

The X-ray crystallographic structure of the disulfide-containing HCAII (dsHCAII) has been solved to 1.77 Å resolution and revealed that successful oxidation of the cysteine bond was achieved while also retaining desirable active-site geometry.
Keywords: human carbonic anhydrase II, thermal stability, disulfide bridges
Abstract
The carbonic anhydrases (CAs) are a family of mostly zinc metalloenzymes that catalyze the reversible hydration of CO2 to bicarbonate and a proton. Recently, there has been industrial interest in utilizing CAs as biocatalysts for carbon sequestration and biofuel production. The conditions used in these processes, however, result in high temperatures and acidic pH. This unfavorable environment results in rapid destabilization and loss of catalytic activity in CAs, ultimately resulting in cost-inefficient high-maintenance operation of the system. In order to negate these detrimental industrial conditions, cysteines at residues 23 (Ala23Cys) and 203 (Leu203Cys) were engineered into a wild-type variant of human CA II (HCAII) containing the mutation Cys206Ser. The X-ray crystallographic structure of the disulfide-containing HCAII (dsHCAII) was solved to 1.77 Å resolution and revealed that successful oxidation of the cysteine bond was achieved while also retaining desirable active-site geometry. Kinetic studies utilizing the measurement of 18O-labeled CO2 by mass spectrometry revealed that dsHCAII retained high catalytic efficiency, and differential scanning calorimetry showed acid stability and thermal stability that was enhanced by up to 14 K compared with native HCAII. Together, these studies have shown that dsHCAII has properties that could be used in an industrial setting to help to lower costs and improve the overall reaction efficiency.
1. Introduction
The carbonic anhydrases (CAs; EC 4.2.1.1) are metalloenzymes that catalyze the reversible hydration of CO2 to bicarbonate and a proton. The CAs consist of several evolutionarily unrelated structural families, the best studied of which are the α-CAs, which are found primarily in animals; β-CAs, which are found predominantly in plants; and γ-CAs, which have only been identified in several species of archaea (Lindskog, 1997 ▶; Aggarwal et al., 2013 ▶). In humans, 12 active α-CA isozymes (HCAI, HCAII, HCAIII, HCAIV, HCAV, HCAVA, HCAVB, HCAVI, HCAVII, HCAIX, HCAXII, HCAXII and HCAXIV) are present. Human CA II (HCAII), the best-characterized isoform to date, is cytosolic and is expressed in nearly all tissues (Sly & Hu, 1995 ▶). It is highly soluble and monomeric in solution, with a molecular weight of approximately 29 kDa, and is one of the fastest CAs known, with a k cat of 1 × 106 s−1 and a k cat/K m of 1.5 × 108 M −1 s−1 that approaches the diffusion limit (Khalifah, 1971 ▶; Steiner et al., 1975 ▶). The catalytic mechanism of HCAII proceeds via a two-step ping-pong mechanism,
In the hydration direction, nucleophilic attack on CO2 by the zinc-bound hydroxide results in the formation of bicarbonate ion, which is subsequently released from the active site, leaving a water molecule bound to the zinc metal (1). In the next step (2), the zinc-bound hydroxide is regenerated for the next round of catalysis via the transfer of a proton from the zinc-bound water, via the proton-shuttle residue His64, to an endogenous H+ acceptor in the bulk solvent, B (Silverman & Lindskog, 1988 ▶; Tu et al., 1989 ▶).
The relative ease of expression, solubility and fast kinetic properties of HCAII have made it a favorable candidate to serve as a biocatalyst for carbon sequestration of the flue gas emitted during the burning of fossil fuels (Savile & Lalonde, 2011 ▶), as well as for utilization in algae to capture and convert CO2 into biofuels and other valuable products (Fulke et al., 2010 ▶; Ramanan et al., 2010 ▶). However, current use of HCAII as a carbon sequester in the industrial setting is limited by the harsh conditions (temperatures in excess of 343 K and a pH of less than 6.0) employed by these techniques, such that the stability of the free enzyme in solution is significantly compromised, ultimately resulting in lowered recycling, recovery and cost-efficiency of the overall process (Kanbar & Ozdemir, 2010 ▶). As such, there have been several studies (Kanbar & Ozdemir, 2010 ▶; Vinoba et al., 2011 ▶) that have immobilized HCAII onto a variety of surfaces in an attempt to overcome these stability and recovery issues. However, these studies have largely been unsuccessful as a significant loss of catalytic activity for these immobilized enzyme arrays is observed at temperatures above 328 K.
A conserved disulfide bridge has been observed between residues 23 and 203 (HCAII numbering) in all of the membrane-bound human isozymes of CA (HCAIV, HCAIX, HCAXII and HCAXIV) as well as in the secreted isoform HCAVI (Stams & Christianson, 2000 ▶). These disulfide bridges are thought to confer extra stability in the harsh extracellular environment to which these isoforms are exposed and could provide an explanation for their resistance to denaturation by SDS (Whitney & Briggle, 1982 ▶). Additionally, a CA isolated from Neisseria gonorrhoeae also contains a disulfide bridge at the aforementioned position (Huang et al., 1998 ▶) and has been shown to remain globular up to 2.1 M concentration of guanidine hydrochloride (GuHCl; Elleby et al., 2001 ▶).
Given the potential industrial need for a highly stable variant of CA that can withstand the high temperatures and acidic environment of current carbon-sequestration protocols, we have extended previous studies (Mårtensson et al., 2002 ▶) via the engineering of a disulfide linkage at residues 23 and 203 (Ala23Cys/Leu203Cys) into a pseudo-wild-type HCAII (HCAIIpwt) containing the Cys206Ser mutation. This disulfide-containing variant (dsHCAII) has previously been shown by tryptophan fluorescence measurements (Mårtensson et al., 2002 ▶) to have an approximately twofold increased resistance to GuHCl-induced denaturation compared with HCAIIpwt (1.7 versus 0.9 M, respectively), corresponding to a net stabilization of 6.6 kcal mol−1 (1 cal = 4.186 J). However, chemical and thermal denaturation have been shown to be uncorrelated in some proteins (Wang et al., 2011 ▶). As such, in this study differential scanning calorimetry (DSC) was used to assess the thermal stability of dsHCAII and revealed that this variant has a significantly enhanced thermal tolerance under near-physiological conditions as well as in an acidic environment compared with HCAII. Moreover, depletion of an 18O label from CO2 measured by mass spectrometry revealed that dsHCAII has a comparable catalytic efficiency to HCAII but has a higher tolerance for elevated temperatures. Finally, the X-ray crystal structure of dsHCAII was solved to 1.77 Å resolution and showed the successful formation of a disulfide linkage in a cytosolic CA.
2. Materials and methods
2.1. Enzyme expression and purification
HCAII cDNA containing Ala23Cys/Leu203Cys/Cys206Ser mutations was prepared from an expression vector containing the enzyme-coding region (Forsman et al., 1988 ▶) via site-directed mutagenesis using the Stratagene QuikChange II kit and primers from Invitrogen. The mutant cDNA was transformed into Escherichia coli XL1-Blue super-competent cells, which were then confirmed by DNA sequencing of the entire coding region. The dsHCAII cDNA was transformed in E. coli BL21(DE3) cells in 1 l 2× Luria broth medium containing ∼0.1 mg ml−1 ampicillin and grown at 310 K to a turbidity of ∼0.6 at 600 nm. Protein production was induced by the addition of ∼0.1 mg ml−1 isopropyl β-d-1-thiogalactopyranoside (IPTG) and ∼1 mM zinc sulfate (final concentrations). The cells were incubated for an additional 3 h and harvested by centrifugation.
A suspension of cells in 200 mM sodium sulfate, 100 mM Tris–HCl pH 9.0 was lysed by the addition of hen egg-white lysozyme and DNaseI with subsequent removal of cellular debris by centrifugation. The dsHCAII enzyme was purified on an affinity column containing agarose resin coupled with p-(aminomethyl)benzenesulfonamide, a tight-binding inhibitor of HCAII (Khalifah et al., 1977 ▶). The bound dsHCAII was eluted with 400 mM sodium azide, 100 mM Tris pH 7.0 followed by extensive dialysis in 50 mM Tris–HCl pH 7.8 to remove the azide. Oxidized glutathione was then added to the purified sample to a final concentration of ∼0.1 mM to induce disulfide formation (Mårtensson et al., 2002 ▶). The oxidized sample was then concentrated to ∼10 mg ml−1 via centrifugal ultrafiltration using a 10 kDa molecular-weight cutoff filter (Amicon). Possible intermolecular disulfide dimeric complexes were then removed via size-exclusion chromatography on a Superdex 75 column using the dialysis buffer and a flow rate of 0.5 ml min−1. The presence of dimeric dsHCAII complexes was checked via visual inspection of native gel electrophoresis.
2.2. Crystallization and diffraction data collection
Crystals of the dsHCAII variant were obtained after one week by the hanging-drop vapor-diffusion method using a 1:1 mixture of protein solution and reservoir solution consisting of 1.3 M sodium citrate pH 9.0. Diffraction data were collected at 100 K using an in-house Rigaku R-AXIS IV++ image-plate detector on an RU-H3R rotating Cu anode (Cu Kα, 1.5418 Å) operating at 50 kV and 22 mA with a crystal-to-detector distance of 80 mm. The X-rays were focused via Osmic optics followed by a helium-purged beam path. Diffraction data were collected as 1° oscillations with an exposure time of 300 s. The HKL-2000 software package (Otwinowski & Minor, 1997 ▶) was used to integrate, merge and scale the data in the monoclinic space group P21 to a resolution of 1.77 Å. A summary of the data statistics is provided in Table 1 ▶.
Table 1. Refinement and final model statistics for dsHCAII.
Values in parentheses are for the highest resolution bin.
| PDB code | 4hba |
| Wavelength (Å) | 1.5418 |
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 42.3, b = 41.2, c = 71.6, β = 104.2 |
| Total No. of measured reflections | 96044 |
| Total No. of unique reflections | 23307 |
| Resolution (Å) | 20.0–1.77 (1.83–1.77) |
| R merge † (%) | 7.0 (50.0) |
| 〈I/σ(I)〉 | 13.5 (2.7) |
| Completeness (%) | 98.2 (98.9) |
| Multiplicity | 4.1 (4.2) |
| R cryst ‡ (%) | 15.0 |
| R free § (%) | 18.9 |
| No. of residues | 257 |
| No. of protein atoms (including alternate conformations) | 2301 |
| No. of water molecules | 203 |
| R.m.s.d. | |
| Bond lengths (Å) | 0.010 |
| Angles (°) | 1.297 |
| Ramachandran statistics (%) | |
| Allowed | 100.0 |
| Most favored | 96.8 |
| Outliers | 0.0 |
| Average B factors (Å2) | |
| All | 19.5 |
| Main chain | 18.7 |
| Side chain | 20.7 |
| Solvent | 29.2 |
| Cα r.m.s.d.¶ (Å) | 0.2 |
R
merge = 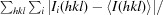
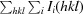 × 100.
× 100.
R
cryst = 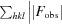 −
− 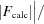
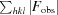 × 100.
× 100.
R free is calculated in the same way as R cryst except that it is calculated for data that were omitted from refinement (5% of reflections).
Root-mean-square deviation of the Cα backbone compared with HCA II (PDB entry 3ks3).
2.3. Structure refinement
Initial phases for the dsHCAII variant were calculated using the coordinates of the high-resolution isomorphous HCAII structure (PDB entry 3ks3; Avvaru et al., 2010 ▶). The PHENIX suite of programs (Adams et al., 2010 ▶) was used in cycles of restrained refinement of the molecular model, alternating with manual building using Coot (Emsley & Cowtan, 2004 ▶). The electron-density map was weak for the disordered N-terminal residues 1–3, but the disulfide linkage at residues 23 and 203 as well as the pseudo-wild-type Cys206Ser mutation were evident in the initial F o − F c difference map, with subsequent refinement showing excellent electron density in the 2F o − F c maps at these positions. Solvent molecules that refined with B factors of greater than 50 Å2 were excluded from the final model. The R cryst and R free values for the final model were 15.0 and 18.9%, respectively. The MolProbity algorithm (Chen et al., 2010 ▶) was used to assess the quality of the final model. The final model statistics are included in Table 1 ▶. All structural figures were generated in PyMOL (DeLano, 2002 ▶). Experimental data and structural coordinates have been deposited in the Protein Data Bank (http://www.rcsb.org) with accession code 4hba.
2.4. Differential scanning calorimetry (DSC)
DSC experiments were performed to assess the thermal stability of the dsHCAII variant under near-physiological and acidic conditions using a VP-DSC calorimeter (Microcal Inc., Northampton, Massachusetts, USA) with a cell volume of ∼0.5 ml. HCAII and dsHCAII samples (6–10 µM) were extensively buffer-exchanged into 50 mM Tris–HCl pH 7.8 or 80 mM sodium citrate, 20 mM citric acid pH 5.6. The samples and buffers were degassed while stirring at 289 K for 20 min prior to data collection. DSC scans were collected from 293 to 373 K at a scan rate of 60 K h−1. The calorimetric enthalpies of unfolding were calculated by integrating the area under the peaks in the thermograms after adjusting the pre-translation and post-translation baselines. The thermograms were fitted to a two-state reversible unfolding model to obtain van’t Hoff enthalpies (ΔH vH) of unfolding.
The melting temperature (T m) values of dsHCAII and HCAII at neutral and acidic pH were obtained from the midpoints of the DSC curves, indicating a two-state transition. The difference in Gibbs free energy (ΔG°) at a given temperature T was calculated via (Becktel & Schellman, 1987 ▶)
where ΔH°m is the calorimetric enthalpy at T m and ΔC p is the observed change in heat capacity between the folded and the unfolded states. The denaturation enthalpies (ΔH°) and entropies (ΔS°) were calculated at a given temperature using the Kirchoff’s law equations (Avvaru et al., 2010 ▶)
Thermograms for all samples were obtained in triplicate and were then averaged to obtain the final profile which was used for reference subtraction and data analysis.
2.5. Kinetics
The catalytic rates of the samples at chemical equilibrium were calculated utilizing 18O exchange between CO2 and water measured by mass spectrometry (Silverman, 1982 ▶). The 18O isotopic content of CO2 in solution was measured as it passed through a semipermeable membrane into an Extrel EXM-200 mass spectrometer. The catalytic process involves two distinct steps which produce two measurable catalytic rates: R
1 and 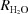 (Silverman, 1982 ▶). The first step entails the random probability of reversibly labeling the zinc-bound hydroxide with 18O (4). In the following reaction, the 18OH− can be protonated, resulting in release of H2
18O to the bulk solvent, where it is essentially infinitely diluted by unlabeled water (7). The residue His64 acts as the proton shuttle in this process (Tu et al., 1989 ▶).
(Silverman, 1982 ▶). The first step entails the random probability of reversibly labeling the zinc-bound hydroxide with 18O (4). In the following reaction, the 18OH− can be protonated, resulting in release of H2
18O to the bulk solvent, where it is essentially infinitely diluted by unlabeled water (7). The residue His64 acts as the proton shuttle in this process (Tu et al., 1989 ▶).
The catalytic rate of conversion between CO2 and HCO3 − is given by R 1 (8). In (8), k cat ex is the rate constant for maximal conversion between substrate and product, while K S eff is the effective binding constant of substrate ([S] is its concentration); the substrate can be either CO2 or HCO3 − depending on the direction of the reaction. The ratio k cat ex/K S eff expressed in (8) is in principle the same as k cat/K m obtained under steady-state conditions and is referred to as k cat/K m in describing our results.
The rate of release of H2
18O from the enzyme into the bulk solvent is given by . This rate is dependent on proton transfer from His64 to the isotopically labeled zinc-bound hydroxide (the dehydration direction in equation 7) through a network of water molecules in the active site (Fig. 1 ▶
d; Tu et al., 1989 ▶). The relationship between the rate constant for proton transfer to the zinc-bound solvent molecule, k
B, and the ionization constants of the proton donor, (K
a)donor, and acceptor, 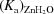 , is shown in (9),
, is shown in (9),
Measurements of R
1 and were performed at 298 K in the absence of buffer using a total substrate concentration (all possible isotope-labeled species of CO2) of 25 mM and an enzyme concentration of ∼1 mg ml−1. The enzymatic activity was also studied in the temperature range 283–343 K in 100 mM HEPES and 10 mM substrate at pH 7.6. After the reaction had been equilibrated at each temperature, a small sample of enzyme [∼0.2%(v/v)] was added to the reaction vessel. Measurements of the 18O content of CO2 were made over the following 1–5 min. The kinetic constants and ionization constants shown in (8) and (9) were determined through nonlinear least-squares methods using EnzFitter (Biosoft).
Figure 1.

X-ray crystallographic structure of dsHCAII. (a) Cartoon view of the overall topology of dsHCAII. The Cα backbone is shown in silver, with N atoms colored blue, O atoms red and S atoms yellow, and the Zn2+ ion represented as a magenta sphere. The coordinating histidine residues to the zinc metal, His64, Cys23, Cys203 and Ser206 are shown in stick view. (b) Enlarged view of the disulfide bridge formed between residues 23 and 203 with the 2F o − F c map contoured at 1.4σ shown in gray. (c) Enlarged view of the dual conformation of Ser206 with the 2F o − F c map contoured at 1.4σ shown in gray with the distance between the hydroxyl group of Ser206 and the backbone carbonyl of Val135 indicated. (d) Comparison of the active-site side chains involved in forming the water network (dashed lines) for dsHCAII (silver) and HCAII (green). The zinc metal is colored blue for dsHCAII and green for HCAII. The figures were generated and rendered with PyMOL (DeLano, 2002 ▶).
3. Results
3.1. X-ray crystallographic structure analysis
The dsHCAII variant crystallized in the monoclinic space group P21, with unit-cell parameters a = 42.3, b = 41.2, c = 71.6 Å, β = 104.2°, and diffracted to a maximum resolution of 1.77 Å. The final model (Fig. 1 ▶ a) refined to an R cryst and R free of 15.0 and 18.9%, respectively. Table 1 ▶ shows a summary of the collected diffraction data set and final refinement statistics; the calculated 2F o − F c map surrounding the disulfide bridge is shown in Fig. 1 ▶(b).
There is very little deviation in the overall structure of the dsHCAII variant when aligned with the Cα backbone of HCAII (0.2 Å; Table 1 ▶); however, there are some local perturbations around the mutation sites. Interestingly, the Cα atom of residue 23 in dsHCAII is shifted by ∼0.8 Å compared with HCAII, whereas the Cα atom at residue 203 is the same in both structures (Fig. 1 ▶ b). This shift in the backbone around residue 23 is presumably a direct result of the formation of the disulfide. In addition, the relative identity in the backbone of residues proximal to residue 203 in dsHCAII compared with HCAII is advantageous in that the residues that are known to be important in the catalytic activity of the enzyme are not distorted (Fig. 1 ▶ d). As such, the crystal structure suggests that the kinetic activity of dsHCAII should correlate well with that of HCAII, as no significant variations are observed in either the backbone or the side chains of the residues lining the catalyic site. The most significant deviation can be seen in the microenvironment surrounding the mutation at residue 206. Ser206 is observed to form a potential hydrogen bond to the carbonyl O atom of Val135, which has been shifted by ∼0.9 Å compared with that in the HCAII structure (Fig. 1 ▶ c). The final F o − F c map shows evidence of a weak secondary conformation of Ser206 (<10% occupancy) that is within ∼2.8 Å of the carbonyl group of Val135. However, refinement of this secondary conformation of Ser206 shows poor electron density in the 2F o − F c map as low as 1.0σ and thus it was not included in the final model. These local deviations do not have an impact on the overall topology of the enzyme and reveal no evidence for a loss in catalytic efficiency, but they do indicate possible sources of enhanced stability (Fig. 1 ▶ a). Interestingly, His64 is predominately observed in the inward configuration (Figs. 1 ▶ a and 1 ▶ d), that is towards the active site, under these crystallization conditions.
The overall geometry of the engineered disulfide bridge in HCAII at residues 23 and 203 correlates well with previous prediction models (Mårtensson et al., 2002 ▶) as well as with X-ray crystallographic models of other CAs in which this disulfide linkage is conserved (Supplementary Fig. S2 and Table S11). The estimated values of the torsion angle (χ 3; Supplementary Fig. S1) and bond length of the engineered disulfide linkage from the prediction model were 91.6° and 2.0 Å, respectively. Additionally, the distance between the Cα atoms of residues 23 and 203 was predicted to be 5.1 Å (Mårtensson et al., 2002 ▶). The observed values from the X-ray crystallographic model of dsHCAII are not significantly different from those predicted above, with corresponding values of 79.8°, 2.1 Å and 5.2 Å, respectively. Gly25 was predicted to undergo a conformational change in its dihedral angles, with a change in the ϕ angle from −69.0° for HCAII to −84.4° for dsHCAII and a change in the ψ angle from 176.7° to −150.9°. The observed ϕ and ψ angles around Gly25 in the X-ray crystallographic model are −77.0° and −172.3°, respectively. This predicted and observed conformational freedom around Gly25 can provide an explanation of the shift observed in the peptide backbone around Cys23.
3.2. Differential scanning calorimetry
The thermal unfolding transitions of dsHCAII and HCAII at neutral and acidic pH values were studied in triplicate using DSC. A major unfolding transition, the observed dominant peak, was observed in all samples (Fig. 2 ▶). The transitions were calculated to be endothermic and were centered at the T m, with the maximum heat capacity (C p) occurring at the midpoint of the peak. The results of DSC analysis are summarized in Table 2 ▶. The T m of dsHCAII at pH 7.8 was measured to be 344.1 ± 0.1 K, revealing remarkable stability in comparison with HCAII, which showed an unfolding transition at 330.2 ± 0.1 K (Figs. 2 ▶ a and 2 ▶ b). This enhanced stability was conserved at the more acidic pH of 5.6, for which dsHCAII showed a T m of 332.7 ± 0.1 K, whereas HCAII was more acid labile with a T m of 318.5 ± 0.1 K (Figs. 2 ▶ c and 2 ▶ d). The increased thermal and acid tolerance of dsHCAII correlates well with its observed chemical stability at high concentrations of GdnHCl in comparison with HCAIIpwt (Mårtensson et al., 2002 ▶). The calorimetric enthalpy at the transition midpoint (ΔH°m) was calculated via integration of the area under the unfolding peak, normalized to the protein molar concentrations, and revealed values of approximately 310 and 220 kcal mol−1 for dsHCAII and HCAII at pH 7.8, respectively. This increased enthalpic contribution to the overall stability of dsHCAII compared with HCAII was also observed at pH 5.6, with approximate ΔH°m values of 200 and 150 kcal mol−1, respectively.
Figure 2.

DSC thermograms showing the specific heat as a function of temperature with the buffer-subtracted and baseline-corrected data shown in black and the fit to the curves shown in red. (a) dsHCAII at pH 7.8 showing three major peaks at 324, 339 and 344 K. (b) HCAII at pH 7.8 showing a single transition at 333 K. (c) dsHCAII at pH 5.6 showing a single transition at 333 K. (d) HCAII at pH 5.6 showing a single transition at 318 K.
Table 2. Thermodynamics of unfolding for dsHCAII and HCAII.
| dsHCAII | HCAII | |||
|---|---|---|---|---|
| pH 7.8 | pH 5.6 | pH 7.8 | pH 5.6 | |
| T m † (K) | 344.1 ± 0.1 | 332.7 ± 0.1 | 330.4 ± 0.1 | 318.5 ± 0.1 |
| ΔH°m † (kcal mol−1) | 310 ± 20 | 201 ± 7 | 223 ± 5 | 152 ± 2 |
| ΔH vH ‡ (kcal mol−1) | 290 ± 20 | 190 ± 10 | 195 ± 6 | 135 ± 2 |
| ΔC p § (kcal mol−1 K−1) | 0.91 | 0.60 | 0.68 | 0.48 |
| ΔG T ¶ (kcal mol−1) | 6.2 | 5.7 | −4.7 | −3.4 |
| ΔH T †† (kcal mol−1) | 310 | 179 | 230 | 155 |
| ΔS T (kcal mol−1) | 0.87 | 0.52 | 0.70 | 0.50 |
Calorimetric parameters determined by DSC.
The van’t Hoff enthalpy (ΔH vH) was determined by fitting the thermograms to a two-state reversible unfolding model.
Heat capacity (ΔC p) of protein unfolding obtained by plotting calorimetric enthalpy (ΔH°m) versus melting temperature (T m).
Thermodynamic parameters extrapolated to reference temperatures of 337 K for samples at pH 7.8 and 325 K for samples at pH 5.8 using (3).
Thermodynamic parameters extrapolated to reference temperatures of 337 K for samples at pH 7.8 and 325 K for samples at pH 5.8 using (4).
Thermodynamic parameters extrapolated to reference temperatures of 337 K for samples at pH 7.8 and 325 K for samples at pH 5.8 using (5).
3.3. 18O kinetics
The pH profiles of the two rate constants, k
cat/K
m and , were determined by measuring the exchange of 18O between CO2 and water by mass spectrometry. The rate of catalyzed interconversion between CO2 and bicarbonate as measured by the k
cat/K
m (8) (Fig. 3 ▶
b; Table 3 ▶) for dsHCAII was similar to that reported previously for HCAII (Fisher et al., 2007 ▶). The determination of k
cat/K
m (Fig. 3 ▶
b) using (8) revealed similar maximal values for both dsHCAII and HCAII, assuming a single ionization pK
a for the zinc-bound solvent molecule. These results are expected as the disulfide location is more than 11 Å away from the catalytic zinc-bound hydroxide and no perturbations of the Cα backbone lining the active site were observed on overlaying it with the HCAII crystal structure (Fig. 1 ▶
d).
Figure 3.

Catalytic constants for dsHCAII and HCAII determined by 18O exchange. The results for dsHCAII are shown as solid black squares and those for HCAII are shown as open circles. (a) The pH profile for the rate constant /[E]. (b) The pH profile of k
cat/K
m. Solutions contained 10 mM of all species of CO2 and 100 mM HEPES pH 7.6.
Table 3. Kinetic properties of dsHCAII and HCAII measured by 18O mass spectrometry at 298 K.
The value of k B, the rate constant for proton transfer, from (9) indicates a slight reduction for the dsHCAII variant in comparison with HCAII (Fig. 3 ▶ a; Table 3 ▶). This decrease in proton transfer has been measured previously via spectrophotometric measurements of the dsHCAII variant (Mårtensson et al., 2002 ▶). Furthermore, comparison of the observed pK a values for the proton-shuttle residue His64 and the zinc-bound solvent molecule of dsHCAII are not significantly different from those observed for HCAII (Table 3 ▶).
To assess the stability of the enzyme at higher temperatures, the rate constant for the dehydration of bicarbonate to CO2, k cat/K m, was measured in 5 K increments from 283 to 353 K (Fig. 4 ▶), giving an approximation of the thermal inactivation temperature. The thermal inactivation temperature, T inact, was estimated as the data point at which the rate constant k cat/K m for solution containing enzyme was found to decrease to a value close to that for the uncatalyzed reaction. Following these guidelines, HCAII showed a significant loss in catalytic activity at 333 K, whereas the dsHCAII variant showed no loss of activity up to 343 K, the temperature limit of the experiment (Fig. 4 ▶; Table 3 ▶). These values also correlate well with the T m values measured by DSC under similar conditions (pH 7.8; Table 2 ▶) and show that the thermal unfolding temperature is also the thermal inactivation temperature for HCAII.
Figure 4.

The rate constant k cat/K M as a function of temperature for the interconversion of CO2 and bicarbonate. Solutions contained 10 mM of all species of CO2 and 100 mM HEPES pH 7.6. The results for dsHCAII are shown as solid black squares and those for HCAII are shown as open circles.
4. Discussion
The configuration of the disulfide bridge between residues 23 and 203 can be described as a (+) right-handed hook, which is one of the most common conformations of disulfide bridges, when analyzing the gauche magnitudes of the χ angles that form the bridge (Supplementary Table S1; Schmidt et al., 2006 ▶). The dihedral strain energy (DSE) of the disulfide can be calculated from the values of the five χ angles (Supplementary Fig. S1) using the relationship (Weiner et al., 1984 ▶; Katz & Kossiakoff, 1986 ▶)
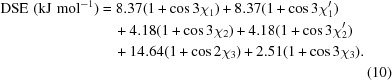 |
Although (10) only accounts for values of the dihedral angles of a disulfide bridge and not other factors such as bond lengths and van der Waals contacts, this relationship has been used to provide a semi-quantitative analysis of the strain in a disulfide bond (Wells & Powers, 1986 ▶; Kuwajima et al., 1990 ▶; Wetzel et al., 1988 ▶; Pjura et al., 1990 ▶). Using the observed χ angles in the crystallographic model, the DSE of the disulfide in dsHCAII is calculated to be 21.5 kJ mol−1. This DSE value is well within the possible values of 2.0–84.5 kJ mol−1 allowed for disulfide bridges and is comparable to the mean DSE for other right-handed hook disulfide formations (20.8 kJ mol−1) found in the PDB (Schmidt et al., 2006 ▶). Comparison of the geometry of the engineered disulfide bond in dsHCAII with those in other X-ray crystallographic models of CAs in which this linkage is conserved (HCAIV, HCAVI, HCAIX, murine CAXIV and N. gonorrhoeae CA) show that the right-handed hook conformation is retained, albeit with varying magnitudes of gauche values for χ1 and χ1′ and DSE values (Supplementary Table S1). It is interesting to note that HCAIV and N. gonorrhoeae CA have higher DSE values and also contain substitutions at position 25 (Glu and Phe, respectively), thereby further suggesting the importance of having a flexible Gly residue in this position for a less strained disulfide linkage.
The thermograms obtained from DSC for dsHCAII and HCAII at acidic and near-physiological pH (Fig. 2 ▶) were fitted to a two-state reversible unfolding model to yield the van’t Hoff enthalpies (Table 2 ▶). Most of the transition curves fitted well to this model, showing a single narrow endothermic event at the T m. However, the thermogram for dsHCAII at pH 7.8 shows a predominant peak at 344 K but does not fit as precisely to a two-state model owing to minor transition peaks at approximately 326 and 339 K (Fig. 2 ▶ a). A molten-globule intermediate (<10% of the total population) that unfolds near the major unfolding transition for this dsHCAII variant has been reported previously (Mårtensson et al., 2002 ▶). Interestingly, the minor transition at 326 K for dsHCAII at pH 7.8 is not observed in the thermograms at pH 5.6, which reveal only a single transition event at ∼333 K (Fig. 2 ▶ c). This suggests that a nonspecific aggregation event involving deprotonated residues occurs at pH 7.8 and could be the molten-globule dimer intermediate that has been suggested to provide a nucleation site for further aggregation (Cleland & Wang, 1990 ▶). Indeed, FPLC elution profiles and SDS–PAGE analysis of affinity-column purified HCAII samples at pH 7.8 do suggest very minor levels (<10%) of dimer formation and higher forms of oligomerization (data not shown).
To compare the thermodynamic properties (ΔG, ΔH and ΔS) of dsHCAII and HCAII, a reference temperature in between the major unfolding transitions for each variant was used (337 and 325 K for pH 7.8 and 5.6, respectively), with the results shown in Table 2 ▶. The measured calorimetric enthalpy of unfolding (ΔH°m) for dsHCAII was significantly increased by 50–80 kJ mol−1 at both pH values in comparison with the ΔH°m of HCAII. This increase in enthalpy may be owing to enhanced favorable electrostatic and van der Waals interactions among the N-terminal residues, which are among the first regions of HCAII to undergo unfolding (Gershenson et al., 2000 ▶; Matthews et al., 1987 ▶) and are tethered together in close proximity via formation of the disulfide covalent linkage. Furthermore, the ΔΔG T=337 K between the two variants at pH 7.8 has a value of 10.9 kcal mol−1, which correlates well with the 6.6 kcal mol−1 gain in stabilization observed from tryptophan fluorescence experiments under similar conditions using dsHCAII and HCAIIpwt (Mårtensson et al., 2002 ▶). The additional 4.3 kcal mol−1 gain in free energy observed between the dsHCAII and the HCAII calorimetric data could be explained by the possible formation of a hydrogen bond between Ser206 and the backbone carbonyl group of Val135 that was observed in the X-ray crystallographic structure. The addition of Ser206, however, has no significant effect on the stability of the variant compared with that of the native state (ΔT m < 1 K; data not shown). This gain in free energy is also present under the more acidic conditions with pH 5.6, with a ΔΔG T=324 K of 9.1 kcal mol−1.
The kinetic data revealed that dsHCAII and HCAII have comparable maximum k cat/K m values (Table 3 ▶). Similarily, the rate of maximum proton transfer, k B, was not significantly different for the two enzymes and correlates well with the previously reported hydration activity of dsHCAII (Mårtensson et al., 2002 ▶). The comparable catalytic rates are further supported by the absence of significant structural and solvent perturbations within the active site of dsHCAII compared with HCAII (Fig. 1 ▶ d) and additionally suggest that this variant can be implemented with mutations within the active site that have been shown to enhance the proton-transfer rate of HCAII (Fisher et al., 2007 ▶).
This study provides the first X-ray crystallographic structure of an engineered disulfide bridge in HCAII (Figs. 1 ▶ a and 1 ▶ b). Moreover, the 14–15 K enhanced denaturing temperature of this dsHCAII variant (Table 2 ▶; Fig. 2 ▶) provides an active and stable carbonic anhydrase that could better withstand the harsh conditions employed by current industrial protocols of atmospheric carbon sequestration (temperatures greater than 343 K and pH less than 6.0; Kanbar & Ozdemir, 2010 ▶) without a loss in catalytic efficiency (Table 3 ▶; Fig. 4 ▶). This designed disulfide linkage can be used in conjuction with other variants of HCAII that show enhanced thermal stability via hydrophobic-to-hydrophilic surface mutations resulting in a gain of enthalpic contributions from hydrogen bonding (Fisher et al., 2012 ▶). This report demonstrates that the rational design of a moderately conserved disulfide bridge into an isoform lacking this linkage can significantly enhance the stability of the enzyme without a loss in the overall catalytic activity.
Supplementary Material
PDB reference: human carbonic anhydrase II, 4hba
Supplementary material file. DOI: 10.1107/S0907444913008743/be5229sup1.pdf
Acknowledgments
This work was supported by a grant from the NIH (GM25154) awarded to DS and RM.
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: BE5229). Services for accessing this material are described at the back of the journal.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Aggarwal, M., Boone, C. D., Kondeti, B. & McKenna, R. (2013). J. Enzyme Inhib. Med. Chem. 28, 267–277. [DOI] [PubMed]
- Alterio, V., Hilvo, M., Di Fiore, A., Supuran, C. T., Pan, P., Parkkila, S., Scaloni, A., Pastorek, J., Pastorekova, S., Pedone, C., Scozzafava, A., Monti, S. M. & De Simone, G. (2009). Proc. Natl Acad. Sci. USA, 106, 16233–16238. [DOI] [PMC free article] [PubMed]
- Avvaru, B. S., Kim, C. U., Sippel, K. H., Gruner, S. M., Agbandje-McKenna, M., Silverman, D. N. & McKenna, R. (2010). Biochemistry, 49, 249–251. [DOI] [PMC free article] [PubMed]
- Becktel, W. J. & Schellman, J. A. (1987). Biopolymers, 26, 1859–1877. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Cleland, J. L. & Wang, D. I. C. (1990). Biochemistry, 29, 11072–11078. [DOI] [PubMed]
- DeLano, W. L. (2002). PyMOL http://www.pymol.org.
- Elleby, B., Chirica, L. C., Tu, C., Zeppezauer, M. & Lindskog, S. (2001). Eur. J. Biochem. 268, 1613–1619. [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Fisher, Z., Boone, C. D., Biswas, S. M., Venkatakrishnan, B., Aggarwal, M., Tu, C., Agbandje-McKenna, M., Silverman, D. & McKenna, R. (2012). Protein Eng. Des. Sel. 25, 347–355. [DOI] [PMC free article] [PubMed]
- Fisher, S. Z., Tu, C., Bhatt, D., Govindasamy, L., Agbandje-McKenna, M., McKenna, R. & Silverman, D. N. (2007). Biochemistry, 46, 3803–3813. [DOI] [PubMed]
- Forsman, C., Behravan, G., Osterman, A. & Jonsson, B. H. (1988). Acta Chem. Scand. B, 42, 314–318. [DOI] [PubMed]
- Fulke, A. B., Mudliar, S. N., Yadav, R., Shekh, A., Srinivasan, N., Ramanan, R., Krishnamurthi, K., Devi, S. S. & Chakrabarti, T. (2010). Bioresour. Technol. 101, 8473–8476. [DOI] [PubMed]
- Gershenson, A., Schauerte, J. A., Giver, L. & Arnold, F. H. (2000). Biochemistry, 39, 4658–4665. [DOI] [PubMed]
- Huang, S., Xue, Y., Sauer-Eriksson, E., Chirica, L., Lindskog, S. & Jonsson, B. H. (1998). J. Mol. Biol. 283, 301–310. [DOI] [PubMed]
- Kanbar, B. & Ozdemir, E. (2010). Biotechnol. Prog. 26, 1474–1480. [DOI] [PubMed]
- Katz, B. A. & Kossiakoff, A. (1986). J. Biol. Chem. 261, 15480–15485. [PubMed]
- Khalifah, R. G. (1971). J. Biol. Chem. 246, 2561–2573. [PubMed]
- Khalifah, R. G., Strader, D. J., Bryant, S. H. & Gibson, S. M. (1977). Biochemistry, 16, 2241–2247. [DOI] [PubMed]
- Kuwajima, K., Ikeguchi, M., Sugawara, T., Hiraoka, Y. & Sugai, S. (1990). Biochemistry, 29, 8240–8249. [DOI] [PubMed]
- Lindskog, S. (1997). Pharmacol. Ther. 74, 1–20. [DOI] [PubMed]
- Mårtensson, L. G., Karlsson, M. & Carlsson, U. (2002). Biochemistry, 41, 15867–15875. [DOI] [PubMed]
- Matthews, B. W., Nicholson, H. & Becktel, W. J. (1987). Proc. Natl Acad. Sci. USA, 84, 6663–6667. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Pjura, P. E., Matsumura, M., Wozniak, J. A. & Matthews, B. W. (1990). Biochemistry, 29, 2592–2598. [DOI] [PubMed]
- Ramanan, R., Kannan, K., Deshkar, A., Yadav, R. & Chakrabarti, T. (2010). Bioresour. Technol. 101, 2616–2622. [DOI] [PubMed]
- Savile, C. K. & Lalonde, J. J. (2011). Curr. Opin. Biotechnol. 22, 818–823. [DOI] [PubMed]
- Schmidt, B., Ho, L. & Hogg, P. J. (2006). Biochemistry, 45, 7429–7433. [DOI] [PubMed]
- Silverman, D. N. (1982). Methods Enzymol. 87, 732–752. [DOI] [PubMed]
- Silverman, D. N. & Lindskog, S. (1988). Acc. Chem. Res. 21, 30–36.
- Sly, W. S. & Hu, P. Y. (1995). Annu. Rev. Biochem. 64, 375–401. [DOI] [PubMed]
- Stams, T., Chen, Y., Boriack-Sjodin, P. A., Hurt, J. D., Liao, J., May, J. A., Dean, T., Laipis, P., Silverman, D. N. & Christianson, D. W. (1998). Protein Sci. 7, 556–563. [DOI] [PMC free article] [PubMed]
- Stams, T. & Christianson, D. W. (2000). The Carbonic Anhydrases: New Horizons, edited by W. R. Chegwidden, N. D. Carter & Y. H. Edwards, pp. 159–174. Basel: Birkhauser Verlag.
- Steiner, H., Jonsson, B. H. & Lindskog, S. (1975). Eur. J. Biochem. 59, 253–259. [DOI] [PubMed]
- Tu, C. K., Silverman, D. N., Forsman, C., Jonsson, B. H. & Lindskog, S. (1989). Biochemistry, 28, 7913–7918. [DOI] [PubMed]
- Vinoba, M., Lim, K. S., Lee, S. H., Jeong, S. K. & Alagar, M. (2011). Langmuir, 27, 6227–6234. [DOI] [PubMed]
- Wang, Q., Christiansen, A., Samiotakis, A., Wittung-Stafshede, P. & Cheung, M. S. (2011). J. Chem. Phys. 135, 175102. [DOI] [PubMed]
- Weiner, S. J., Kollman, P. A., Case, D. A., Singh, U. C., Ghio, C., Alagona, G., Profeta, S. & Weiner, P. (1984). J. Am. Chem. Soc. 106, 765–784.
- Wells, J. A. & Powers, D. B. (1986). J. Biol. Chem. 261, 6564–6570. [PubMed]
- Wetzel, R., Perry, L. J., Baase, W. A. & Becktel, W. J. (1988). Proc. Natl Acad. Sci. USA, 85, 401–405. [DOI] [PMC free article] [PubMed]
- Whitney, P. L. & Briggle, T. V. (1982). J. Biol. Chem. 257, 12056–12059. [PubMed]
- Whittington, D. A., Grubb, J. H., Waheed, A., Shah, G. N., Sly, W. S. & Christianson, D. W. (2004). J. Biol. Chem. 279, 7223–7228. [DOI] [PubMed]
- Whittington, D. A., Waheed, A., Ulmasov, B., Shah, G. N., Grubb, J. H., Sly, W. S. & Christianson, D. W. (2001). Proc. Natl Acad. Sci. USA, 98, 9545–9550. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: human carbonic anhydrase II, 4hba
Supplementary material file. DOI: 10.1107/S0907444913008743/be5229sup1.pdf