

The structure of the functional interaction of NRPS adenylation and carrier protein domains, trapped with a mechanism-based inhibitor, is described. Crystals exhibit translational non-crystallographic symmetry, which challenged structure determination and refinement.
Keywords: nonribosomal peptide synthetases, EntE, EntB, adenylation domain, carrier protein domain, noncrystallographic translational symmetry
Abstract
The nonribosomal peptide synthetases (NRPSs) are a family of modular proteins that contain multiple catalytic domains joined in a single protein. Together, these domains work to produce chemically diverse peptides, including compounds with antibiotic activity or that play a role in iron acquisition. Understanding the structural mechanisms that govern the domain interactions has been a long-standing goal. During NRPS synthesis, amino-acid substrates are loaded onto integrated carrier protein domains through the activity of NRPS adenylation domains. The structures of two adenylation domain–carrier protein domain complexes have recently been determined in an effort that required the use of a mechanism-based inhibitor to trap the domain interaction. Here, the continued analysis of these proteins is presented, including a higher resolution structure of an engineered di-domain protein containing the EntE adenylation domain fused with the carrier protein domain of its partner EntB. The protein crystallized in a novel space group in which molecular replacement and refinement were challenged by noncrystallographic pseudo-translational symmetry. The structure determination and how the molecular packing impacted the diffraction intensities are reported. Importantly, the structure illustrates that in this new crystal form the functional interface between the adenylation domain and the carrier protein domain remains the same as that observed previously. At a resolution that allows inclusion of water molecules, additional interactions are observed between the two protein domains and between the protein and its ligands. In particular, a highly solvated region that surrounds the carrier protein cofactor is described.
1. Introduction
The nonribosomal peptide synthetases (NRPSs) are a family of large multi-domain enzymes that use a modular architecture to produce important biological peptides (Condurso & Bruner, 2012 ▶; Hur et al., 2012 ▶). The NRPSs contain multiple catalytic domains on a single polypeptide chain that can be thousands of residues in length. The domains are organized into modules; an individual module is generally responsible for all of the catalytic steps necessary for the incorporation of a single amino acid into the peptide product. Each module also contains an integrated peptidyl carrier protein (PCP) to which the amino acid and peptide are covalently bound. The PCP domains, which are homologous to acyl carrier proteins (ACPs) in fatty-acid synthesis and transport (Crosby & Crump, 2012 ▶), transfer the amino acid and peptide substrate to neighboring catalytic domains. The final module also catalyzes release of the covalently bound peptide product. The fusion of multiple modules results in large protein machines that function in an assembly-line fashion to produce the final product. Crystallographic structures of multidomain NRPSs (Strieker et al., 2010 ▶), as well as of the functionally similar polyketide synthases (Keatinge-Clay, 2012 ▶), remain important targets for understanding the fascinating enzymatic synthesis of these important biomolecules. Detailed understanding of the rules that govern the interaction of the catalytic and carrier domains may ultimately enable the engineering of NRPS pathways (Baltz, 2009 ▶) for the production of novel NRPS peptides.
The reactions catalyzed by standard NRPS domains (Fig. 1 ▶) are now well understood (Fischbach & Walsh, 2006 ▶; Marahiel & Essen, 2009 ▶). Amino acids are first activated through the activity of an adenylation domain that uses the energy derived from ATP hydrolysis to form an acyl-adenylate and then loads the amino acid on the thiol of the pantetheine cofactor of the PCP domain (Gulick, 2009 ▶). Most often, the adenylation and PCP domains are joined in a multi-domain protein; however, self-standing adenylation domains are not uncommon. Activated amino acids on PCP domains in neighboring modules are then joined by a condensation domain that catalyzes peptide-bond formation and transfers the upstream amino acid or peptide to the downstream substrate. Finally, because the peptide is covalently bound as a thioester, most NRPS termination modules harbor a thioesterase domain that releases the peptide, often in a cyclized form. These core catalytic (adenylation, condensation and thioesterase) domains are additionally joined by tailoring domains that can catalyze epimerization, methylation or other chemical modifications of the amino-acid building blocks or the growing peptide (Samel & Marahiel, 2008 ▶).
Figure 1.

Schematic diagram of a prototypical three-module NRPS. The NRPS is represented as a single rod, which is divided to represent the three separate modules, with the adenylation (A), condensation (C), thioesterase (TE) and thiolation/carrier protein (T) domains shown. The pantetheine cofactor of the PCP is represented by a line and the thiol group. In the first step, the three adenylation domains load the PCP with the specific amino acid. The condensation domain of module 2 then transfers the first amino acid from the first module to the amino acid loaded onto the second carrier domain. Similarly, the condensation domain of module 3 then transfers this dipeptide to the third amino acid that was previously loaded onto module 3. Finally, the thioesterase domain cleaves the tripeptide from the third PCP domain, freeing the enzyme for another cycle of catalysis.
Many structures of individual catalytic domains have been determined, including adenylation (Drake et al., 2010 ▶; Du et al., 2008 ▶; Lee et al., 2010 ▶; May et al., 2002 ▶; Yonus et al., 2008 ▶), condensation (Keating et al., 2002 ▶) and thioesterase domains (Bruner et al., 2002 ▶; Samel et al., 2006 ▶). More recently, insights into the interactions of catalytic and carrier domains have been provided, including the interactions of PCP domains with thioesterase domains (Frueh et al., 2008 ▶; Koglin et al., 2008 ▶; Liu et al., 2011 ▶) and adenylation domains (Mitchell et al., 2012 ▶; Sundlov, Shi et al., 2012 ▶). A complete termination module, composed of a condensation–adenylation–PCP–thioesterase domain architecture, was structurally characterized in 2008 (Tanovic et al., 2008 ▶). In this structure, the PCP domain was positioned to interact with the condensation domain. However, the PCP domain was located 60 and 45 Å from the active sites of the adenylation and thioesterase domains, respectively, suggesting that large conformational rearrangements were required for the domains to be delivered properly to the alternate catalytic domains.
The NRPS adenylation domains are part of a large superfamily of adenylate-forming enzymes (Gulick, 2009 ▶). This ANL superfamily additionally contains acyl-CoA synthetases and beetle luciferase enzymes. Enzymes of all three subfamilies contain two subdomains and catalyze two-step reactions. A carboxylate substrate and ATP react in an initial adenylate-forming step to form an acyl-adenylate. In the NRPS adenylation domains and acyl-CoA synthetases, a second partial reaction results in the formation of a thioester with either the pantetheine cofactor of the PCP domain or with CoA. The structures of many members of the ANL family (Yonus et al., 2008 ▶; Gulick et al., 2003 ▶; Kochan et al., 2009 ▶; Reger et al., 2007 ▶, 2008 ▶; Sundlov, Fontaine et al., 2012 ▶) demonstrate that the smaller C-terminal domain rotates by 140° to adopt two different conformations that are used to catalyze the two partial reactions. We have proposed (Gulick, 2009 ▶; Gulick et al., 2003 ▶; Reger et al., 2007 ▶) that this large domain rotation in the NRPS adenylation domains could be one of the conformational changes that deliver the carrier domain to different active sites.
We recently determined the structures of two adenylation–PCP domain complexes (Mitchell et al., 2012 ▶; Sundlov, Shi et al., 2012 ▶). These structures required the use of a mechanism-based inhibitor (Qiao et al., 2007 ▶) that trapped the functional interaction between the PCP pantetheine cofactor and an analog of the adenylate intermediate. One of the proteins used in these studies was EntE-B (Sundlov, Shi et al., 2012 ▶), an engineered fusion protein composed of the EntE free-standing adenylation domain with the PCP1 domain from a distinct protein EntB (Fig. 2 ▶ a). EntE and EntB are part of the Escherichia coli enterobactin synthetic cluster (Gehring et al., 1998 ▶). This fusion protein was designed to simulate the adenylation–PCP di-domain constructs that are commonly present in multi-domain NRPS enzymes.
Figure 2.

The reaction of EntE and EntB. (a) The reaction catalyzed by the adenylation domain EntE proceeds in two steps. An initial adenylate is formed from ATP and 2,3-dihydroxybenzoate (DHB). The pantetheine cofactor of EntB then attacks this intermediate to release AMP, which results in loading of the DHB onto the EntB domain. (b) The domain-swapped organization of the chimeric EntE-B di-domain protein. One EntE-B molecule is shown in blue; the N- and C-terminal domains of EntE are shown in light blue and medium blue, while the PCP domain is shown in dark blue. Similarly, the other chain contains pale wheat and yellow N- and C-terminal domains of EntE, with an orange PCP. This model was constructed from chains C and H of PDB entry 3rg2 (Sundlov, Shi et al., 2012 ▶).
The crystals of EntE-B diffracted to only 3.1 Å resolution and the protein crystallized as a domain-swapped dimer in which two proteins come together to share their respective carrier proteins (Fig. 2 ▶ b). The crystallographic asymmetric unit contained five dimers. The limited resolution and the presence of multiple copies also limited analysis of the interface. However, the structure provided the foundation for directed engineering experiments to improve the activity of BasE, an EntE homolog from Acinetobacter baumannii (Sundlov, Shi et al., 2012 ▶). This supported our conclusion that the crystallographic interface represented the true biological complex and was not influenced by the domain-swapped dimerization.
During crystallization of EntE-B, other crystal forms were screened in an effort to find a crystal that diffracted to higher resolution and potentially contained fewer protein chains in the asymmetric unit. Here, we report a structure determined from an orthorhombic crystal form that contains a single domain-swapped dimer in the asymmetric unit. The structure determination was challenged by noncrystallographic translational symmetry, a feature that can make both structure determination by molecular replacement and refinement difficult (Chook et al., 1998 ▶; Guarné et al., 1998 ▶; Oksanen et al., 2006 ▶; Read et al., 2013 ▶; Rudolph et al., 2004 ▶). Our strategy for structure determination is presented here and we present this higher resolution view of the domain interface. Although the protein crystallized in a new space group, the interface between the domains is conserved. As was observed for the multiple copies in the previous structure (Sundlov, Shi et al., 2012 ▶), the linker that joins the adenylation and PCP domains adjusts to allow the functional interface to remain constant. The observation of two distinct EntE-B crystal forms that exhibit the same adenylation–PCP domain interface, as well as the structural similarity to a second adenylation–PCP domain protein structure (Mitchell et al., 2012 ▶) and the mutagenesis analysis in our previous study (Sundlov, Shi et al., 2012 ▶), supports our contention that the observed interface is used functionally by the EntE adenylation and EntB carrier protein domains and serves as a model for other NRPS domain interactions. We present here this higher resolution view along with the structure-determination protocol and analysis of the translational noncrystallographic symmetry (NCS).
2. Methods
2.1. Protein expression and crystallization
EntB is a two-domain protein that contains an N-terminal isochorismatase domain, which is used in the production of the enterobactin building block 2,3-dihydroxybenzoate, and a C-terminal acyl carrier protein domain (Drake et al., 2006 ▶; Gehring et al., 1997 ▶). To generate a two-domain adenylation–PCP domain construct that we could use to model the natural two-domain NRPS proteins, we created a fusion protein between the adenylation domain of EntE and the carrier protein domain of EntB (Sundlov, Shi et al., 2012 ▶). We fused the two cDNAs and incorporated into the linker between the two domains the coding sequence for four residues, Gly-Arg-Ala-Ser, that were modeled on the similar linker region of the EntF NRPS protein.
The EntE-B protein was produced as described previously (Sundlov, Shi et al., 2012 ▶) using the pET15bTEV expression plasmid (Kapust et al., 2001 ▶). Cells were grown in minimal medium to induce the genomic enterobactin operon including the pantetheinyltransferase EntD, which converts the EntB carrier domain from apo to holo (Drake et al., 2006 ▶). Unlike the previous experiments (Sundlov, Shi et al., 2012 ▶), the His5 purification tag and the intervening TEV protease site were left in place and the protein was of sufficient purity after a single metal ion-affinity step to allow crystallization. The final protein was concentrated to 16 mg ml−1 and dialyzed into 10 mM Tris pH 7.5, 25 mM NaCl, 0.3 mM TCEP. For crystallization experiments, the protein was incubated overnight at room temperature with a twofold molar excess of the mechanism-based inhibitor 5′-amino-5′-deoxy-5′-N-{[2-(2,3-dihydroxyphenyl)ethenyl]sulfonyl}adenosine. This compound binds to the EntE adenylation domain active site and reacts covalently through the vinyl linker region with the pantetheine thiol, forming a covalent analog of the reaction intermediate (Sundlo, Shi et al., 2012 ▶; Qiao et al., 2007 ▶).
Crystallization screens were performed by the Center for High Throughput Structural Biology (CHTSB) using the microbatch-under-oil technique (Luft et al., 2003 ▶). Optimized crystals were grown via a modified vapor-diffusion setup. A precipitant consisting of 24% PEG monomethyl ether 2000 and 0.1 M bis(2-hydroxyethyl)amino-tris(hydroxymethyl)methane (bis-tris pH 6.5) was prepared. The hanging-drop experiment consisted of 2 µl of this precipitant solution and 2 µl protein solution. For the crystallization reservoir, 300 µl precipitant solution was diluted in a 1:1 ratio with 300 µl of the protein buffer above. The final crystallization experiment thus consisted of 600 µl diluted precipitant in the well and a 4 µl hanging drop. Under these conditions, the drop solution is expected to change very little over the course of crystallization and the experiment could also be described as a modified microbatch experiment. We have found this unusual approach to be successful at reproducing the rapidly growing crystals that were originally identified in the microbatch-under-oil conditions used in the CHTSB. The crystals grew within 24 h at 293 K and were harvested after one week. The crystal was prepared for data collection by sequential transfer through three solutions consisting of 12% PEG MME 2000, 50 mM bis-tris supplemented with 2-methyl-2,4-propanediol to concentrations of 8, 16 and 24%, respectively, for cryoprotection.
2.2. Data collection and structure determination
Diffraction data were collected remotely on beamline 11-1 of SSRL using the Blu-Ice software package (McPhillips et al., 2002 ▶). The crystal used was 0.15 × 0.15 × 0.1 mm in size and a beam size of 0.2 mm was used. Diffraction data were collected at 113 K. The data were processed and scaled with iMosflm (Battye et al., 2011 ▶) and converted to structure-factor amplitudes with TRUNCATE (Winn et al., 2011 ▶). The unit cell was determined to be primitive orthorhombic. Initial analysis of the systematic absences suggested that three screw axes were present. Initial attempts at molecular replacement using MOLREP (Vagin & Teplyakov, 1997 ▶, 2010 ▶), EPMR v.10.09 (Kissinger et al., 2001 ▶) and Phaser v.2.3 (McCoy et al., 2007 ▶) failed to give a satisfactory solution using multiple search models, including a dimer of EntE-B or a single EntE-B chain. Inspection of a native Patterson function indicated the potential presence of pseudotranslational symmetry.
As described below, the structure was ultimately solved with MOLREP through manually solving the rotation function using just the EntE adenylation domain as the search model and applying the translational vector determined from the Patterson function to create a model with two protein chains. This model was then used for the final translation function with the eight possible primitive orthorhombic space groups containing all combinations of pure twofold rotation or twofold screw axes. The model was completed through manual model building (Emsley & Cowtan, 2004 ▶) and refined with PHENIX (Adams et al., 2010 ▶). Diffraction and refinement statistics are described in Table 1 ▶. The final molecule was submitted to the MolProbity server (Chen et al., 2010 ▶), where it received an overall score of 2.26, placing it in the 83rd percentile for structures of comparable resolution. Five residues were listed as outliers in the Ramachandran plot: Leu338 and Glu422 in chain A, Asp173 in chain B and Gly337 in both chains. All five residues lie just outside the boundaries for allowed regions in the Ramachandran plot and inspection of the electron density shows no obvious reason why they adopt the slightly disfavored orientation.
Table 1. Data-collection and refinement statistics for EntE-B.
Values in parentheses are for the highest resolution shell.
| PDB code | 4iz6 |
| Beamline | SSRL 11-1 |
| Wavelength (Å) | 1.00 |
| Resolution (Å) | 30.0–2.6 |
| Space group | P21212 |
| Unit-cell parameters (Å) | a = 111.0, b = 119.1, c = 99.8 |
| R merge (%) | 9.4 (40.2) |
| R p.i.m. (%) | 6.2 (28.8) |
| Completeness (%) | 98.0 (91.1) |
| 〈I/σ(I)〉† | 7.5 (2.1) |
| No. of observations | 190335 |
| No. of reflections | 51261 |
| Refinement | |
| R cryst (%) | 25.1 (32.8) |
| R free (%) | 31.2 (39.0) |
| Wilson B factor (Å2) | 31.6 |
| No. of water molecules | 205 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.25 |
| Ramachandran plot | |
| Favored (%) | 97.0 |
| Allowed (%) | 2.6 |
| Outliers (%) | 0.4 |
| MolProbity clashscore/percentile | 13.95/85th |
| MolProbity overall score/percentile | 2.26/83rd |
The overall 〈I/σ(I)〉 calculated with the weak reflections removed, as defined as in §3.3, is 8.8 for 40 936 reflections.
The failed attempts at molecular replacement were all performed with Phaser v.2.3 or earlier. Read and coworkers have recently described an algorithm to estimate the effects of translational NCS on diffraction intensities (Read et al., 2013 ▶). A recent update to Phaser, which is available as part of the PHENIX suite, uses a correction term that accounts for pseudo-translational noncrystallographic symmetry in molecular replacement (Randy Read, personal communication). Subsequent to the structure determination and analysis described here, the P212121 reflection file was used along with the single chain of the EntE adenylation domain with Phaser v.2.5 embedded within PHENIX v.1.8.1. This version of Phaser identified the presence of noncrystallographic symmetry and the proper space group, and rapidly identified the molecular-replacement solution.
3. Results and discussion
3.1. Data collection and analysis of pseudotranslational symmetry
In our efforts to find an improved crystal of the chimeric EntE-B protein, we identified a new form that indexed in a primitive orthorhombic space group with unit-cell parameters a = 99.8, b = 111.0, c = 119.1 Å. Analysis of systematic absences showed h00, 0k0 and 00l absences for odd reflections, suggesting that the space group was P212121. Matthews coefficient analysis suggested there would be two molecules in the asymmetric unit, resulting in a V M value of 2.4 Å3 Da−1 and 48% solvent content. Diffraction statistics are shown in Table 1 ▶.
Initial molecular-replacement searches with either the domain-swapped dimer of EntE-B (PDB entry 3rg2) or with one complete chain failed to give a solution. Therefore, a model of the EntE adenylation domain alone (Sundlov, Shi et al., 2012 ▶), lacking the carrier protein domain, was used as a search model for molecular replacement with MOLREP (Vagin & Teplyakov, 1997 ▶, 2010 ▶) and Phaser (McCoy et al., 2007 ▶). MOLREP showed a high peak in the rotation function; however, the solutions of the translation function did not refine properly. Inspection of the log file, as well as a native Patterson map (Fig. 3 ▶ a), indicated a substantial pseudotranslation peak that was greater than one half the size of the origin peak at (0.5, 0.427, 0.5). This raised concerns that the absences observed on the crystallographic axes might result from the pseudotranslation and that the true space group may not contain screw axes along all three axes.
Figure 3.

Analysis of the diffraction data illustrates the presence of noncrystallographic translational symmetry. (a) A native Patterson map, calculated at 3.0 Å resolution, is shown for the z = 0.5 section. The peak at (0.5, 0.427, 0.5) represents the translational vector between the two molecules in the asymmetric unit. Intensity analysis was performed as described by Padilla & Yeates (2003 ▶). (b) The cumulative intensity distribution normalized for resolution shells is shown, where z = I/〈I〉. Theoretical plots are shown for acentric (solid) and centric (dashed) data. The observed values for acentric data are shown by the dotted line. (c) The cumulative probability distribution of the Yeates local intensity difference statistics are shown for acentric data, where L = (I 1 − I 2)/(I 1 + I 2). The theoretical distributions for acentric data (solid) and perfectly twinned data (dashed) are compared with the observed acentric data.
The phenix.xtriage module of the PHENIX software suite (Adams et al., 2010 ▶) noted the high Patterson peak as well as deviations from expected values in the intensity distributions. The 〈I 2〉/〈I〉2 for acentric reflections was 3.2, which is higher than the expected values for either untwinned or twinned data (2.0 or 1.5, respectively). The intensity probability distribution additionally illustrated the impact of pseudotranslation on the diffraction intensities (Fig. 3 ▶ b). Finally, applying the L-test of Padilla & Yeates (2003 ▶) showed there was no twinning (Fig. 3 ▶ c).
3.2. Molecular replacement
The ambiguity in the space group led us to perform molecular replacement in all eight primitive orthorhombic space groups. No satisfactory solutions were obtained with MOLREP, Phaser or EPMR (Kissinger et al., 2001 ▶). Additionally, the pseudotranslation vector search of MOLREP was employed, which again did not identify a solution.
We reasoned that the pseudotranslational symmetry was causing difficulty with the molecular replacement. We therefore attempted to solve the problem in individual steps. The rotation search was performed using the EntE protein molecule from PDB entry 3rg2, providing a model that was in the correct orientation. We applied the translation vector to this chain, resulting in a second protein molecule parallel to the first and related to it by the pseudotranslation vector. The two protein chains were then combined into a single file that was used as a search model with MOLREP in all eight primitive orthorhombic space groups. The best solution gave an R factor of 50.4% and a score of 0.668, while the other seven solutions resulted in R factors ranging from 54.5 to 59.6% and MOLREP scores ranging from 0.44 to 0.62. The best solution was achieved in space group P22121, suggesting that the a axis was not a true screw axis and that the observed h00 absences derived from the pseudotranslational symmetry.
We reindexed the data in standard space group P21212 with unit-cell parameters a = 111.0, b = 119.1, c = 99.8 Å. We repeated the above strategy to ensure that the solution was in the proper position with the reindexed data and continued to refinement. An initial cycle of refinement with REFMAC (Murshudov et al., 2011 ▶) reduced the crystallographic R factor to 35% and R free to 40%; electron density in the active site of each EntE molecule showed the presence of the pantetheine group and the vinylsulfonamide ligand (Fig. 4 ▶). Density for the EntB carrier protein was also apparent. The EntB residues were manually built using the previous complex as a guide. The refinement continued, with ligands and water molecules added as the refinement progressed.
Figure 4.
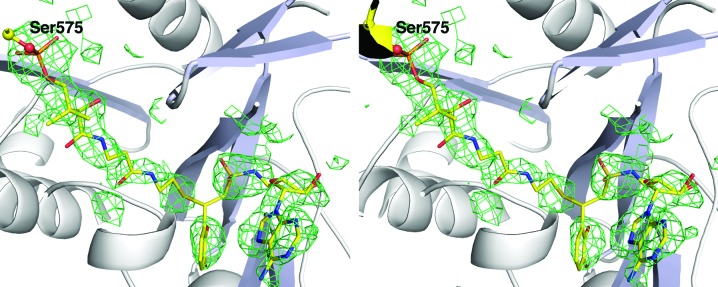
Stereo representation of electron density of the panthetheine cofactor and inhibitor. Unbiased electron density calculated with coefficients of the form F o − F c generated after molecular replacement and prior to inclusion of any ligands or water molecules in the structure. Density is contoured at 2.5σ and is displayed with a carve radius of 4 Å around the ligands. The discontinuity in the density near the covalent attachment was not apparent in the final 2F o − F c map; however, it may represent some fractional incomplete reaction of the pantetheine and inhibitor.
3.3. Refinement and analysis of the molecular-replacement solution
During the refinement, the R factors failed to converge to generally acceptable values. Despite the nearly complete model, with or without application of NCS restraints on the two protein chains the R factors remained at ∼25% for the working R factor and ∼32% for R free. Several reports have described a failure to reduce R factors resulting from pseudotranslational symmetry and systematically weak reflections (Chook et al., 1998 ▶; Guarné et al., 1998 ▶; Oksanen et al., 2006 ▶; Rudolph et al., 2004 ▶). We therefore examined the structure-factor intensities more closely in order to understand the nature of the difficult refinement.
The reindexed P21212 data set should exhibit systematic absences only for the h00 and 0k0 axes. Visual examination of the 0kl zone with HKLVIEW from the CCP4 suite illustrated that k + l = odd reflections were missing in the low-resolution region of the zone. At higher resolution, the k + l = odd reflections were present in the h = 0 zone but were notably weaker than the k + l = even reflections.
To determine whether the translational NCS caused systematically weak data and therefore resulted in higher than usual R factors, we first calculated the overall 〈F〉 and 〈F/σ(F)〉 values for h + k + l = odd and even for all data. Surprisingly, there was relatively little difference in these values, with h + k + l = 2n (even) reflections exhibiting an overall 〈F〉 of 320.3 and h + k + l = 2n + 1 (odd) reflections having an overall 〈F〉 of 316.5. The 〈F/σ(F)〉 values for both parity groups were 15.13 and 15.16. Visual inspection of the various zones about the h axis with HKLVIEW showed that, in addition to the h = 0 zone (0kl), other zones showed alternating absences (representative zones are shown in Fig. 5 ▶ a). The h = 1 (1kl) zone also showed weak alternating reflections, although the effect was not as dramatic as in the 0kl zone. The 2kl, 3kl, 4kl and 5kl zones showed no systematically weak reflections, but they reappeared in the 6kl, 7kl and 8kl zones. Thus, multiple zones showed systematic absences when examining the hkl zones at values of h. However, upon careful analysis of this feature, we realised that in the 0kl and 1kl zones the h + k + l = odd reflections were weak, while in the 6kl, 7kl and 8kl zones the h + k + l = even reflections were weak and the h + k + l = odd reflections were strong. This cycling continued through the data with a period of 7 as we monitored the nkl layers of reciprocal space along the h axis (Fig. 5 ▶ b).
Figure 5.

Impact of pseudotranslational symmetry on diffraction intensities. (a) Pseudo-precession images to illustrate structure-factor amplitudes generated with the CCP4 program HKLVIEW (Winn et al., 2011 ▶). The 0kl, 4kl and 7kl zones are shown, using a circular cutoff at a d min of approximately 7 Å. Note that in the 0kl zone h + k + l = odd reflections are weak, while in the 7kl zone h + k + l = odd reflections are strong. (b) Mean F/σ(F) values of hkl were calculated for h + k + l = odd (solid line, filled circles) and h + k + l = even (dashed line, open circles) for the nkl zones of reciprocal space and plotted as a function of h. The values were calculated using all low-resolution data to 3 Å.
The pattern of alternating absences between odd and even parity groups in different regions of the data explains why 〈F odd〉 approximates 〈F even〉 for all data. In some zones h + k + l = odd reflections are weak or absent, while in other zones this is true for the h + k + l = even reflections and the overall impact cancels.
An understanding of the basis for systematic absences in standard crystallographic symmetry provides an explanation for this observation. We realised that the noncrystallographic translational vector of our data (0.427, 0.5, 0.5) is strikingly close to (3/7, 1/2, 1/2), as 3/7 = 0.42857. Summing the structure-factor equation over n/2 atoms and applying the translational symmetry, as one would normally determine systematic absences for a standard crystallographic symmetry element, we observe
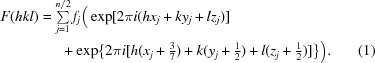 |
This equation reduces to
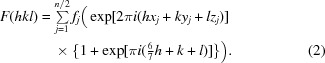 |
Expanding the second exponential term via Euler’s formula, we obtain
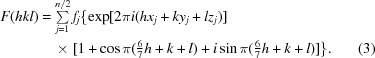 |
Examination of (3) provides an explanation of the observed diffraction intensities. The sine term is zero when (6/7)h + k + l sums to an integer. Additionally, if (6/7)h + k + l is odd the cosine term is −1, resulting in an absent reflection. In the h = 0 zone (or h = 14, 28 or 42), (6/7)h + k + l is odd when h + k + l is odd. In the h = 7 zone (or h = 21 or 35), (6/7)h + k + l is odd when h + k + l is even. These alternating effects on the h + k + l data mask the appearance of the systematically weak data on the overall statistics. (3) is a specific example of how the effect of the observed translational NCS impacts the structure factors and intensities in the case of EntE-B. A general consideration of this phenomenon has been presented (Tsai et al., 2009 ▶) that relates the observed intensities as the product of the ‘true’ crystallographic intensities that result in the absence of NCS and the transform of the translational vectors.
In summary, although it was not apparent when determining the overall absences in parity groups h + k + l = odd or h + k + l = even for all data, a significant fraction of reflections are missing or weak, resulting in higher R factors than expected. To confirm the negative impact of the inclusion of the weak data, we created a reflection file from which the h + k + l = odd reflections were removed from the h = 0, 1, 13, 14, 15, 27, 28, 29, 41, 42 and 43 zones and the h + k + l = even reflections were removed from the h = 6, 7, 8, 20, 21, 22, 34, 35 and 36 zones. We used PHENIX to calculate a crystallographic R factor of 22.3% and an R free of 28.4% for our final model against this resulting reflection file, which contained 76% of all data.
3.4. Description of the structure of the EntE-B protein
Examination of the molecular-replacement solution showed that a suitable choice of crystallographic symmetry mates could form a true domain-swapped dimer. Thus, chains A and B, which were related by the pure translation symmetry, each interacted with the crystallographic symmetry mate of the other chain. We therefore chose a different symmetry-related molecule for chain B so that the refined asymmetric unit contained two chains that interact to form a domain-swapped dimer, in which the PCP domain of chain A donates to the EntE adenylation domain of chain B and the PCP domain of chain B interacts with the EntE domain of chain A (Fig. 6 ▶ a).
Figure 6.

Structure of EntE-B in the new crystal form. (a) Ribbon diagram showing two molecules of EntE-B in the asymmetric unit. Each EntE adenylation domain interacts with the PCP domain from the EntB portion of the alternate chain. The pantetheine and inhibitor molecules are highlighted in blue. (b) Superposition of the current structure of EntE-B (pink and red) upon the EntE residues of PDB entry 3rg2, the previous crystal structure. The adenylation domain of the original structure is shown in white and the PCP domain is shown in green. The C-terminal linker helix and the EntB domain of 3rg2 are rotated by 26° relative to the current structure. (c) Although the relative positions of the EntE and EntB domains are distinct in the two crystal forms, the two structures form the same intermolecular interaction that represents the functional interaction between NRPS adenylation and PCP domains. The surface represents the EntE adenylation domain from the current structure, with the N-terminal subdomain colored white and the C-terminal subdomain colored pink. The interacting PCP domains are shown in red for the new structure and in green for the original structure 3rg2.
The model contains residues 2–615 of both chains. Two flexible regions that are often disordered in other family members are poorly ordered in the current structures. The phosphate-binding loop at Ser190-Gly-Gly-Thr-Thr-Gly-Thr196 is poorly ordered in both chains, with Thr193 missing in chain A and Gly192 and Thr193 missing in chain B. Additionally, the linker sequence that joins the adenylation domain to the PCP, Gly537-Arg-Ala-Ser-Ile-Pro542, which was engineered to mimic the interdomain linker in the related two-domain protein EntF (Sundlov, Shi et al., 2012 ▶), is poorly ordered in both chains. In chain A, Arg538–Ile541 were not modeled. In chain B, the density of these residues was weak; however, it was deemed to be of sufficient quality for all of the linker residues to be included in the final model.
The two chains of the model superimposed with an r.m.s. displacement of 0.3 Å for all Cα atoms. Despite the inherent conformational flexibility of the interdomain interactions, both chains adopt the same overall structure. As in the previous structure of EntE-B, the protein adopts a domain-swapped dimeric structure with the PCP domain of chain A interacting with the EntE adenylation domain of chain B and vice versa (Fig. 6 ▶ a). The C-terminal domain of EntE ends with a long α-helix that is pulled away from the rest of the C-terminal domain, presenting the PCP domain to the neighboring subunit. In the previous structure of EntE-B, this helix adopted different angles to allow the PCP domain to maintain consistent interactions with the neighboring EntE domain (Sundlov, Shi et al., 2012 ▶).
The current structure, which is derived from an alternate space group, extends this observation further to the new crystal form. Aligning the EntE portion of the new 2.4 Å resolution crystal structure with the original 3.1 Å resolution structure, the r.m.s. displacement over all Cα positions is 0.5 Å. (All analyses with PDB entry 3rg2 were performed with chains C and H as these chains both had complete density for the interdomain linker.) Similarly, comparison of the PCP domains yields values of 0.3–0.4 Å. However, alignment of the entire chain results in an r.m.s. displacement of 3.0 Å, demonstrating a change in the relative orientation of the domains. Indeed, superposition of the EntE adenylation domains of the current and the previous structures (Fig. 6 ▶ b) illustrates movement of the C-terminal helix and the PCP relative to the EntE molecule. Analysis of the relative change with DynDom (Hayward & Lee, 2002 ▶) shows that the C-terminal helix and PCP domain rotate by 26° around the pivot point at Lys519, a residue that is part of a conserved catalytic motif (Gulick, 2009 ▶). This region of the protein, which extends from the C-terminal subdomain in the thioester-forming conformation and is frequently poorly ordered in crystal structures, may thus represent an additional hinge region joining the adenylation and PCP domains that enables some degree of flexibility between these domains.
Despite differences in the intramolecular domain orientation between the previous and current EntE-B structures, both the original monoclinic and the new orthorhombic models illustrate identical interactions in the intermolecular interface between the EntE adenylation domain of one chain and the PCP domain of the alternate chain (Fig. 6 ▶ c). Along with the biochemical analyses that allowed us to use this structure to guide the optimization of activity with an EntE homolog (Sundlov, Shi et al., 2012 ▶) and the subsequent determination of an additional crystal structure of a native adenylation–PCP di-domain protein (Mitchell et al., 2012 ▶), this striking conservation of the domain interface supported the biological relevance of the observed intermolecular adenylation–PCP domain interaction.
3.5. Analysis of the ligand interactions and domain interface
The observation of an identical domain interaction in a second crystal form strongly supports the hypothesis that the structural interface represents an enzymatically relevant conformation. Additionally, the current structure provides a higher resolution view of the domain interactions and the active site (Fig. 7 ▶). The inclusion of water molecules in the current structure identifies a more highly solvated ligand environment, particularly in the region of the cofactor phosphate moiety. These waters form a network of interactions in both chains that include the phosphate O atoms.
Figure 7.

Stereo representation of the active site of the EntE adenylation domain. (a) View of the adenylate-binding pocket and the interactions with the DHB-adenosylvinylsulfonamide inhibitor. The β-cysteamine portion of the pantetheine cofactor is also shown. Both ligands are shown in ball-and-stick representation with green C atoms, blue N atoms, red O atoms and yellow S atoms. Key interacting residues are indicated. (b) View of the pantetheine tunnel and the network of waters near the phosphate moiety of the phosphopantetheine cofactor. Atoms are colored as in (a); however, the PCP domain of chain B is represented as yellow sticks while the EntE residues of chain A are shown in white.
The active site of the EntE adenylation domain is similar to the structures of other 2,3-dihydroxybenzoate-activating enzymes that have been determined previously (Drake et al., 2010 ▶; May et al., 2002 ▶). The sulfonamide moiety is rotated slightly compared with the previous structures. The position of the central S atom can be confidently placed by the highest peak of the unbiased difference map and is moved by ∼1 Å relative to the previous structures. This may reflect a new position imposed by the covalent trisubstrate analog at the active site. The 3-OH of DHB interacts through a hydrogen bond with the side chain of Ser240, distinguishing it from the salicyl-based inhibitor of the previous EntE-B structure. The pantetheine cofactor of the EntB PCP domain enters the EntE domain through a pantetheine tunnel that forms between the EntE N- and C-terminal subdomains. The pantetheine group makes several hydrogen-bond interactions through its amide groups. The amine of the cysteamine moiety hydrogen-bonds to the main-chain carbonyl of Gly439, and the β-alanine carbonyl forms a water-mediated interaction with the carbonyl of Pro231. The carbonyl and hydroxyl of the pantoate moiety both interact with water molecules, leading to the network of waters that surround the phosphate (Fig. 7 ▶).
As noted, the EntE and EntB proteins adopt the same functional interaction as was observed in the previous structure (Fig. 6 ▶ c). PCP domains are generally composed of four α-helices (Crosby & Crump, 2012 ▶). The conserved serine that serves as the site of phosphopantetheinylation is positioned at the start of helix 2. Two regions of the EntB carrier protein domain contribute to the interface with the adenylation domain. The first group of residues lie between helix 1 and helix 2, a motif known as loop 1, and interact with residues from the C-terminal subdomain of EntE. Additionally, residues from helix 2 interact with a helix on the N-terminal subdomain of EntE (Fig. 8 ▶ a).
Figure 8.

View of the interface between the adenylation (EntE) and PCP (EntB) domains. (a) Cartoon representation of the EntE (white) and EntB (cyan) interface. The pantetheine cofactor bound to Ser575 and the vinylsulfonamide inhibitor are shown in stick representation with yellow C atoms. The interface is composed of two regions of the PCP: loop 1 and helix 2. Cα positions in these two regions are highlighted with spheres. (b) Interactions made with the helix 2 residues include a hydrophobic region at the start of this helix as well as several ionic or polar interactions. (c, d) Interactions made between the loop 1 residues and regions from the dynamic C-terminal subdomain of EntE. The orientation in (b)–(d) is approximately the same as in (a).
Interactions between helix 2 and the EntE include a hydrophobic patch near the N-terminus of the helix (Fig. 8 ▶ b). Val576 and Met579 from this helix interact with Leu469, Met470 and Leu485 of EntE. The carbonyl O atom of Met579 also forms a water-mediated hydrogen bond to the side chain of Thr262. At the other end of the helix, Arg584 forms an ionic interaction with Glu292 and the side chain of Lys587 interacts with the carbonyl O atom of Glu292. The loop 1 interactions include both direct and new water-mediated bonds that were not observed in the previous low-resolution structure (Figs. 8 ▶ c and 8 ▶ d). Four aspartic acid residues of EntB loop 1 form ionic interactions with residues from the mobile C-terminal subdomain of EntE. Asp557 interacts with Arg490. Asp566 interacts with Arg491. Asp570 interacts with the side chains of both Arg491 and Arg494. Asp560 forms a hydrogen bond to the amide N atom of Val487. This valine residue lies at the N-terminus of an α-helix and therefore may carry a partial positive charge owing to the helix dipole. From the EntE side of the interface, Arg437, Lys473 and Asp505 interact with the network of water molecules that includes the cofactor phosphate and the carbonyl O atom of Gly572.
4. Conclusions
Here, we provide an example of a challenging molecular-replacement structure determination involving a pure translational symmetry between the two protein chains in the asymmetric unit. We also present a retrospective analysis of the impact of the translational symmetry on the diffraction and the imposed absences, which were not obvious until the diffraction patterns were analyzed visually.
More importantly, the new structure provides a higher resolution view of the interface between two protein domains from the NRPS biosynthetic proteins. The intermolecular interaction between EntE of one protein chain and EntB of the other is conserved in the current structure and the multiple copies in the previous lower resolution structure. This offers confidence that the crystallographically observed interface reflects the biological complex and is not altered by the constraints of the crystal lattice. The structures we have determined of adenylation–PCP domain interfaces therefore provide a view of the complex and offer guidance for efforts to engineer new NRPS clusters that require heterologous interaction of non-native NRPS domains. Continued structural and functional studies of multiple structures of NRPS domain interactions will remain valuable for the understanding of these assembly-line enzymes.
Supplementary Material
PDB reference: EntE-B, 4iz6
Acknowledgments
We thank Dr Robert H. Blessing for carefully reading the manuscript and helpful suggestions from the Co-editor and reviewers. This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award No. R01-GM068440 to AMG. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the US Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393) and the National Center for Research Resources (P41RR001209). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS, NCRR or NIH.
Footnotes
The EntB carrier protein domain has been variously termed an acyl carrier protein, an aryl carrier protein and a peptidyl carrier protein. Although aryl carrier protein is most correct, we will use peptidyl carrier protein (PCP) as a generic term for all NRPS carrier domains.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Baltz, R. H. (2009). Curr. Opin. Chem. Biol. 13, 144–151. [DOI] [PubMed]
- Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. (2011). Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed]
- Bruner, S. D., Weber, T., Kohli, R. M., Schwarzer, D., Marahiel, M. A., Walsh, C. T. & Stubbs, M. T. (2002). Structure, 10, 301–310. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Chook, Y. M., Lipscomb, W. N. & Ke, H. (1998). Acta Cryst. D54, 822–827. [DOI] [PubMed]
- Condurso, H. L. & Bruner, S. D. (2012). Nat. Prod. Rep. 29, 1099–1110. [DOI] [PMC free article] [PubMed]
- Crosby, J. & Crump, M. P. (2012). Nat. Prod. Rep. 29, 1111–1137. [DOI] [PubMed]
- Drake, E. J., Duckworth, B. P., Neres, J., Aldrich, C. C. & Gulick, A. M. (2010). Biochemistry, 49, 9292–9305. [DOI] [PMC free article] [PubMed]
- Drake, E. J., Nicolai, D. A. & Gulick, A. M. (2006). Chem. Biol. 13, 409–419. [DOI] [PubMed]
- Du, L., He, Y. & Luo, Y. (2008). Biochemistry, 47, 11473–11480. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Fischbach, M. A. & Walsh, C. T. (2006). Chem. Rev. 106, 3468–3496. [DOI] [PubMed]
- Frueh, D. P., Arthanari, H., Koglin, A., Vosburg, D. A., Bennett, A. E., Walsh, C. T. & Wagner, G. (2008). Nature (London), 454, 903–906. [DOI] [PMC free article] [PubMed]
- Gehring, A. M., Bradley, K. A. & Walsh, C. T. (1997). Biochemistry, 36, 8495–8503. [DOI] [PubMed]
- Gehring, A. M., Mori, I. & Walsh, C. T. (1998). Biochemistry, 37, 2648–2659. [DOI] [PubMed]
- Guarné, A., Tormo, J., Kirchweger, R., Pfistermueller, D., Fita, I. & Skern, T. (1998). EMBO J. 17, 7469–7479. [DOI] [PMC free article] [PubMed]
- Gulick, A. M. (2009). ACS Chem. Biol. 4, 811–827. [DOI] [PMC free article] [PubMed]
- Gulick, A. M., Starai, V. J., Horswill, A. R., Homick, K. M. & Escalante-Semerena, J. C. (2003). Biochemistry, 42, 2866–2873. [DOI] [PubMed]
- Hayward, S. & Lee, R. A. (2002). J. Mol. Graph. Model. 21, 181–183. [DOI] [PubMed]
- Hur, G. H., Vickery, C. R. & Burkart, M. D. (2012). Nat. Prod. Rep. 29, 1074–1098. [DOI] [PMC free article] [PubMed]
- Kapust, R. B., Tözsér, J., Fox, J. D., Anderson, D. E., Cherry, S., Copeland, T. D. & Waugh, D. S. (2001). Protein Eng. 14, 993–1000. [DOI] [PubMed]
- Keating, T. A., Marshall, C. G., Walsh, C. T. & Keating, A. E. (2002). Nature Struct. Biol. 9, 522–526. [DOI] [PubMed]
- Keatinge-Clay, A. T. (2012). Nat. Prod. Rep. 29, 1050–1073. [DOI] [PubMed]
- Kissinger, C. R., Gehlhaar, D. K., Smith, B. A. & Bouzida, D. (2001). Acta Cryst. D57, 1474–1479. [DOI] [PubMed]
- Kochan, G., Pilka, E. S., von Delft, F., Oppermann, U. & Yue, W. W. (2009). J. Mol. Biol. 388, 997–1008. [DOI] [PubMed]
- Koglin, A., Löhr, F., Bernhard, F., Rogov, V. V., Frueh, D. P., Strieter, E. R., Mofid, M. R., Güntert, P., Wagner, G., Walsh, C. T., Marahiel, M. A. & Dötsch, V. (2008). Nature (London), 454, 907–911. [DOI] [PMC free article] [PubMed]
- Lee, T. V., Johnson, L. J., Johnson, R. D., Koulman, A., Lane, G. A., Lott, J. S. & Arcus, V. L. (2010). J. Biol. Chem. 285, 2415–2427. [DOI] [PMC free article] [PubMed]
- Liu, Y., Zheng, T. & Bruner, S. D. (2011). Chem. Biol. 18, 1482–1488. [DOI] [PMC free article] [PubMed]
- Luft, J. R., Collins, R. J., Fehrman, N. A., Lauricella, A. M., Veatch, C. K. & DeTitta, G. T. (2003). J. Struct. Biol. 142, 170–179. [DOI] [PubMed]
- Marahiel, M. A. & Essen, L.-O. (2009). Methods Enzymol. 458, 337–351. [DOI] [PubMed]
- May, J. J., Kessler, N., Marahiel, M. A. & Stubbs, M. T. (2002). Proc. Natl Acad. Sci. USA, 99, 12120–12125. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- McPhillips, T. M., McPhillips, S. E., Chiu, H.-J., Cohen, A. E., Deacon, A. M., Ellis, P. J., Garman, E., Gonzalez, A., Sauter, N. K., Phizackerley, R. P., Soltis, S. M. & Kuhn, P. (2002). J. Synchrotron Rad. 9, 401–406. [DOI] [PubMed]
- Mitchell, C. A., Shi, C., Aldrich, C. C. & Gulick, A. M. (2012). Biochemistry, 51, 3252–3263. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Oksanen, E., Jaakola, V.-P., Tolonen, T., Valkonen, K., Åkerström, B., Kalkkinen, N., Virtanen, V. & Goldman, A. (2006). Acta Cryst. D62, 1369–1374. [DOI] [PubMed]
- Padilla, J. E. & Yeates, T. O. (2003). Acta Cryst. D59, 1124–1130. [DOI] [PubMed]
- Qiao, C., Wilson, D. J., Bennett, E. M. & Aldrich, C. C. (2007). J. Am. Chem. Soc. 129, 6350–6351. [DOI] [PMC free article] [PubMed]
- Read, R. J., Adams, P. D. & McCoy, A. J. (2013). Acta Cryst. D69, 176–183. [DOI] [PMC free article] [PubMed]
- Reger, A. S., Carney, J. M. & Gulick, A. M. (2007). Biochemistry, 46, 6536–6546. [DOI] [PMC free article] [PubMed]
- Reger, A. S., Wu, R., Dunaway-Mariano, D. & Gulick, A. M. (2008). Biochemistry, 47, 8016–8025. [DOI] [PMC free article] [PubMed]
- Rudolph, M. G., Wingren, C., Crowley, M. P., Chien, Y. & Wilson, I. A. (2004). Acta Cryst. D60, 656–664. [DOI] [PubMed]
- Samel, S. A., Marahiel, M. A. & Essen, L.-O. (2008). Mol. Biosyst. 4, 387–393. [DOI] [PubMed]
- Samel, S. A., Wagner, B., Marahiel, M. A. & Essen, L.-O. (2006). J. Mol. Biol. 359, 876–889. [DOI] [PubMed]
- Strieker, M., Tanović, A. & Marahiel, M. A. (2010). Curr. Opin. Struct. Biol. 20, 234–240. [DOI] [PubMed]
- Sundlov, J. A., Fontaine, D. M., Southworth, T. L., Branchini, B. R. & Gulick, A. M. (2012). Biochemistry, 51, 6493–6495. [DOI] [PMC free article] [PubMed]
- Sundlov, J. A., Shi, C., Wilson, D. J., Aldrich, C. C. & Gulick, A. M. (2012). Chem. Biol. 19, 188–198. [DOI] [PMC free article] [PubMed]
- Tanovic, A., Samel, S. A., Essen, L.-O. & Marahiel, M. A. (2008). Science, 321, 659–663. [DOI] [PubMed]
- Tsai, Y., Sawaya, M. R. & Yeates, T. O. (2009). Acta Cryst. D65, 980–988. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst. 30, 1022–1025.
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yonus, H., Neumann, P., Zimmermann, S., May, J. J., Marahiel, M. A. & Stubbs, M. T. (2008). J. Biol. Chem. 283, 32484–32491. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: EntE-B, 4iz6