TP53.
The analysis was undertaken using PCR-based 3730 capillary sequencing at Beckman Coulter Genomics, as previously described 64. Putative SNVs and indel variants were detected by SNPdetector25 65. Non-silent coding variations present in tumor, but absent in normal tissue, were considered somatic mutations after manual review using the program consed. To remove additional germline variations from the dataset generated by sequencing tumors without a matching germline sample, novel non-silent mutations were compared to the 5K exomes data (NHLBI GO Exome Sequencing Project) and to a database of germline variations identified in the PCGP 66. Novel variants that passed this germline filter were manually reviewed and presented in two groups; those at a site of known somatic sequence mutation or that caused a truncation mutation were grouped with somatic mutations, while others were considered variants of unknown origin.
Mutated genes – analysis of significance
In order to assess the significance of validated non-silent sequence mutations across the entire cohort, we used the Significantly Mutated Gene test 67, which identifies genes with significantly higher mutation rates than the background mutation rate.
Experimental validation of genetic aberrations identified in WGS
All sequence mutations in exons (tier1 SNVs / indels) discovered in WGS were validated experimentally by Sanger, 454, or MiSeq sequencing. Of 89 high-quality tier1 SNVs tested, 86 were validated at a rate of 96.6%. All three high quality somatic indels were validated. All SVs affecting coding regions were validated by Sanger sequencing.
Validation by 454 or Sanger sequencing was as previously described 60. For MiSeq sequencing, primer pairs were designed with Primer3 to bracket the genomic regions containing putative SNVs / indels. These regions were amplified using Accuprime GC-rich DNA polymerase (Life Technologies), using DNA amplified from genomic DNA as PCR template (Qiagen). Amplicons were barcoded and prepared for sequencing using the Nextera XT DNA Sample Prep Kit (Illumina). Libraries were sequenced on MiSeq using the paired-end 150-cycle protocol, followed by variant analysis (MiSeq Reporter). Further evaluation of SNVs and indels was performed by manual review of the BAM files using Bambino 68.
Mutation hotspot analysis by Sanger sequencing
Mutational hotspots in BRAF, KRAS, FGFR1, IDH1, IDH2 and H3F3A were sequenced in genomic DNA from the entire series of tumors using previously published primers 11,22,42.
Validation of structural variations (SVs)
SVs were validated by PCR in cDNA, using specific primers (Supplementary Table 11). Primer combinations yielding an SV-specific product were validated by direct sequencing. Further primers were then designed for specific SVs, covering the majority of exons within each gene partner, and these were used to screen the tumor cohort for potential SVs: QKI-RAF1, FXR1-BRAF, FGFR1-TACC1, ETV6-NTRK3, KIAA1549-BRAF, SRGAP3-RAF1, and within FGFR1.
Copy number analysis and expression profiling by array
Affymetrix™ arrays were utilized for the analysis of copy number alterations (SNP6) and expression profiling (U133plus2) as previously described 69.
Construction of FGFR1 duplication vectors and FGFR1 retroviral production
Full-length open reading frame cDNA for human wild-type FGFR1 and three FGFR1 duplication variants were amplified by reverse transcription-PCR from human brain RNA pools or LGG RNA from SJLGG006_D, SJLGG008_D, and SJLGG044_D, with forward primer FGFR1ex2-for (aactgggatgtggagctgga) and reverse primer FGFR1ex18-rev (cagtcagcggcgtttgagtc). PCR products were cloned in PCR2.1 using a TA-cloning kit (Invitrogen) and verified by sequencing. After introducing 5’ BamHI and 3’ XhoI restriction sties by PCR, fragments encoding wt and duplication variants were sub-cloned into BamHI-XhoI digested retroviral vector MSCV-ires-GFP (MIG) to generate MIG-FGFR1-wt, MIG-FGFR1-Dp006, MIG-FGFR1-Dp008 and MIG-FGFR1-Dp044 constructs 70. A single aspartic acid to alanine mutation, D623A, was introduced into the FGFR1-wt fragment by site-directed mutagenesis to make a kinase-inactive construct (MIG-FGFR1-KD). The same kinase-dead (KD) mutation was also introduced into FGFR1 TKD-duplicated variants, either at the proximal site (MIG-FGFR1-Dp006KD-prox) or at a corresponding site within the distal fragment (MIG-FGFR1-Dp006KD-dist). In addition, a dual TKD construct (NLK) was prepared with a truncated linker of 22 amino acids between the two TKDs.
FGFR1 transfection studies
For inhibition assays, FGFR1-transfected 293T cells or FGFR1-transfected MCF7 cells were treated with serum-free DMEM for 12 or 18 hours, respectively, followed by incubation with inhibitors for 3 or 2 hours, respectively. The FGFR1 inhibitors BGJ398 and PD173074, MEK inhibitor PD0325901, and PI3K/mTOR inhibitor BEZ235 were dissolved in DMSO and added to cell cultures at a concentration of 100nM when used as single agents. For dual agent inhibition, PD0325901 and BEZ235 were each added at a concentration of 50nM.
Primary astrocyte cell culture and tumorigenesis
FGFR1 retroviral constructs (MIG-FGFR1-wt, MIG-FGFR1-Dp006, MIG-FGFR1-Dp008) or control GFP retrovirus (MIG) were used to transduce p53-null early passage primary mouse astrocytes (PMAs) established from 2-day old GFAPcre;Trp53 mice, as previously described 71-73. For tumorigenesis studies, 2×106 transduced PMAs were implanted into CD1-nude mouse brains 72.
Tissue collection, immunohistochemistry and fluorescence in situ hybridization (FISH)
Tumors from mice were processed and evaluated histopathologically as previously described 72. Immunohistochemistry using heat-mediated antigen retrieval in citrate buffer employed antibodies to GFAP (Z0334 at 1:1500) from Dako, Carpinteria, CA and phospho-Akt Ser473 (#9271 at 1:50), phospho-MAPK (#4370 at 1:400), and FGFR1 (#9740 at 1:500) from Cell Signaling, Beverly, MA.
Immunohistochemistry on human tumors employed an antibody to the N-terminal region of MYB (Abcam EP769Y; Cambridge, MA) 22.
Dual-color interphase FISH was undertaken as previously described 74. FISH probes (Supplementary Table 11) were derived from BAC clones (BACPAC Resources, Oakland, CA), labeled with either AlexaFluor-488 or Rhodamine fluorochromes, and validated on normal control metaphase spreads.
Immunoblot analysis and phosphoprotein multiplex immunoassay
For immunoblotting, transfected cells were lysed and extracts clarified by centrifugation. Western blotting was performed as previously described with antibodies to: FGFR1 (Epitomics, Burlingame, CA), phospho-FGFR1 (Y653-654) (MBL, Woburn, MA & Santa Cruz Biotechnology, Santa Cruz, CA), p44/42 ERK, phospho-p44/42 ERK (Thr202/Tyr204), phospho-AKT (Ser473), pan-AKT (C67E7), phospho-GSK-3β (Ser9), phospho-S6 ribosomal protein (Ser240/244), and GAPDH (14C10), all from Cell Signaling, Beverly, MA 75.
Proteins were assayed using the Bio-Plex™ detection array (Bio-Rad, Hercules, CA), following extraction from tumor samples using the Bio-Plex™ Cell Lysis Kit (Bio-Rad). The immunoassay utilized antibodies to the following phosphoproteins: p-ERK1/2, p-MEK1, p-AKT, p-GSK3α/β, p-c-JUN, p-P70 S6 kinase, and p-NF-κB p65. Protein extracts from a control cell line and a phosphatase-treated HeLa cell lysate served as positive and negative controls, respectively.
Supplementary Material
Figure 3. Genetic alterations in infratentorial LGGs.
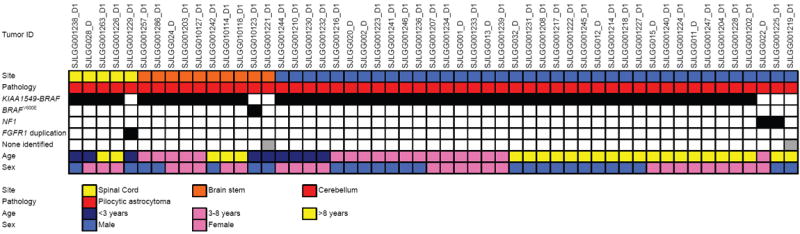
Associations between genetic alterations and clinicopathological characteristics for LGGs in the cerebellum, brain stem or spinal cord.
Acknowledgments
We are grateful for support from Anatomic Pathology and the Hartwell Center of Biotechnology and Bioinformatics at St. Jude Children’s Research Hospital, and Beckman Coulter Genomics. We acknowledge the St. Jude Children’s Research Hospital tissue resource facility, from which tissue samples were obtained, in accordance with Institutional Review Board approval for the Pediatric Cancer Genome Project. We thank Dr. William Evans for advice and support, and Mr. Panduka Nagahawatte for submitting the genomic data to EBI. This work was supported by the St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project and the American Lebanese Syrian Associated Charities (ALSAC) of St. Jude Children’s Research Hospital and by a grant from the US National Institutes of Health (NIH) (CA096832).
Footnotes
Author contributions
D.W.E., J.Z., R.G.T., B.T., L.D., R.K., D.S., R.J.G., E.R.M., R.K.W., J.R.D., and S.J.B. designed experiments or supervised research. I.Q., F.A.B., S.S., and A.G., provided samples or clinical data. D.W.E. undertook all pathologic evaluations. J.Z., G.W., R.G.T., J.D., B.T., W.O., C.P., C.P.M., C.L., C.K., L.D., M.P., R.L., R.H., X.C., E.H., P.N., M.R., K.B., J.C., J.B., J.M., G.S., L.W., J.W., J.E., D.Z., R.S.F., L.L.F., B.V., H.L.M. C.T., C.G.M., R.K., D.S., S.J.B. and D.W.E. performed experiments, analysed data or prepared tables and figures. D.J.D, K.O. contributed reagents, materials, or analysis tools. D.W.E. and J.Z. wrote the paper, with contributions from G.W., R.G.T., and S.J.B.
URLs
NCBI RefSeq; http://www.ncbi.nlm.nih.gov/RefSeq/
NCBI reference human genome; http://www.ncbi.nlm.nih.gov/genome/51
NHLBI GO Exome Sequencing Project; ESP, http://evs.gs.washington.edu/EVS/
The European Genome-phenome Archive (EGA), https://www.ebi.ac.uk/ega/studies/EGAS00001000255
Accession codes
Our entire genomic dataset, including WGS, whole exome sequencing, mRNA-seq, and SNP or gene expression array data have been deposited at the European Bioinformatics Institute (EBI) with accession number EGAS00001000255.
References
- 1.Armstrong GT, et al. Survival and long-term health and cognitive outcomes after low-grade glioma. Neuro-oncology. 2011;13:223–34. doi: 10.1093/neuonc/noq178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arora RS, et al. Age-incidence patterns of primary CNS tumors in children, adolescents, and adults in England. Neuro Oncol. 2009;11:403–13. doi: 10.1215/15228517-2008-097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qaddoumi I, Sultan I, Gajjar A. Outcome and prognostic features in pediatric gliomas: a review of 6212 cases from the Surveillance, Epidemiology, and End Results database. Cancer. 2009;115:5761–70. doi: 10.1002/cncr.24663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bouffet E, et al. Phase II study of weekly vinblastine in recurrent or refractory pediatric low-grade glioma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:1358–63. doi: 10.1200/JCO.2011.34.5843. [DOI] [PubMed] [Google Scholar]
- 5.Duffner PK, Cohen ME, Myers MH, Heise HW. Survival of children with brain tumors: SEER Program, 1973-1980. Neurology. 1986;36:597–601. doi: 10.1212/wnl.36.5.597. [DOI] [PubMed] [Google Scholar]
- 6.Fisher PG, et al. Outcome analysis of childhood low-grade astrocytomas. Pediatric blood & cancer. 2008;51:245–50. doi: 10.1002/pbc.21563. [DOI] [PubMed] [Google Scholar]
- 7.Gajjar A, et al. Low-grade astrocytoma: a decade of experience at St. Jude Children’s Research Hospital. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1997;15:2792–9. doi: 10.1200/JCO.1997.15.8.2792. [DOI] [PubMed] [Google Scholar]
- 8.Gnekow AK, et al. Long-term follow-up of the multicenter, multidisciplinary treatment study HIT-LGG-1996 for low-grade glioma in children and adolescents of the German Speaking Society of Pediatric Oncology and Hematology. Neuro Oncol. 2012 doi: 10.1093/neuonc/nos202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merchant TE, Conklin HM, Wu S, Lustig RH, Xiong X. Late effects of conformal radiation therapy for pediatric patients with low-grade glioma: prospective evaluation of cognitive, endocrine, and hearing deficits. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:3691–7. doi: 10.1200/JCO.2008.21.2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stokland T, et al. A multivariate analysis of factors determining tumor progression in childhood low-grade glioma: a population-based cohort study (CCLG CNS9702) Neuro-oncology. 2010;12:1257–68. doi: 10.1093/neuonc/noq092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forshew T, et al. Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol. 2009;218:172–81. doi: 10.1002/path.2558. [DOI] [PubMed] [Google Scholar]
- 12.Jones DT, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68:8673–7. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pfister S, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest. 2008;118:1739–49. doi: 10.1172/JCI33656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sievert AJ, et al. Duplication of 7q34 in pediatric low-grade astrocytomas detected by high-density single-nucleotide polymorphism-based genotype arrays results in a novel BRAF fusion gene. Brain Pathol. 2009;19:449–58. doi: 10.1111/j.1750-3639.2008.00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Listernick R, Charrow J, Gutmann DH. Intracranial gliomas in neurofibromatosis type 1. American journal of medical genetics. 1999;89:38–44. doi: 10.1002/(sici)1096-8628(19990326)89:1<38::aid-ajmg8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 16.Listernick R, Ferner RE, Liu GT, Gutmann DH. Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations. Ann Neurol. 2007;61:189–98. doi: 10.1002/ana.21107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeClue JE, et al. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell. 1992;69:265–73. doi: 10.1016/0092-8674(92)90407-4. [DOI] [PubMed] [Google Scholar]
- 18.Dias-Santagata D, et al. BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PLoS One. 2011;6:e17948. doi: 10.1371/journal.pone.0017948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin A, et al. BRAF alterations in primary glial and glioneuronal neoplasms of the central nervous system with identification of 2 novel KIAA1549:BRAF fusion variants. J Neuropathol Exp Neurol. 2012;71:66–72. doi: 10.1097/NEN.0b013e31823f2cb0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schindler G, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta neuropathologica. 2011;121:397–405. doi: 10.1007/s00401-011-0802-6. [DOI] [PubMed] [Google Scholar]
- 21.Tatevossian RG, et al. MAPK pathway activation and the origins of pediatric low-grade astrocytomas. J Cell Physiol. 2010;222:509–14. doi: 10.1002/jcp.21978. [DOI] [PubMed] [Google Scholar]
- 22.Tatevossian RG, et al. MYB upregulation and genetic aberrations in a subset of pediatric low-grade gliomas. Acta neuropathologica. 2010;120:731–43. doi: 10.1007/s00401-010-0763-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stephens PJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Louis DN, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pollack IF. The role of surgery in pediatric gliomas. Journal of neuro-oncology. 1999;42:271–88. doi: 10.1023/a:1006107227856. [DOI] [PubMed] [Google Scholar]
- 26.Wisoff JH, et al. Primary neurosurgery for pediatric low-grade gliomas: a prospective multi-institutional study from the Children’s Oncology Group. Neurosurgery. 2011;68:1548–54. doi: 10.1227/NEU.0b013e318214a66e. discussion 1554-5. [DOI] [PubMed] [Google Scholar]
- 27.Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol. 2006;1:97–117. doi: 10.1146/annurev.pathol.1.110304.100043. [DOI] [PubMed] [Google Scholar]
- 28.Ohgaki H, Kleihues P. Genetic profile of astrocytic and oligodendroglial gliomas. Brain tumor pathology. 2011;28:177–83. doi: 10.1007/s10014-011-0029-1. [DOI] [PubMed] [Google Scholar]
- 29.Riemenschneider MJ, Jeuken JW, Wesseling P, Reifenberger G. Molecular diagnostics of gliomas: state of the art. Acta neuropathologica. 2010;120:567–84. doi: 10.1007/s00401-010-0736-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cin H, et al. Oncogenic FAM131B-BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol. 2011;121:763–74. doi: 10.1007/s00401-011-0817-z. [DOI] [PubMed] [Google Scholar]
- 31.Jones DT, et al. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene. 2009;28:2119–23. doi: 10.1038/onc.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ciesielski MJ, Fenstermaker RA. Oncogenic epidermal growth factor receptor mutants with tandem duplication: gene structure and effects on receptor function. Oncogene. 2000;19:810–20. doi: 10.1038/sj.onc.1203409. [DOI] [PubMed] [Google Scholar]
- 33.Lin WM, et al. Modeling genomic diversity and tumor dependency in malignant melanoma. Cancer research. 2008;68:664–73. doi: 10.1158/0008-5472.CAN-07-2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rand V, et al. Sequence survey of receptor tyrosine kinases reveals mutations in glioblastomas. Proc Natl Acad Sci U S A. 2005;102:14344–9. doi: 10.1073/pnas.0507200102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh D, et al. Transforming Fusions of FGFR and TACC Genes in Human Glioblastoma. Science. 2012 doi: 10.1126/science.1220834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li F, Zhai YP, Tang YM, Wang LP, Wan PJ. Identification of a novel partner gene, TPR, fused to FGFR1 in 8p11 myeloproliferative syndrome. Genes, chromosomes & cancer. 2012;51:890–7. doi: 10.1002/gcc.21973. [DOI] [PubMed] [Google Scholar]
- 37.Turner N, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer research. 2010;70:2085–94. doi: 10.1158/0008-5472.CAN-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nature reviews Cancer. 2010;10:116–29. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 39.Wang M, et al. Monomorphous angiocentric glioma: a distinctive epileptogenic neoplasm with features of infiltrating astrocytoma and ependymoma. J Neuropathol Exp Neurol. 2005;64:875–81. doi: 10.1097/01.jnen.0000182981.02355.10. [DOI] [PubMed] [Google Scholar]
- 40.Schwartzentruber J, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–31. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 41.Sturm D, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–37. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 42.Wu G, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nature genetics. 2012;44:251–3. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muller W, Afra D, Schroder R. Supratentorial recurrences of gliomas. Morphological studies in relation to time intervals with astrocytomas. Acta Neurochir (Wien) 1977;37:75–91. doi: 10.1007/BF01401927. [DOI] [PubMed] [Google Scholar]
- 44.Okamoto Y, et al. Population-based study on incidence, survival rates, and genetic alterations of low-grade diffuse astrocytomas and oligodendrogliomas. Acta Neuropathol. 2004;108:49–56. doi: 10.1007/s00401-004-0861-z. [DOI] [PubMed] [Google Scholar]
- 45.Peraud A, Kreth FW, Wiestler OD, Kleihues P, Reulen HJ. Prognostic impact of TP53 mutations and P53 protein overexpression in supratentorial WHO grade II astrocytomas and oligoastrocytomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2002;8:1117–24. [PubMed] [Google Scholar]
- 46.Pollack IF, Claassen D, al-Shboul Q, Janosky JE, Deutsch M. Low-grade gliomas of the cerebral hemispheres in children: an analysis of 71 cases. J Neurosurg. 1995;82:536–47. doi: 10.3171/jns.1995.82.4.0536. [DOI] [PubMed] [Google Scholar]
- 47.Jones DT, et al. Adult grade II diffuse astrocytomas are genetically distinct from and more aggressive than their paediatric counterparts. Acta neuropathologica. 2011;121:753–61. doi: 10.1007/s00401-011-0810-6. [DOI] [PubMed] [Google Scholar]
- 48.Korshunov A, et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol. 2009;118:401–5. doi: 10.1007/s00401-009-0550-z. [DOI] [PubMed] [Google Scholar]
- 49.Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.von Deimling A, Korshunov A, Hartmann C. The next generation of glioma biomarkers: MGMT methylation, BRAF fusions and IDH1 mutations. Brain Pathol. 2011;21:74–87. doi: 10.1111/j.1750-3639.2010.00454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yan H, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–73. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ohgaki H, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004;64:6892–9. doi: 10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 53.Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170:1445–53. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bettegowda C, et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science. 2011;333:1453–5. doi: 10.1126/science.1210557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yip S, et al. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. The Journal of pathology. 2012;226:7–16. doi: 10.1002/path.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Flaherty KT, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. The New England journal of medicine. 2012;367:107–14. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 57.Katoh M. Genetic alterations of FGF receptors: an emerging field in clinical cancer diagnostics and therapeutics. Expert Rev Anticancer Ther. 2010;10:1375–9. doi: 10.1586/era.10.128. [DOI] [PubMed] [Google Scholar]
- 58.Ren M, Qin H, Ren R, Cowell JK. Ponatinib suppresses the development of myeloid and lymphoid malignancies associated with FGFR1 abnormalities. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, U K. 2012 doi: 10.1038/leu.2012.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang J, et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature. 2012;481:329–34. doi: 10.1038/nature10733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang J, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–63. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nature methods. 2011;8:652–4. doi: 10.1038/nmeth.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McPherson A, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput Biol. 2011;7:e1001138. doi: 10.1371/journal.pcbi.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mullighan CG, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–9. doi: 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang J, et al. SNPdetector: a software tool for sensitive and accurate SNP detection. PLoS computational biology. 2005;1:e53. doi: 10.1371/journal.pcbi.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Downing JR, et al. The Pediatric Cancer Genome Project. Nat Genet. 2012;44:619–22. doi: 10.1038/ng.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dees ND, et al. MuSiC: identifying mutational significance in cancer genomes. Genome Res. 2012;22:1589–98. doi: 10.1101/gr.134635.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Edmonson MN, et al. Bambino: a variant detector and alignment viewer for next-generation sequencing data in the SAM/BAM format. Bioinformatics. 2011;27:865–6. doi: 10.1093/bioinformatics/btr032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mullighan CG. Single nucleotide polymorphism microarray analysis of genetic alterations in cancer. Methods Mol Biol. 2011;730:235–58. doi: 10.1007/978-1-61779-074-4_17. [DOI] [PubMed] [Google Scholar]
- 70.Persons DA, et al. Enforced expression of the GATA-2 transcription factor blocks normal hematopoiesis. Blood. 1999;93:488–99. [PubMed] [Google Scholar]
- 71.Bajenaru ML, et al. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol. 2002;22:5100–13. doi: 10.1128/MCB.22.14.5100-5113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Endersby R, Zhu X, Hay N, Ellison DW, Baker SJ. Nonredundant functions for Akt isoforms in astrocyte growth and gliomagenesis in an orthotopic transplantation model. Cancer Res. 2011;71:4106–16. doi: 10.1158/0008-5472.CAN-10-3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jonkers J, et al. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29:418–25. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 74.Ellison DW, et al. Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol. 2011;29:1400–7. doi: 10.1200/JCO.2010.30.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tang B, et al. Characterization of signal transduction through the TCR-zeta chain following T cell stimulation with analogue peptides of type II collagen 260-267. Journal of immunology. 1998;160:3135–42. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.