

Abstract
Aberrant glycosylation of human glycoproteins is related to various physiological states, including the onset of diseases such as cancer. Consequently, the search for glycans that could be markers of diseases or targets of therapeutic drugs has been intensive. Here, we describe a high-throughput ion mobility spectrometry/mass spectrometry analysis of N-linked glycans from human serum. Distributions of glycans are assigned according to their m/z values, while ion mobility distributions provide information about glycan conformational and isomeric composition. Statistical analysis of data from 22 apparently healthy control patients and 39 individuals with known diseases (20 with cirrhosis of the liver and 19 with liver cancer) shows that ion mobility distributions for individual m/z ions appear to be sufficient to distinguish patients with liver cancer or cirrhosis. Measurements of glycan conformational and isomeric distributions by IMS–MS may provide insight that is valuable for detecting and characterizing disease states.
Keywords: ion mobility, electrospray ionization, glycans, cancer, conformers, isomers, principal component analysis
Introduction
The process of discovering biomarkers is being transformed by detailed proteomic analyses that involve separation or affinity techniques, such as two-dimensional polyacrylamide gel electrophoresis, high-performance liquid chromatography, or surface-enhanced laser desorption ionization, to decrease complexity of tissue extracts prior to mass spectrometic (MS) detection.1 A common feature of these strategies is the comparison of proteomic data from cohorts of individuals that express specific pathology with control individuals in order to find reproducible differences in protein expression between groups. While it is difficult to overstate the potential of such studies (especially those involving detailed analysis of plasma and serum), such experiments usually lack sufficient resolution and/or throughput because of analytical challenges associated with characterizing a large number of proteins (and their modified forms) that may exist over a wide range of concentrations.2 There is substantial interest in developing techniques that are capable of examining specific biomolecules in a high-throughput fashion.3
During the recent years, several groups have coupled gas-phase ion mobility separations with mass spectrometry (IMS–MS).4 In this approach, a mixture of analytes is ionized and an ion packet is introduced into a drift tube containing a buffer gas (often a few torr of helium). Ions migrate through the gas under the influence of a weak electric field and individual components in the mixture separate because of differences in their mobilities through the gas.5 IMS–MS is similar in many ways to electrophoretic migration in the condensed phase; however, because the number density of gases is much lower than condensed phases, it is possible to carry out separations in only a few milliseconds. Although this separation time is very short, it is sufficient to allow hundreds of independent mass spectra to be recorded, and IMS-MS has been developed as a two-dimensional, high-throughput approach for analyzing complex mixtures.
In the present paper, we describe a high-throughput IMS–MS analysis of human samples that is aimed at characterizing disease states. We have focused on the analysis of a relatively small sample set corresponding to N-linked glycans that are enzymatically released from human serum glycoproteins. Several research groups have analyzed glycans and glycoproteins from human serum via matrix-assisted laser desorption ionization mass spectrometry (MALDI–MS) and electrospray ionization mass spectrometry (ESI–MS),6–8 and a few putative cancer biomarkers have been assigned.9–11 The specificity of these MS methods is often based on comparison of the raw intensities of glycan ions between healthy and diseased tissue, while structural and isomeric composition of glycans is not considered. However, because IMS separates ions based on differences in their mobility, it is possible to obtain insight about the number (and abundances) of different isomers and conformers. We have recently combined this approach with liquid chromatography in order to characterize the human plasma proteome,12 and we also developed a three-dimensional IMS-IMS–MS technique that allows specific ion conformations to be selected according to their mobilities and further activated in order to change ion conformations or induce fragmentation.13,14 This approach (IMS–IMS) has similarities with MS–MS;13 however, it aims to characterize changes in structure based on production of new conformations rather than generation of ion fragments (although the latter is also possible). The ability to rapidly distinguish conformers and isomers from large numbers of samples is likely to complement insights gained from the MS analyses alone.
Experimental Procedures
Materials
The enzyme Peptide-N-Glycosidase F (PNGase F, ≥95% purity) was obtained from Sigma (St. Louis, MO). Water, methanol, and acetonitrile (ACN) (HPLC grade) were purchased from EMD Chemicals (Gibbstown, NJ). Human serum samples were collected for a study of hepatocellular carcinoma in an Egyptian population as described previously.15,16 The study protocols were approved by the institutional review committees of all participating institutions and conformed to the ethical guidelines of the 1975 Helsinki Declaration.
Preparation of Glycans from Serum
Glycans from 10 μL-aliquots of human blood serum were prepared as described previously.11 Briefly, a 10-μL aliquot of serum was added to 150 μL of 25 mM ammonium bicarbonate and 2.5 μL of 200 mM DTT prior to incubation at 60 °C for 45 min. A 10-μL aliquot of 200 mM IAA was added and allowed to react at room temperature for 1 h in the dark. Subsequently, a 2.5-μL aliquot of 200 mM DTT was added to react with the excess IAA. The reaction mixture was diluted with 100 μL of ammonium bicarbonate to adjust the pH to 7.5–8.0 for the enzymatic release of N-glycans using PNGase F. Next, a 5 mU aliquot of PNGase F was added to the mixture prior to incubation overnight (18–22 h) at 37 °C.
The volume of enzymatically released glycans was adjusted to 1 mL with deionized water and applied to a C18 Sep-Pak cartridge (Waters, Milford, MA), which was preconditioned with ethanol and deionized water as described previously.11 The reaction mixture was circulated through the cartridge five times to retain peptides and O-linked glycopeptides. Glycans were present in the pass-through and the 0.25 mL deionized water washes. The combined eluents were then passed over activated charcoal microcolumns (Harvard Apparatus, Holliston, MA) preconditioned with 1 mL of ACN and 1 mL aqueous solution of 0.1% trifluroacetic acid (TFA). The microcolumn was washed with 1 mL of 0.1% TFA, and samples were eluted with 1 mL of 50% aqueous ACN with 0.1% TFA. The purified N-glycans were evaporated to dryness using vacuum CentriVap Concentrator (Labconco Corporation, Kansas City, MO) prior to solid-phase permethylation. Purified glycans were permethylated by using a recently developed spin-column permethylation procedure.17 Prior to IMS–MS analysis, glycans were dissolved in 15 μL of 50%:50% (v/v) CH3OH/H2O containing 0.1% formic acid and 1 mM sodium acetate; 5 μL aliquots were transferred into a 96-well plate. Prior to IMS–IMS analysis, glycans were dissolved and diluted in 50%:50% (v/v) AcCN/H2O containing 0.1% of formic acid and 1 mM sodium acetate to produce a final concentration of ~0.25 mg/mL.
Automated Sample Analysis and ESI Source Conditions
Ninety glycan samples were electrosprayed from a 96-well plate through a 400-nozzle ESI chip of a NanoMate autosampler (Advion Biosciences, Inc., Ithaca, NY) using a flow rate of ~0.25 μL/min and voltage of 1.5 kV. For the IMS–IMS analysis of glycans, the flow rate was set at 0.25 μL/min by the syringe pump (KD Scientific, Holliston, MA), and a PEEK microtee was used for the coupling of the capillary tip, syringe, and platinum electrode. The glycan mixture was electrosprayed through a pulled capillary tip (75 μm i.d. × 360 μm o.d.) held at a voltage of ~5 kV.
IMS–MS Instrumentation and Experimental Approach
The two IMS–MS instruments used in this study were similar to the instruments described in our previous studies.13,14,18 One of them, shown in Figure 1, consists of two drift tube sections (D1 and D2) each being ~1 m in length. The incorporation of a NanoMate autosampler in front of this instrument enabled the throughput necessary for analysis of 90 glycan samples. Prior to the analysis of the samples of interest, a study was done to find the optimal acquisition time, that is, the minimum time that is required to record ion mobility profiles with a good signal-to-noise ratio. A glycan test sample was analyzed using acquisition times that ranged from 10 s to 4 min; the 2 min-acquisition time was chosen as optimal for the analysis of the 90 glycan samples. Samples were electrosprayed, alternating among groups, from a 96-well plate. Because the sample plate was at the room temperature, samples were analyzed in 10 batches (first batch contained 15 samples, next eight batches contained 9 samples each, and the last batch contained 3 samples) in order to prevent evaporation of methanol from the samples.
Figure 1.
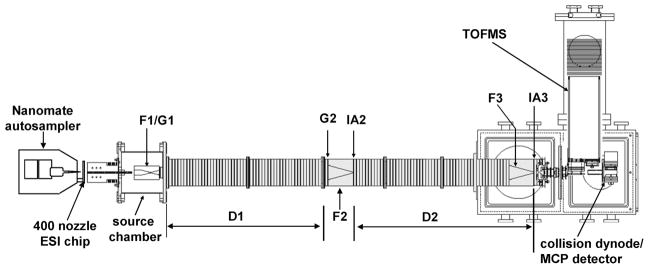
Schematic diagram of the NanoMate-IMS-TOF instrument.
Electrosprayed ions were introduced into the first ion funnel (F1) and trapped by applying a DC voltage of 45 V. Periodically, ions were gated into the first drift tube (D1), which contained ~2.25 Torr of He buffer gas (ultra high purity, Airgas, Radnor, PA), and separated based on their ion mobilities through the gas. After exiting the first drift tube, ions entered another ion funnel (F2) that focused and transmitted ions into the second drift tube (D2). A gate (G2) and an activation region (IA2) at the entrance of the second drift tube are used to mobility-select and energize selected components. The activation region was set at 20 V during the experiments to improve ion intensities. After exiting the drift tube, ions were focused into a time-of-flight (TOF) mass analyzer and detected by a multichannel plate (MCP) detector.
For the analysis of serum glycans, we also used an IMS-IMS-IMS–MS instrument, which incorporates an additional ~1m-long drift tube section and provides for higher drift time resolution than the instrument described above. Glycans were separated in a buffer gas containing ~3 Torr of He and 0.03 Torr of N2 (ultra high purity, Airgas, Radnor, PA). Nitrogen was included in order to increase energy deposition during activation studies. Drift regions were operated at a field of 9 V · cm−1, while the fields through the funnels were 11 V · cm−1. RF field amplitudes and frequencies ranged from 110 to 250 V and 250 to 460 kHz, respectively. Electrosprayed ions were trapped in the first ion funnel (F1) by applying a DC voltage of 80 V. After exiting the first drift tube, ions of particular mobilities were selectively gated by raising the fields in the gate region at delay times that were controlled by a digital pulse-delay generator (Stanford Research Systems, Sunnyvale, CA). In activation experiments, ions were activated by raising the voltage between the last two lenses of the second funnel to 140 V. Ion m/z values were further measured by an orthogonal TOF mass spectrometer.
Data Evaluation
Origin 6.1 (OriginLab Corp., Northampton, MA) was used to make three-dimensional plots showing ion intensities as function of drift time and m/z values [tD(m/z) distribution]. Slicing software, written in-house, was used to extract ion mobility distributions of glycan ions from the three-dimensional data sets. Principal component analysis (PCA) of ion mobility profiles was performed using MarkerView software (ABI, Framingham, MA). The PCA was done on a total of 61 samples, that is, 19 samples from serum of patients with liver cancer (HC), 22 samples from healthy patients (NC), and 20 samples from patients with the liver cirrhosis (QT). Other samples were not included in the PCA analysis due to occasional problems with the sample delivery on our prototype system (clogging of the NanoMate chip nozzles and/or solvent evaporation). Supervised PCA was employed using prior knowledge about which sample groups were healthy or diseased. MS data were weighted using the natural logarithm of ion intensities across the drift time distributions. The intensities were also scaled using the pareto option, in which each value is subtracted by the average and divided by the square root of the standard deviation. The pareto option is suitable for MS data since it prevents intense peaks from completely dominating the PCA process, thus, allowing any peak with a good signal-to-noise ratio to contribute to PCA scores. Single-factor analysis of variances (ANOVA) test between sample groups was done using Microsoft Excel.
Results and Discussion
IMS–MS Distributions of Glycans from Serum
To test the possibility that glycan conformer and isomer distributions may be correlated with diseased states, we examined serum samples from 90 individuals: 30 that were diagnosed with hepatocellular carcinoma (HC), 30 with liver cirrhosis (QT), and 30 healthy individuals (NC). These samples were treated to obtain glycans as described above. Samples were analyzed by the prototype IMS–MS techniques also outlined above. With this approach, the mobility distributions and mass spectra associated with all ions that are transmitted through the source are simultaneously obtained.
Figure 2 shows an example of two-dimensional data set for one of the samples, which was recorded using our IMS-IMS-IMS–MS instrument. The IMS separation reduces spectral congestion by separating components into mobility families, as described previously.18 One outcome of this separation is a reduction of background chemical noise. In the data shown in Figure 2, chemical noise (that appears as a baseline in a single dimension MS measurement) is dispersed across a wide range of drift times and can be recognized as a broad, light-blue region in Figure 2. Integrating the data over narrow regions where peaks appear makes it possible to reduce contributions due to the background noise.18
Figure 2.

Two-dimensional tD(m/z) plot of mobility-dispersed glycan ions originating from human serum. Glycan structures were assigned by comparison of measured m/z values with m/z values cited in the literature. S represents sialic acid, H represents a hexose (galactose or mannose), and N represents N-acetyl glucosamine. In cartoon images, solid squares represent N-acetyl glucosamine, open circles represent mannose, solid circles represent galactose, and diamonds represent sialic acid. Two structural isomers of S1H5N4 have sialic acid added to the antenna either on the α-1,3-linked mannose residues or α-1,6-linked mannose residues.
Peaks in the spectra are assigned to specific ions by comparing the experimental m/z values with theoretical values that are calculated for expected glycans. In many separate experiments, the m/z values of glycans that are present in the sample have been assigned at high resolution. Although the m/z values associated with the automated studies are somewhat less accurate (because calibration was carried out only at the beginning of the analysis), the overall shapes of the two-dimensional distributions allow little doubt in these assignments. As an example, peaks observed at m/z = 825.9 and 946.7 correspond to [S1H5N4 + 3Na]3+ and [S2H5N4 + 3Na]3+, respectively, having calculated m/z values of 825.7 and 946.1, respectively (Figure 2). Other peaks in the spectrum are assigned in a similar fashion. Over the m/z range between 700 and 1500, the primary peaks correspond to multiply sodiated glycan ions. Many of the peaks are assigned to the known bi-, tri-, and tetra-antennary glycan structures, with varying degrees of sialylation and fucosylation.
Table 1 provides a list of 22 ions (including charge states and assignments) that are observed in this sample. We focus on these assignments because the peaks associated with these ions are observed in all of the samples examined. Moreover, a number of these ions correspond to glycans where there is evidence for only a single-ion structure, while other glycans are expected to exist in more than one isomer form.19,20 As discussed in more detail below, glycans that exist as a single positional isomer may exhibit multiple peaks corresponding to stable gas-phase ion conformations; however, the ratio of these peaks should not vary from sample to sample. Thus, they can be used as controls. On the other hand, the IMS distributions of glycans that may exist as isomers might change depending upon differences in the abundances of different isomers.
Table 1.
Assignments of Glycans from a QT Serum Samplea
| m/z measured | charge | m/z calculated | proposed glycan | putative structure |
|---|---|---|---|---|
| 705.3 | 3+ | 705.3 | H5N4 |
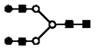
|
| 746.0 | 3+ | 746.0 | H8N2 |
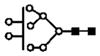
|
| 763.6 | 3+ | 763.4 | F1H5N4 |
|
| 801.6 | 2+ | 801.4 | H5N2 |
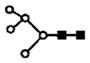
|
| 814.0 | 3+ | 814.1 | H9N2 |
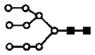
|
| 826.0 | 3+ | 825.7 | S1H5N4 |
|
| 884.1 | 3+ | 883.8 | S1F1H5N4 |
|
| 903.7 | 2+ | 903.4 | H6N2 |
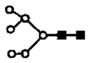
|
| 907.8 | 3+ | 907.4 | S1H5N5 |
|
| 946.7 | 3+ | 946.1 | S2H5N4 |
|
| 1004.8 | 3+ | 1004.2 | S2F1H5N4 |
|
| 1005.8 | 2+ | 1005.5 | H7N2 |
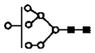
|
| 1086.5 | 3+ | 1085.9 | S2F1H5N5 |
|
| 1096.4 | 3+ | 1095.9 | S2H6N5 |
|
| 1108.0 | 2+ | 1107.5 | H8N2 |
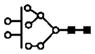
|
| 1209.5 | 2+ | 1209.6 | H9N2 |
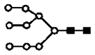
|
| 1216.9 | 3+ | 1216.3 | S3H6N5 |
|
| 1227.4 | 2+ | 1227.1 | S1H5N4 |
|
| 1275.0 | 3+ | 1274.3 | S3F1H6N5 |
|
| 1314.5 | 2+ | 1314.1 | S1F1H5N4 |
|
| 1366.3 | 3+ | 1366.0 | S3H7N6 |
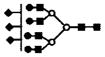
|
| 1407.9 | 2+ | 1407.7 | S2H5N4 |
|
S represents sialic acid (diamonds); F represents fucose (triangles); H represents hexose (mannose open circles, galactose solid circles); N-represents N-acetyl glycosamine (squares).
Example Glycan IMS Distributions
Additionally, Figure 2 shows two insets that are typical of the IMS profiles of many of the glycan ions that are detected. The insets that are shown correspond to [S1H5N4 + 3Na]3+ and [S2H5N4 + 3Na]3+. The latter ion, [S2H5N4 + 3Na]3+, is chosen because it is one of the species that is expected (based on prior work)20 to contain only a single positional structural isomer, whereas the former distribution, corresponding to [S1H5N4 + 3Na]3+, is chosen because this glycan is known to exist as two different positional structural isomers (i.e., we have not included linkage isomers).19,20 Interestingly, the IMS distributions for both of these ions show multiple features. The [S2H5N4 + 3Na]3+ distribution shows a main peak at 42.4 ms, with shoulders on each side at 41.0 and 43.4 ms; the [S1H5N4 + 3Na]3+ ion shows a sharp peak at 38.3 ms, and broad, unresolved features corresponding to species with higher mobility that extend from ~34.0 ms to approximately ~37.9 ms. The observation of multiple peaks in the IMS data could be explained by the existence of multiple isomeric forms, and/or the separation of different conformations of the glycan ions that are stable in the gas phase during the millisecond time scale of the mobility separation.
Example MS Distributions for Different Samples
It is possible to obtain mass spectral data by integrating the signals that are obtained for all drift times. It is often useful to visually inspect mass spectra since it provides direct information about the abundances of different ions. MS distributions are dominated by the same peaks for all data sets. Figure 3 illustrates this overall similarity for the mass spectra obtained from analysis of samples from three individuals of the HC, NC, or QT cohorts (chosen randomly). These spectra also help provide some insight about the variations in signals associated with different ions. We stress that others have shown that differences that are apparent upon careful analysis of these types of mass spectral data are sufficient to group disease states.21 However, here we are interested in examining the possibility that information contained in the IMS distributions might also be valuable for grouping such individuals.
Figure 3.

Overlap of normalized total mass spectra for a set (three samples) of glycans isolated from human serum of a patient with liver cancer (bottom), a healthy patient (middle), and a patient with a liver cirrhosis (top). Glycan structures were assigned by comparison of measured m/z values with m/z values of glycans found in the literature, and the assignment is shown on the top of the plot. S represents sialic acid, H represents hexose (mannose or galactose), N represents N-acetyl glucosamine, and F represents fucose.
Principal Component Analysis
With this in mind, we carried out a PCA of IMS distributions for individual m/z values. For an informative statistical analysis of the acquired glycomic profiles, the PCA procedure was employed, as is commonly used in microarray research for cluster analysis. PCA is designed to capture a variance in the given data sets in terms of their principal components, meaning a set of variables which defines a projection encapsulating the maximum amount of variation in a data set. It is orthogonal (and, therefore, uncorrelated) to the previous principal component.22,23 Principal components are linear combinations of the original variables that explain the variance in the data, that is, PC1 = p1x1 + p2x2 + p3x3 + … + pnxn, where loading (pn) represents the contribution of variable xn to the principal components. In our experiment, the variables are intensities across glycan ion mobility profiles, and the highest loadings correspond to the most variable features (peaks) in the ion mobility distributions.
We began by using a clustering algorithm to examine the ion mobility profiles of the [S1H5N4 + 3Na]3+ ion (as mentioned above, S1H5N4 is known to exist as two positional isomers)19,20 from the serum. Figure 4A shows a plot of principal component scores for the different samples. PCA scores are shown commonly for first two principal components because they show maximum variances in the data. This analysis shows that data points from individuals making up the QT and NC groups are intermixed primarily across the left side of the plot. On the other hand, data points from the individuals corresponding to the HC group appear to cluster into a region of the plot with higher values of PC1 than the other groups. That the HC samples cluster in a region of the PCA plot suggests that there are common differences in IMS profiles for the HC group. The aim of this supervised PCA is to use prior knowledge of the sample groups to find a rule for allocating individuals from unknown group.24 Although the appearance of false positives or false negatives cannot be ruled out completely, from our analysis, we would expect other HC individuals to fall into the clustering region shown in Figure 4A. This in effect may allow us to distinguish patients having liver cancer from the healthy and liver cirrhosis groups.
Figure 4.

(A) PCA of drift time profiles for glycan [S1H5N4 + 3Na]3+ (m/z = 826.0). (B) PCA of drift time profiles for glycan [S2H5N4 + 3Na]3+ (m/z = 946.7). Both analyses were done a total of 61 samples, that is, 19 glycan samples from serum of patients with liver cancer (HC samples, green circles), 22 glycan samples from healthy patients (NC-samples, red squares), and 21 glycan sample from patients with liver disease cirrhosis (QT-samples, purple triangles).
In contrast, ion mobility profiles of [S2H5N4 + 3Na]3+ that originate from the same sets of glycan samples failed to show this clustering (Figure 4B). This lack of statistical correlation immediately implies that the ion mobility profiles for this particular glycan cannot be used to distinguish patient groups. Moreover, the finding is interesting because there is no evidence in the literature associated with multiple positional structural isomers for this glycan.20 Thus, it would appear that the IMS profile, which shows a peak with shoulders on each side, arises from a distribution of conformations rather than covalent isomers.
Features in the ion mobility profiles of these glycans were also statistically analyzed using the ANOVA test. The results are summarized in Table 2. The ANOVA test was applied to four features in the ion mobility profiles of [S1H5N4 + 3Na]3+ (recorded on the IMS instrument shown in Figure 1), indicating a greater than 95% probability of significance in the difference between the HC group and other two groups of samples. This is also true for the doubly sodiated ion of this glycan. Hence, both PCA and ANOVA assign S1H5N4 as a putative biomarker of liver cancer. The PCA and ANOVA tests were also applied to the analysis of ion mobility profiles of several other glycans (Tables 2 and 3). It is worth noting that both PCA and ANOVA of ion mobility profiles of the glycan ion [S2H6N5 + 3Na]3+ (m/z = 1096.4) can differentiate between all three sample groups, assigning this glycan as a putative biomarker of both liver cancer and cirrhosis. Also, the PCA of the drift time profiles of the glycan [S3F1H6N5 + 3Na]3+ (m/z = 1274.9) shows that profiles of this glycan could readily distinguish both cancer and cirrhosis patients from healthy individuals (data not shown). In addition, the PCA of several other glycan ions shows clustering of data corresponding to diseased and healthy groups (Table 3).
Table 2.
ANOVAa Test of Features Across Drift Time Profiles of Selected Glycan Ions
| glycan structureb | measured m/z | charge | featurec drift time (ms) |
P-valuesd,e HC vs NC |
P-valuesd,e NC vs QT |
P-valuesd,e HC vs QT |
|---|---|---|---|---|---|---|
| H5N2 | 801.6 | 2+ | 19.231 | 0.00005 | 0.0007 | 0.6NS |
| 19.595 | 0.00001 | 0.001 | 0.2NS | |||
| S1H5N4 | 826.0 | 3+ | 17.417 | 0.004 | 0.09NS | 0.04 |
| 18.324 | 0.002 | 0.05 | 0.05 | |||
| 19.171 | 0.005 | 0.1NS | 0.03 | |||
| 19.504 | 0.003 | 0.3NS | 0.007 | |||
| S1F1H5N4 | 884.1 | 3+ | 18.657 | 0.6NS | 0.2NS | 0.04 |
| 19.201 | 0.7NS | 0.3NS | 0.1NS | |||
| 19.988 | 0.4NS | 0.007 | 0.03 | |||
| 20.351 | 0.5NS | 0.009 | 0.03 | |||
| S2H5N4 | 946.7 | 3+ | 20.291 | 0.3NS | 0.1NS | 0.003 |
| 21.440 | 0.3NS | 0.08NS | 0.0008 | |||
| S2H6N5 | 1096.4 | 3+ | 22.408 | 0.0002 | 0.04 | 0.003 |
| S3H6N5 | 1216.9 | 3+ | 24.253 | 0.02 | 0.1NS | 0.00001 |
| S1H5N4 | 1227.4 | 2+ | 26.129 | 0.003 | 0.07NS | 0.02 |
| 27.157 | 0.01 | 0.2NS | 0.02 | |||
| S3F1H6N5 | 1275.0 | 3+ | 24.586 | 0.05 | 0.3NS | 0.003 |
| S2H5N4 | 1407.9 | 2+ | 29.123 | 0.8NS | 0.0001 | 0.0003 |
Analysis of variances.
S represents sialic acid; F represents fucose; H represents hexose (mannose or galactose); N represents N-acetyl glycosamine.
Drift time of a peak in the ion mobility distribution of a glycan.
NS, not significant, P > 0.05.
HC-glycans from liver cancer serum; NC-glycans from control serum; QT-glycans from cirrhosis serum.
Table 3.
Supervised PCA Analysis of Selected Glycan Ionsa
| glycan structureb | charge | number of positional isomers | HC clustering | NC clustering | QT clustering |
|---|---|---|---|---|---|
| H5N2 | 2+ | 1 | Yes | Yes | Yes |
| S1H5N4 | 3+ | 2 | Yes | No | No |
| S1F1H5N4 | 3+ | 2 | Yes | No | No |
| S2H5N4 | 3+ | 1 | No | No | No |
| S2H6N5 | 3+ | 3 | Yes | Yes | Yes |
| S3H6N5 | 3+ | 1 | Yes | Yes | Yes |
| S1H5N4 | 2+ | 2 | Yes | Yes | Yes |
| S3F1H6N5 | 3+ | 1 | Yes | Yes | Yes |
| S2H5N4 | 2+ | 1 | No | No | No |
The PCA was done on a total of 61 samples, that is, 19 glycan samples from serum of patients with liver cancer (HC samples), 22 glycan samples from healthy patients (NC-samples), and 20 glycan samples from patients with the liver disease cirrhosis (QT-samples).
S represents sialic acid; F represents fucose; H represents hexose (mannose or galactose); N represents N-acetyl glycosamine.
In considering the tabulated results, it is interesting to note that the glycoprotein alpha-fetoprotein (AFP) is currently used as a tumor marker for hepatocellular carcinoma (HCC).25 Additional work has previously identified N-linked glycans that are bound to this 591 amino acid–protein as H5N4, F1H5N4, S1H5N4, S1F1H5N4, S2H5N4, and S2F1H5N4.26,27 These glycans are among the ones that we detected in both healthy and diseased samples, while the level of S1H5N4 glycan structure is significantly different between the three sets of samples as depicted in Table 2. Increased or decreased amounts of some of these in the cirrhosis or cancer serum could be indicative of an aberrant expression of diagnostic proteins such as AFP. It has been shown that increased fucosylation might be a sign of hepatocellular carcinoma,28–30 and the analysis of ion mobility profiles bolsters the evidence that glycosylation changes with the development of cancer.
According to the World Health Organization, liver cancer was the third common cancer in the world in 2005 causing the death of 662 000 people.31 Liver cirrhosis is a disease, which often precedes liver cancer. Because 5-year survival rate of patients with liver cancer is very low (approximately 10% for the period from 1975 to 2002),32 any advancement in the early diagnosis of cirrhosis and liver cancer should prove to be very beneficial. Glycomic analysis of serological biomarkers seems to be a reasonable approach in the search for biomarkers of liver cancer and cirrhosis because many serum glycoproteins are produced by the liver. Serological analysis of glycans may be less appropriate for the analysis of other cancer types due to the lack of tissue-specific glycoproteins released into the bloodstream. However, the method described may be also useful for the monitoring metastasis of a particular cancer to the liver, or for the analysis of glycans released from bodily tissues other than serum.
Further Delineation of Isomers and Conformers
One of the complicating factors of the IMS approach is that, in the gas-phase, isomers may exist as a distribution of stable conformations. The degree to which these distributions overlap in drift time influences the characterization of isomers. It is possible to test whether multiple peaks in an IMS spectrum are associated with a single glycan covalent structure by selecting different regions of the spectrum. If upon activation the different selected regions reestablish the same distribution of peaks, then the isolated species must be capable of reaching a “quasi equilibrium” with the other states. Such behavior is indicative of a mixture of conformers which have a common covalent framework, that is, a single isomeric form. On the other hand, the inability of a selected region of the spectrum to reestablish equilibrium with other peaks in the IMS distribution indicates that there are kinetic restrictions associated with reaching all conformations. While such barriers could arise from tight transition states required to reach specific conformers, the behavior is also consistent with the coexistence of multiple isomeric forms. In the latter case, we would not expect isomers to be in equilibrium in the gas phase because of the need to rearrange covalent bonds.
The IMS–IMS capabilities shown in Figure 2 enable us to test these ideas. That is, we set a selection gate in order to select specific regions of the distribution, activate the ions by exposing them to a high-field region, and then measure the new distribution that is formed in the second drift tube. We begin by examining the [S2H5N4 + 3Na]3+ ions which are expected not to have positional structural isomers in the serum.20 As mentioned above, multiple features were detected across the drift time profile of [S2H5N4 + 3Na]3+ (Figure 5, left). These features were selected according to their mobilities across this distribution. The positions of three representative selections (out of a total of 7 across the distribution) are shown by dashed lines in Figure 5. Selection and activation of the low-mobility precursors leads to a new IMS distribution that is essentially indistinguishable from the original distribution. This behavior is observed for all 7 selections and indicates that each of the structures that have been separated is capable of reestablishing the other states upon activation. Moreover, these states are formed in the same abundances, indicating that the distribution reflects a system that has reached equilibrium. This behavior is consistent with the presence of a single positional structural isomer of [S2H5N4 + 3Na]3+.
Figure 5.

(A) Probing conformational changes of glycan isomers by IMS-IMS–MS. Precursor and mobility selected drift time distributions for [S2H5N4 + 3Na]3+ (m/z = 946.7) at three different selection times (shown by dashed lines). The drift time distributions for mobility selected precursors are obtained after gentle activation of ions at IA2. (B) Probing conformational changes of isomers by IMS-IMS–MS (continued). Precursor and mobility selected drift time distributions for [S1H5N4 + 3Na]3+ (m/z = 826.0) at three different selection times (shown by dashed lines). The drift time distributions for mobility selected precursors are obtained after gentle activation of ions at IA2. Selections made for low mobility precursors are dominated by similar features with an identical distribution after ion activation indicating presence of one isomer. High-mobility region is dominated by features that do not show similar distribution upon ion activation indicating presence of another isomer.
In contrast, a similar analysis of the [S1H5N4 + 3Na]3+ ion shows that selection and activation of high- and low-mobility regions leads to spectra that appear very different (Figure 5, right). Specifically, activation of such selected high-mobility ions leads to a distribution that is dominated by a broad high-mobility feature, with only a small hint of the sharp peak at longer drift times (38.3 ms). The sharp features are favored, but only when low-mobility ions are selected. From this, we conclude that more than one type of ion may be present.
Similar analyses of other glycans (and a series of controls) lead us to conclude that the behavior associated with the [S1H5N4 + 3Na]3+ ion arises because of the existence of multiple isomers. Additionally, variation in the relative abundances of these isomers across different samples (i.e., different physiological states) would explain the ability of the PCA analysis to distinguish groups. We stress that it is important to follow up this type of study with other analyses, such as the measurements of MS/MS distributions for these ions (which also should reflect differences in relative isomer distributions). However, this analysis provides direct evidence that changes in isomeric composition of glycans may also be an indicator of disease state. Further work is underway to assign different regions of the IMS distributions to specific glycan isomers.
Conclusions
IMS–MS and IMS-IMS–MS techniques were used to study glycans isolated from human serum of healthy and diseased patients. The IMS distributions that are recorded show evidence for multiple stable species in the gas phase corresponding in some cases to what appears to be mixtures of conformers and in others what may be mixtures of different conformers and isomer forms. Statistical analyses of these data suggest that aberrations in isomer distributions (which cannot be measured easily by MS techniques) may be indicative of some disease states. Although this work is still at an early stage, the methodology appears to be especially relevant to studies of glycans because different isomeric forms of these ions may exhibit structures that are readily separated and distinguished as based on differences in three-dimensional geometries. Having said this, we have shown that even a single covalent structure may give rise to a range of different stable conformations that can be separated in the gas phase. Thus, caution must be used in interpreting the meaning of multiple peaks in IMS data.
The PCA and ANOVA approaches used to analyze data show that, in favorable cases, information from the IMS separation of a single m/z ion can be sufficient to cluster individuals into the cohorts with specific physiologies. In the case of S1H5N4, individuals with liver cancer appear to cluster away from cirrhosis and healthy groups. This finding is consistent with analysis of MS data; the ability of an IMS analysis to further characterize these groups (at no additional cost in time of analysis) should complement the MS-based methods by providing an internal cross check, as well as more refined interpretations of the physiological state (from combinations of MS and IMS data). Additional markers would certainly be valuable for diagnosis of hepatocellular carcinoma and other pathological conditions. For example, the specificity of the currently used glycoprotein tumor marker alpha-fetoprotein (AFP) is 34–61%,33 while the specificity of prostate-specific antigen (PSA) is only 25%;34 an IMS glycan analysis would be expected to improve this confidence level of cancer diagnosis. We are presently working on methods to improve fragmentation efficiency of the selected glycan isomers and conformers, since this would substantially impact the characterization of these species.
Acknowledgments
This work was supported by the National Center for Glycomics and Glycoproteomics (NCGG) under grant number RR018942 through the National Institute of Health (NIH) within the National Center for Research Resources (NCRR), and was supported in part by an Associate Membership from NCI’s Early Detection Research Network, NCI grants R03 CA119288 and R01 CA115625-01A2 awarded to R.G.
References
- 1.Hanash S. Nature. 2003;422:226–232. doi: 10.1038/nature01514. [DOI] [PubMed] [Google Scholar]
- 2.Anderson NL, Anderson NG. Mol Cell Proteomics. 2002;1:845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 3.(a) Aebersold R, Mann M. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]; (b) Domon B, Aebersold R. Science. 2006;322:212–217. doi: 10.1126/science.1124619. [DOI] [PubMed] [Google Scholar]; (c) Merenbloom SI, Koeniger SL, Bohrer BC, Valentine SJ, Clemmer DE. Anal Chem. 2007 doi: 10.1021/ac7018602. submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Valentine SJ, Counterman AE, Hoaglund CS, Reilly JP, Clemmer DE. J Am Soc Mass Spectrom. 1998;9:1213–1216. doi: 10.1016/S1044-0305(98)00101-9. [DOI] [PubMed] [Google Scholar]; (b) Henderson SC, Valentine SJ, Counterman AE, Clemmer DE. Anal Chem. 1999;71:291–301. doi: 10.1021/ac9809175. [DOI] [PubMed] [Google Scholar]; (c) Ruotolo BT, Gillig KJ, Stone EG, Russell DH. J Chromatogr, B. 2002;782:385–392. doi: 10.1016/s1570-0232(02)00566-4. [DOI] [PubMed] [Google Scholar]; (d) Steiner WE, Clowers BH, English WA, Hill HH. Rapid Commun Mass Spectrom. 2004;18:882–888. doi: 10.1002/rcm.1419. [DOI] [PubMed] [Google Scholar]; (e) Moon MH, Myung S, Plasencia MD, Hilderbrand AE, Clemmer DE. J Proteome Res. 2003;2:589–597. doi: 10.1021/pr034018v. [DOI] [PubMed] [Google Scholar]; (f) Valentine SJ, Liu X, Plasencia MD, Hilderbrand AE, Kurulugama RT, Koeniger SL, Clemmer DE. Expert Rev Proteomics. 2005;2:553–565. doi: 10.1586/14789450.2.4.553. [DOI] [PubMed] [Google Scholar]
- 5.Clemmer DE, Jarrold MF. J Mass Spectrom. 1997;32:577–592. [Google Scholar]
- 6.Zhang H, Li X, Martin DB, Aebersold R. Nat Biotechnol. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 7.Liu T, Qian WJ, Gritsenko MA, Camp DG, Monroe ME, Moore J, Smith RD. J Proteome Res. 2005;4:2070–2080. doi: 10.1021/pr0502065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qiu R, Regnier FE. Anal Chem. 2005;77:7225–7231. doi: 10.1021/ac050554q. [DOI] [PubMed] [Google Scholar]
- 9.Zhao J, Simeone DM, Heidt D, Anderson MA, Lubman DM. J Proteome Res. 2006;5:1792–1802. doi: 10.1021/pr060034r. [DOI] [PubMed] [Google Scholar]
- 10.An HJ, Miyamoto S, Lancaster KS, Kirmiz C, Li BS, Lam KS, Leiserowitz GS, Lebrilla CB. J Proteome Res. 2006;5:1626–1635. doi: 10.1021/pr060010k. [DOI] [PubMed] [Google Scholar]
- 11.(a) Kyselova Z, Mechref Y, Al Bataineh MM, Dobrolecki LE, Hickey RJ, Vinson J, Sweeney CJ, Novotny MV. J Proteome Res. 2007;6:1822–1832. doi: 10.1021/pr060664t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kyselova Z, Mechref Y, Kang P, Goetz JA, Dobrolecki LE, Hickey RJ, Malkas LH, Novotny MV. Clin Chem. 2007 doi: 10.1373/clinchem.2007.087148. in press. [DOI] [PubMed] [Google Scholar]
- 12.(a) Liu X, Valentine SJ, Plasencia MD, Trimpin S, Naylor S, Clemmer DE. J Am Soc Mass Spectrom. 2007;18:1249–1264. doi: 10.1016/j.jasms.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Valentine SJ, Plasencia MD, Liu X, Krishnan M, Naylor S, Udseth HR, Smith RD, Clemmer DE. J Proteome Res. 2006;5:2977–2984. doi: 10.1021/pr060232i. [DOI] [PubMed] [Google Scholar]; (c) Liu X, Plasencia MD, Ragg S, Valentine SJ, Clemmer DE. Briefings Funct Genomics Proteomics. 2004;3:177–186. doi: 10.1093/bfgp/3.2.177. [DOI] [PubMed] [Google Scholar]
- 13.Koeniger SL, Merenbloom SI, Valentine SJ, Jarrold MF, Udseth HR, Smith RD, Clemmer DE. Anal Chem. 2006;78:4161–4174. doi: 10.1021/ac051060w. [DOI] [PubMed] [Google Scholar]
- 14.Merenbloom SI, Koeniger SL, Valentine SJ, Plasencia MD, Clemmer DE. Anal Chem. 2006;78:2802–2809. doi: 10.1021/ac052208e. [DOI] [PubMed] [Google Scholar]
- 15.Goldman R, Ressom HW, Abdel-Hamid M, Goldman L, Wang A, Varghese RS, An Y, Loffredo CA, Drake SK, Eissa SA, Gouda I, Ezzat S, Moiseiwitchs FS. Carcinogenesis. 2007;28:2149–2153. doi: 10.1093/carcin/bgm177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ezzat S, Abdel-Hamid M, Eissa SA, Mokhtar N, Labib NA, El-Ghory R, Mikhaill NN, Abdel-Hamid A, Hifnay T, Strickland GT, Loffredo CA. Int J Hyg Environ Health. 2005;208:329–339. doi: 10.1016/j.ijheh.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 17.Kang P, Mechref Y, Klouckova I, Novotny MV. Rapid Commun Mass Spectrom. 2005;19:3421–3428. doi: 10.1002/rcm.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trimpin S, Plasencia M, Isailovic D, Clemmer DE. Anal Chem. 2007;79:7965–7974. doi: 10.1021/ac071575i. [DOI] [PubMed] [Google Scholar]
- 19.Kranz C, Ng BG, Sun LW, Sharma V, Eklund EA, Miura Y, Ungar D, Lupashin V, Winkel RD, Cipollo JF, Costello CE, Loh E, Hong W, Freeze HH. Hum Mol Genet. 2007;16:731–741. doi: 10.1093/hmg/ddm028. [DOI] [PubMed] [Google Scholar]
- 20.Consortium for functional glycomics and Nature publishing group. Functional Glycomics Gateway; 2006. available at www.functionalglycomics.org. [Google Scholar]
- 21.Goldman R, Ressom HW, Varghese RS, Goldman L, Bascug G, Loffredo CA, Abdel-Hamid M, Gouda I, Ezzat S, Kyselova Z, Mechref Y, Novotny MV. Clin Cancer Res. 2007 doi: 10.1158/1078-0432.CCR-07-5261. submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hotelling H. J Educat Psych. 1933;24:417–441. [Google Scholar]
- 23.Musumarra G, Barresi V, Condorelli DF, Scire S. Biol Chem. 2003;384:321–327. doi: 10.1515/BC.2003.037. [DOI] [PubMed] [Google Scholar]
- 24.Miller JN, Miller JC. Statistics and Chemometrics for Analytical Chemistry. Pearson Education Limited; Harlow, England: 2000. [Google Scholar]
- 25.Li D, Mallory T, Satomura S. Clin Chim Acta. 2001;313:15–19. doi: 10.1016/s0009-8981(01)00644-1. [DOI] [PubMed] [Google Scholar]
- 26.Yoshima H, Mizuochi T, Ishii M, Kobata A. Cancer Res. 1980;40:4276–4281. [PubMed] [Google Scholar]
- 27.Johnson PJ, Poon TCW, Hjelm NM, Ho CS, Ho SKW, Welby C, Stevenson D, Patel T, Parekh R, Townsend RR. Br J Cancer. 1999;81:1188–1195. doi: 10.1038/sj.bjc.6690828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Comunale MA, Lowman M, Long RE, Krakover J, Philip R, Seeholzer S, Evans AA, Hann HWL, Block TM, Mehta AS. J Proteome Res. 2006;5:308–315. doi: 10.1021/pr050328x. [DOI] [PubMed] [Google Scholar]
- 29.Steel LF, Mattu TS, Mehta A, Hebestreit H, Dwek R, Evans AA, Dai Z, Liu YK, Cui JF, Shen HL, Chen J, Sun RX, Zhang Y, Zhou XW, Yang PY, Tang ZY. Proteomics. 2006;6:5857–5867. [Google Scholar]
- 30.Dai Z, Liu YK, Cui JF, Shen HL, Chen J, Sun RX, Zhang Y, Zhou XW, Yang PY, Tang ZY. Proteomics. 2006;6:5857–5867. doi: 10.1002/pmic.200500707. [DOI] [PubMed] [Google Scholar]
- 31.World Health Organization. Cancer Facts. available at http://www.who.int/mediacentre/factsheets/fs297/en/
- 32.American Cancer Society. Cancer Statistics. 2007 available at www.cancer.org.
- 33.Lopez LJ, Marrero JA. Curr Opin Gastroenterol. 2004;20:248–253. doi: 10.1097/00001574-200405000-00009. [DOI] [PubMed] [Google Scholar]
- 34.Brawer MK. Cancer J Clin. 1999;49:264–281. doi: 10.3322/canjclin.49.5.264. [DOI] [PubMed] [Google Scholar]