

Summary
In this short review, the general principles are described for obtaining microscopic images with resolution beyond the optical diffraction limit with single molecules. Although it has been known for several decades that single-molecule emitters can blink or turn on and off, in recent work the addition of on/off control of molecular emission to maintain concentrations at very low levels in each imaging frame combined with sequential imaging of sparse subsets has enabled the reconstruction of images with resolution far below the optical diffraction limit. Single-molecule active control microscopy provides a powerful window into information about nanoscale structures that was previously unavailable.
Keywords: Single-molecule, super-resolution, subdiffraction microscopy
Introduction
Although the optical study of single molecules in condensed phases is now a relatively mature field (Moerner, 2009), in the past 5 years a clever modification of standard wide-field single-molecule fluorescence microscopy has enabled the optical diffraction limit to be circumvented (Betzig et al., 2006; Hess et al., 2006; Rust et al., 2006), an achievement which is now stimulating a revolution in biological and nanoscopic microscopy. The essential requirements for this process are (1) sufficient sensitivity to enable imaging of single-molecule labels, (2) determination of the position of a single molecule with a precision better than the diffraction limit and (3) the addition of on/off control of the molecular emission to maintain concentrations at very low levels in each imaging frame. When these requirements are met, sequential imaging of sparse subsets of single molecules yields many samples of the underlying structure, enabling reconstruction of a final image with resolution far below the optical diffraction limit, or super-resolution. This new type of imaging provides a powerful window into information about nanoscale structures that was previously unavailable, and each of the required elements for obtaining super-resolution from single-molecule imaging will now be described (Moerner, 2007). Other approaches to obtaining resolution beyond the diffraction limit utilize patterned excitation beams and nonlinear response effects to directly reduce the size of the point spread function (PSF), with two key examples being stimulated emission depletion microscopy (STED) and structured illumination microscopy (Hell, 2007). These two methods do not explicitly require single-molecule imaging and are discussed elsewhere.
The ability to image single molecules in a condensed phase system such as a crystal, polymer, or cell represents the attainment of high signal to noise for individual emitters with reasonable averaging times, i.e. both high probability of photon emission as well as low background levels from the host material. Because high-quantum efficiency detectors and cameras are now available, this can now be achieved by many microscopic configurations, including wide-field epifluorescence, total internal reflection, confocal imaging, multiphoton microscopy and near-field microscopy (Moerner & Fromm, 2003). As a result, optical fluorescence imaging and analysis of single molecules has emerged over the last two decades as a powerful way to study the individual behaviour of biological and complex condensed phase systems, unobscured by ensemble averaging (Gräslund et al., 2010).
An area of intense current interest involves applications of single-molecule optical studies to the interior of living cells (Lord et al., 2010), because optical probing with light at a distance is relatively non-invasive and time-dependent dynamics become accessible. Although tracking of moving single molecules on the plasma membrane or moving in the cytoplasm began more than a decade ago, observing single molecules as they work carrying out a multitude of cellular functions has also been achieved and provides a wealth of additional information about individual behaviours. As is well known, biological fluorescence microscopy depends upon a variety of labelling techniques to light up different structures in cells, but the price often paid for using visible light is the relatively poor spatial resolution compared to x-ray or electron microscopy. The basic problem is that in conventional microscopes, fundamental diffraction effects limit the resolution to a dimension of roughly the optical wavelength λ divided by two times the numerical aperture (NA) of the imaging system, λ/(2 × NA). Because the largest values of NA for state of the art, highly corrected microscope objectives are in the range of about 1.3–1.6, the spatial resolution of optical imaging has been limited to about ~200 nm for visible light of 500 nm wavelength.
Super-localization of single molecules
In fact, the light from single fluorescent molecular labels about 1–2 nm in size provides a way around this problem, that is, a way to provide ‘super-resolution’, or resolution far better than the diffraction limit. How can single emitters help? Figure 1A illustrates schematically how single molecules can still be detected and imaged, using a cell as an example (Moerner, 2007). A focused pumping beam (green) bathes the cell to pump the fluorescent molecules inside the cell, and the cell itself is essentially transparent and not fluorescent if pumping wavelengths longer than about 500 nm are used. The key requirement that must be met is that only one emitting molecule can be present in the pumping volume, which usually means that ultralow concentrations of emitting molecules must be maintained so that molecules are farther apart than about 500 nm to prevent pumping more than one. Each single molecule is a few nanometres in size, far smaller than the focused laser spot, yet, if only one molecule is pumped, information related to one individual molecule and its local ‘nanoenvironment’ can be extracted by detecting the photons from that molecule alone. In terms of spatial resolution, however, when the laser beam is scanned, the observed ‘peak’ from the single nanoscale source of light approximately maps out the PSF of the microscope, because the molecule is a nanoscale light absorber, far smaller than the size of the PSF. More specifically, the molecule absorbs light with a probability proportional to the square of the dot product between the local optical electric field and the molecule’s transition dipole moment. This point was realized at the very beginning of single-molecule imaging, where the fluorescence excitation signal from one molecule was used to map out the size of the focused pumping laser beam (Ambrose et al., 1991).
Fig. 1.
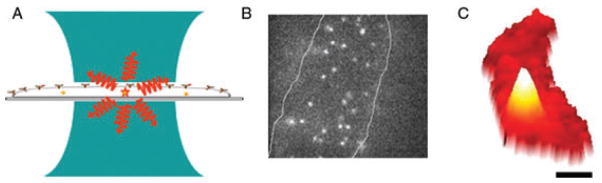
Imaging single molecules. (A) Illustration of single-molecule imaging. A diffraction-limited pumping region (green) illuminates a sample such as a cell containing fluorescent labels. For single-molecule imaging, only one molecule should be emitting (red) within a diffraction-limited pumping volume. (B) Example of wide-field epifluorescence imaging of single fluorescently labelled transmembrane proteins on a cell surface. Frame: 12× 12 μm. (C) Wide-field fluorescence image of a bacterial cell (red) containing a single protein fusion between the bacterial actin protein MreB and EYFP. Rendered in three dimensions with the z-axis as brightness, a single molecule looks like a mountain. A total of 100 ms acquisition time, bar: 0.5 μm. [Adapted and reproduced by permission from (Moerner, 2007); copyright the National Academy of Sciences.]
These considerations apply equally well to both confocal scanning imaging as well as to wide-field imaging, because in the latter, the argument applies to each diffraction-limited voxel. Effectively, then, images of single molecules provide a picture of the PSF of the microscope, as long as subtle effects arising from the dipole emission pattern are not sensed. For the purposes of this short review, we assume that the emitter is rotating sufficiently during the image acquisition that regarding the image as arising from a point source is reasonable. For example, in Fig. 1B, single transmembrane proteins have been labelled with fluorescent dyes, and with the exception of a few molecules that are moving or moving out of focus in the z-direction during the camera imaging time, the individual spots have a diffraction-limited diameter of ~200 nm when reflected back to the sample plane. Similarly, in a much tinier bacterial cell (the red crescent shape), Fig. 1C shows the approximate PSF shape for emission from a single molecule of the bacterial actin protein MreB [the white mountain, labelled by fusion to enhanced yellow fluorescent protein (EYFP)]. It is this mountain-like image from a single molecular emitter that forms one key element for current super-resolution efforts based on single-molecule microscopy.
Recently, several researchers have begun to take advantage of the nanoscale size of single-molecule emitters more directly. Simply by measuring the shape of the PSF using a wide-field image, the (x, y) position of its centre can be determined much more accurately than its width, a process that may be termed “super-localization” (as distinct from super-resolution to be described below). This idea, digitizing and fitting the PSF, a form of simple deconvolution, has been used in many areas of science for many years, but computational deconvolution without exact knowledge of the PSF can generate spurious features in the presence of noise. The knowledge that a single small object is emitting means that a good estimate of the PSF can be extracted almost trivially just by recording the shape of the detected images, illustrated in Fig. 2. Put another way, the knowledge that only one tiny nanoscale emitter is present allows the experimenter to interpret the centre of the PSF as a measurement of the location of the emitter. This idea was applied early on to single nanoscale fluorescent beads labelled with many emitters and then to low-temperature single-molecule images where both spatial information and the secondary variable, laser wavelength, were used to separate molecules (see Refs. in Moerner, 2007).
Fig. 2.
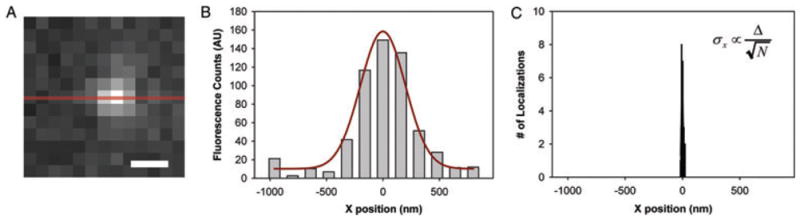
Super-localization of a single fluorescent molecule. (A) Fluorescence image of a single emitting molecule embedded in a polymer thin film on a cover slip, showing camera pixelation. (B) Cross-section in the x-direction through the centre of the image in (A). Each bin is a pixel (160 nm in width), and the counts in the pixel are the digital counts of detected photons coming from the camera. The data is fit to a Gaussian function with a standard deviation of ~200 nm, which is roughly equivalent to the diffraction limit. (C) Distribution of 50 centre position determinations from 50 images plotted on the same spatial scale as the data in (B), showing a drastically smaller width distribution (a standard deviation of 9 nm). [Adapted and reproduced by permission from (Thompson et al., 2010); copyright Academic Press.]
The process proceeds as follows. First, a wide-field fluorescence image of the single molecule must be recorded, with sufficient expansion of the image to cause the light to fall on several pixels of the camera as in Fig. 2A. In this way, the different camera pixels sample different parts of the shape of the diffraction-limited spot. A cross-section of this image is shown in Fig. 2B in the histogram, and the presence of nonzero background signal is evident as in most single-molecule imaging. Then a model function is used to fit the shape of the recorded image, such as a Gaussian function plus a constant, or an Airy function plus a constant. Although the Airy function is more precisely the correct PSF for a circular-aperture limited image of a point source, most situations have insufficient signal to noise to require use of the Airy function. An example two-dimensional fitting function with a Gaussian shape with a standard deviation Δ limited by diffraction is:
| (1) |
where (u, v) are the coordinates in the sample plane, (μx, μy) is the two-dimensional position of the molecule, A is the amplitude or peak value of the function and B is the background level, which generally represents the background photons from other emitters in the sample or from Rayleigh or Raman scattering.
Super-localization works because the parameters μx and μy extracted from the fit to the image have a much narrower probability distribution and therefore can be known with a precision much smaller than Δ, as illustrated in Fig. 2C, which was directly measured by performing the measurement and fitting procedure on the same single molecule multiple times. Theoretically, the precision with which a single molecule’s position can be determined by digitizing the PSF depends fundamentally upon the Poisson process of photon detection. In this case, the PSF of the microscope serves as the probability distribution function for the positions on the detector where photons are detected, which is equivalent to a probability distribution for the position of the molecule. Each photon that arrives at the detector in a particular position is a sample measured from this distribution. If we measure the mean position of the Gaussian image (μx, μy) (the position of the molecule), the precision of the measurement is given by the standard error of the mean with N samples. In one dimension (say x), the standard error of the mean σx is given by (ignoring background and pixelation)
| (2) |
The value of σx in Eq. (2) is taken as the (statistical) localization precision, a quantity of particular importance because it effectively represents the uncertainty associated with one measurement of a molecule’s position extracted from an image with N detected photons. From Eq. (2), it can be seen that σx improves both with a smaller PSF (smaller Δ, which is not really possible due to the diffraction limit) or with more photons collected. Therefore a molecule that emits more photons will be localized with better precision (i.e. organic fluorophores such as Cy3 can be localized much more precisely than fluorescent proteins such as green fluorescent protein (GFP) and its mutants).
In a real measurement, fluctuations in background level B and the division of the photons into pixelated bins both affect the precision. In 2002, Thompson, Larson and Webb (Thompson et al., 2002) derived an expression for the localization precision in the presence of both a finite pixel size, a, and background root mean square noise, b. The theoretical two-dimensional localization precision assuming only Poisson noise fluctuations can then be written as
| (3) |
It is important to note that Eq. (3) overestimates the precision of true measurements by at least 30% mainly because of higher-order terms that are neglected in the derivation. Nevertheless, this expression has been used as a theoretical estimate of the localization precision to good effect in many studies, perhaps some of the most prominent being fluorescence imaging with one nanometre accuracy (see Table 1 and Refs. in Moerner, 2007).
Table 1.
Selected acronymsa.
| Super-Localization Methods | |
| FIONA | Fluorescence Imaging with One Nanometre Accuracy |
| SHREC | Single-Molecule High-Resolution Colocalization of Fluorescent Probes |
| Super-Resolution Methods: Single-Molecule Active Control Microscopies (SMACM) | |
| SHRImP | Single-Molecule High-Resolution Imaging with Photobleaching |
| NALMS | Nanometre Localized Multiple Single-Molecule Fluorescence Microscopy |
| PALM | Photoactivated Localization Microscopy |
| F-PALM | Fluorescence Photoactivation Localization Microscopy |
| STORM | Stochastic Optical Reconstruction Microscopy |
| PAINT | Points Accumulation for Imaging in Nanoscale Topography |
| dSTORM | Direct STORM |
| RESOLFT | Reversible Saturable Optical Fluorescence Transitions |
| GSDIM | Ground State Depletion with Intermittent Return |
For references, see Moerner (2007) and Hell (2007).
A variation on the digitization of the PSF for one single molecule occurs when a variable, such as excitation colour or wavelength, allows different molecules in the same diffraction-limited volume to be separately localized. If this can be done, there is no need to reduce the concentration of single molecules to levels so low that only one molecule is present in the pumped laser volume. This idea was central to the early low-temperature fluorescence excitation work of the early 1990s, where hundreds of molecules in the same volume were separated by excitation wavelength, and was generalized to other variables at room temperature in 1995 by Betzig. By separately imaging two fluorophores (Cy3 and Cy5) attached to two different calmodulin molecules that bind to the ‘legs’ of the same single molecule of myosin V, distance measurements accurate to ~10 nm were achieved, and a another acronym was generated (SHREC, for single-molecule high-resolution colocalization of fluorescent probes).
Super-resolution
However, imaging of complex structures in cells is a more difficult problem than super-localizing just one or a few at ultralow concentrations. It is still necessary to deal with a densely labelled structure, with many labels whose PSFs overlap. A partial solution involves utilizing naturally occurring photobleaching – eventually all molecules will bleach except one, and some information can be obtained from the positions of the last few emitters. Adding further to the exploding menagerie of acronyms, this basic idea was demonstrated by Gordon et al. for Cy3 labels on DNA (SHRImP, for single-molecule high-resolution imaging with photobleaching) and by Qu et al. using Cy3- PNA probes on DNA (NALMS, for nanometre localized multiple single-molecule fluorescence microscopy). Lidke et al. showed in 2005 that position information beyond the diffraction limit can be obtained for several closely spaced quantum dots using the intrinsic blinking of the emitters, but no super-resolution structure of an extended object was provided.
A major advance occurred in 2006 when three groups described and demonstrated a solution to the general problem of effectively resolving closely spaced emitters decorating a structure to be determined, as shown in Fig 3A. The essential idea is that one must arrange that not all emitters are emitting at the same time, and then sequentially image sparse subsets to build up a pointillist reconstruction of the underlying structure. Suppose for the moment that the emitters are photoactivatable, that is they are dark and in a non-emissive precursor state illustrated in Fig. 4, left side. Then weak activating light is used to turn on only a very small number of emitters, so few that their PSFs do not overlap (Fig. 3B). These emitters can then be super-localized as described above and the positions of the single molecules recorded. The emitters eventually photobleach, and then a new sparse subset is photoactivated. Because the turning on of single emitters is stochastic, this approach will sample the underlying structure more and more. After a series of sequential imaging steps, the underlying structure can be reconstructed as shown at the right in Fig. 3B. This idea was termed PALM (photoactivated localization microscopy) by Betzig et al. (2006), where light-induced photoactivation of GFP mutant fusions was used to randomly turn on only a few single molecules at a time in fixed cell sections or fixed cells. [Indeed, reversible photoswitching of the emission of certain GFP mutants was reported in the first single-molecule observations of this amazingly useful cellular label in 1997 (Dickson et al., 1997.)] In the tour de force PALM experiment, individual PSFs were recorded in detail to find their positions to ~20 nm, then were photobleached so that others could be turned on, and so on until many thousands of PSF positions were determined. After 2–12 h of imaging, a high-resolution image was extracted that correlated well with a transmission electron microscopy (TEM) image. Essentially simultaneously, Hess et al. published a nearly identical approach with a very similar acronym, termed F-PALM (fluorescence photoactivation localization microscopy) (Hess et al., 2006), which also utilized a photoactivatable GFP with PSF super-localization to obtain super-resolution. In another report at essentially the same time, Rust et al. utilized photoswitching of a single photoswitchable fluorophore for super-resolution demonstrations in fixed cells with immunofluorescence labelling termed STORM (stochastic optical reconstruction microscopy; Rust et al., 2006).
Fig. 3.
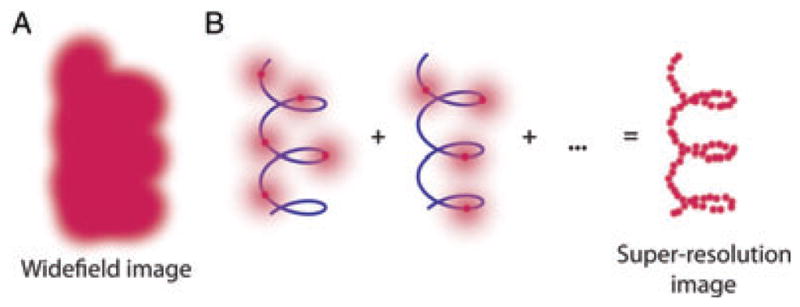
Schematic showing the key idea of super-resolution imaging of a structure by single-molecule super-localization and active control (SMACM). (A) It is not possible to resolve the underlying structure in a conventional wide-field fluorescence image because the fluorescent labels are in high concentration and the images overlap. (B) Using controllable fluorophores, it is possible by either blinking or photoactivation to guarantee that only a sparse subset of molecules are emitting which then can be localized with nanometre precision (blue line is the underlying structure being sampled). Once the first subset of molecules photobleaches or enters a dark state, another subset can be activated or stochastically turned on and localized. This process is repeated and the resulting localizations summed to give a super-resolution reconstruction of the underlying structure. [Adapted and reproduced by permission from (Thompson et al., 2010); copyright Academic Press.]
Fig. 4.
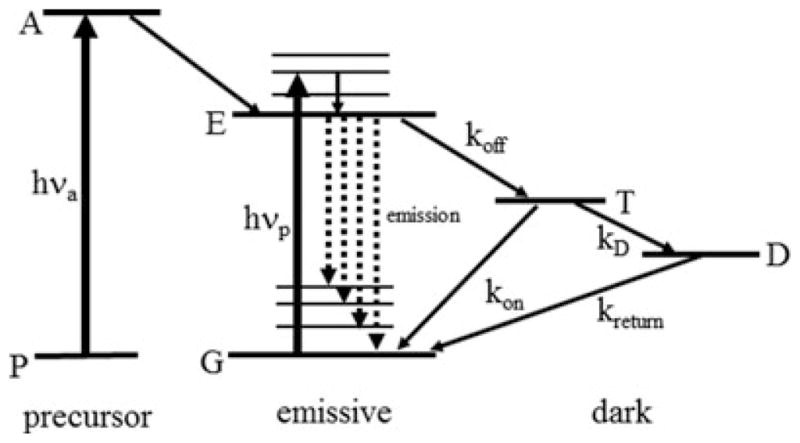
Active control of single-molecule emission from the energy level point of view. The standard emissive form of the molecule is in the centre; pump photons of energy hνp drive transitions from the ground state G typically to vibronic sidebands of the first electronic excited state E. Vibrational relaxation quickly leads to population of state E, from which emission can occur terminating on vibrationally excited levels of the ground state (dashed lines). The molecule can turn off with rate constant koff into a dark state such as a triplet state T for a time period equal to 1/kon before the emission cycle resumes. In some schemes, the molecule can leave the state T with rate constant kD, populating long-lived dark state D for a time (kreturn−1). If photoactivation is involved, then the molecule is not emissive in its precursor form P, but optical activation can occur by pumping to an activated state A with photons of energy hνa, which, with a certain probability, generates the emissive form of the molecule.
It is important to note that all three of these approaches have the common feature that the experimenter must actively control the concentration of emitting single molecules to be at very low levels throughout the experiment. For this reason, the entire class of methods may be termed ‘single-molecule active control microscopy’ or SMACM. The experimenter can use a wide array of photophysical and photochemical effects to achieve control of the emitting concentration. For example, in Fig. 4, photoactivation can be used as noted above, but so can almost any process that turns an emitting molecule off by driving the molecule into a dark state T with rate koff from which the molecule can return to the emissive form at a rate kon. Examples of the dark state can be the molecular triplet state, further dark states produced by photoinduced electron transfer, by intermolecular twists, or by reversible photochemical generation of a dark different molecule from which the emitter can be regenerated by reversal of the photochemical change. In this kind of level structure, higher intensity pumping of the emissive state generates emitted photons at a higher rate, but also the fraction of the time the molecule spends in the dark state increases in a way that is dependent upon the ratio koff/kon and other rate constants. As long as most molecules are in the dark state at any one time, this type of active control enables super-resolution imaging. For example, in the STORM experiments, the presence of a thiol in high concentration near the emitter Cy5 allowed generation of a dark Cy5-thiol adduct.
In recent years, a variety of additional acronyms have appeared denoting alternative mechanisms for actively controlling the emitter concentration. For example, Sharonov et al. presented a scheme based on accumulated binding of diffusible probes, which are quenched in solution yet de-quench in close proximity of the surface of the object to be imaged (PAINT, for points accumulation for imaging in nanoscale topography). The method relies upon the photophysical behaviour of molecules with a twisted intermolecular charge transfer state such as Nile Red. PAINT has the advantages that the object to be imaged need not be covalently labelled and that many individual fluorophores are used for the imaging, thus relaxing the requirement on the total number of photons needed from each single molecule. The feasibility of this approach in the restricted cytoplasm needs to be explored. Additional switching schemes which have been recognized as useful for SMACM imaging include optical saturation of the emission, first envisioned as a method to achieve STED and termed RESOLFT (reversible saturable optical fluorescence transitions), dSTORM (direct STORM) and GSDIM (ground-state depletion with intermittent single-molecule return); for references see Moerner (2007) and Hell (2007, 2009).
Examples
To provide a couple of examples, Fig. 5 shows diffraction-limited and super-resolution images acquired using SMACM methods with resolution in the 40 nm range. In Fig. 5A, B, a bacterial cell has been labelled by fusion of EYFP to a particular cytoskeletal protein called ParA. Without super-resolution, the structure is impossible to determine, but using the intrinsic blinking of EYFP, the fact that ParA forms a tiny 40-nm-wide filament running down the centre of the cell is revealed (Ptacin et al., 2010). In Fig. 5C, D, similar data are shown for microtubules in a BSC-1 mammalian cell. In this situation, the fluorophore was photoactivatable by dim blue light (Lee et al., 2010), and the fluorophores reported the location of tubulin molecules using a genetically directed enzymatic labelling scheme. These examples are just two of the many super-resolution images being reported at the present time (Huang et al., 2010).
Fig. 5.
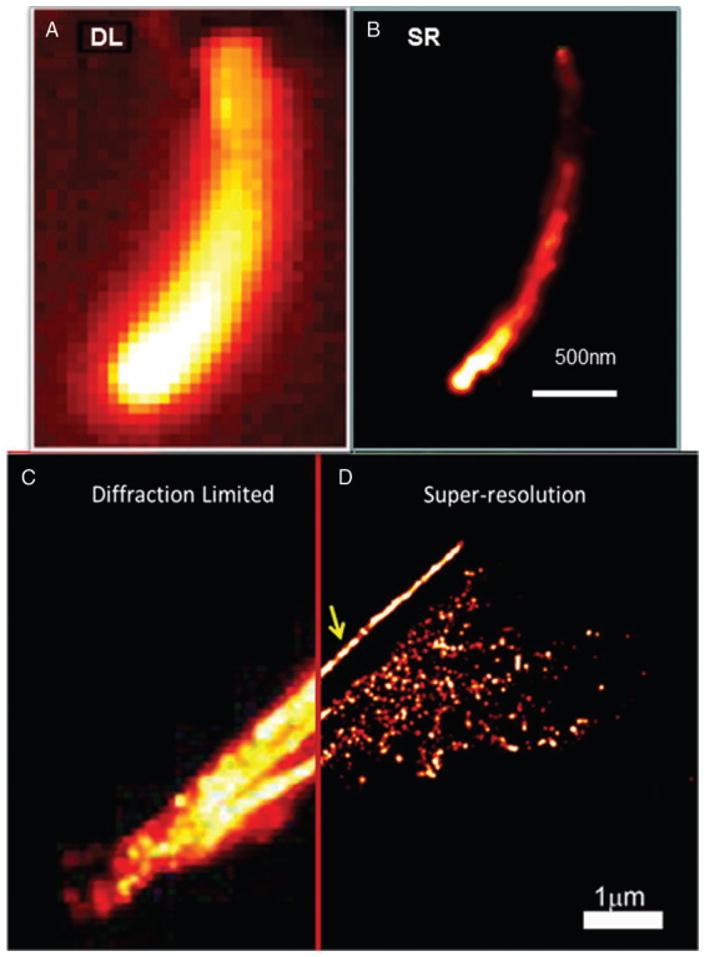
Examples of super-resolution imaging by SMACM. (A) Diffraction-limited and (B) Super-resolution image of EYFP fluorescent protein fusions to the ParA protein in a bacterial cell. Image acquired by blinking of the EYFP. [Adapted and reproduced by permission from (Ptacin et al., 2010); copyright Nature Publishing Group.] and (D) Super-resolution image of microtubules in a mammalian cell. Image acquired by photoactivation of Halo-Tag genetically targeted small molecules. [Adapted and reproduced by permission from (Lee et al., 2010); copyright American Chemical Society.]
All of these tantalizing new approaches to SMACM imaging have advantages and disadvantages, and which method to use can depend upon the questions being asked. First of all, these approaches require sequential imaging of sparse subsets, which means that the time scale for acquiring sufficient numbers of single-molecule locations without physical motion or changes in the underlying structure may be an issue. However, with higher laser intensities and fast on/off switching, imaging times down to the range of tens of seconds to 1 min have been reported, a speed which is fast enough for many cellular experiments of relatively static structures. Next, it is essential when wishing to obtain resolution at a scale of x nm that localizations be recorded down to a separation of x/2 nm (Nyquist criterion) or smaller, so analysis of the data to ensure sufficient sampling is important. Third, in each case, the experimenter can choose a number of ways to display the data: as many tiny points for each position found, as Gaussian spots with a width determined by the precision of the determination and so on. Generally, in each situation, several reconstruction schemes should be explored and validated as carefully as possible to avoid the introduction and unfortunate interpretation of artefacts. Fourth, careful calibration and measurement of the numbers of detected photons is important to provide information on the localization precision achieved (Thompson, 2011). Fifth, there is a subtle distinction between PALM-like methods in which each emitter is turned on only once, and STORM-like methods where each single molecule can blink on and off many times before photobleaching. In the former, one molecule’s information about its position is provided all at once, and there is no confusion regarding multiple determinations of the position of the same molecule (overcounting). On the other hand, considering the blinking methods, one can regard each emission time of the molecule as a new position measurement, so that over time a large number of determinations are made of the position of the molecule. Each of these determinations is performed with fewer photons than if blinking did not occur, thus each has larger mean-squared error. However, taken as a whole, the many position determinations of the same molecule should be equivalent to one long acquisition of all the photons, as long as the molecule does not move and central-limit statistics applies. The situation is roughly analogous to trying to measure a photon emission stream in time, where one can use small time constants and many determinations, or one long time constant. As is well known, if there is any chance of dynamical changes (either unwanted motion or drifts), one would prefer the short time acquisitions, because then dynamical effects (such as motion of the emitters) can be extracted, either directly or by various forms of correlation analysis. Each method has its place, and such considerations may be the topic of future investigations. Of course, more photo stable single-molecule emitters featuring efficient photoactivation and/or fast photoswitching for cellular labelling would certainly help, as precision improves with more photons available from a single molecule before photobleaching (with the exception of PAINT). In addition, as the precision becomes smaller, more detailed analysis of additional subtle optical effects such as image aberration and the influence of the dipole emission pattern will be necessary to push to higher and higher ultimate resolution.
Finally, in comparison to confocal STED imaging, SMACM has precision that improves with the square root of the number of photons detected, whereas the scanning STED method reports on the position of the emitter for every photon detected. At the same time, SMACM methods are experimentally much easier to implement, but require careful computation and analysis of the numbers of detected photons during the image reconstruction process. SMACM methods can also be easily extended to three-dimensional imaging by a variety of approaches (Thompson et al., 2010). It is particularly pleasing, however, that SMACM methods utilize the true digital nature of each single-molecule label to provide direct information on the position of the label, a gratifying concept that provides a way to circumvent the optical diffraction limit by sampling the underlying structure, even inside complex cellular environments.
Acknowledgments
The author is grateful to Julie Biteen, Hsiao-lu D. Lee, Steven F. Lee, Matthew D. Lew, Michael A. Thompson and other past members of the Moerner Lab for fruitful collaborations. This work was supported in part by Award No. R01GM086196 from the National Institute of General Medical Sciences of the National Institutes of Health.
References
- Ambrose WP, Basché T, Moerner WE. Detection and spectroscopy of single pentacene molecules in a p-terphenyl crystal by means of fluorescence excitation. J Chem Phys. 1991;95:7150–7163. [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313(5793):1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Dickson RM, Cubitt AB, Tsien RY, Moerner WE. On/off blinking and switching behavior of single green fluorescent protein molecules. Nature. 1997;388:355–358. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- Gräslund A, Rigler R, Widengren J, editors. Single Molecule Spectroscopy in Chemistry, Physics and Biology: Nobel Symposium 138 Proceedings. Springer; Heidelberg: 2010. [Google Scholar]
- Hell SW. Far-field optical nanoscopy. Science. 2007;316(5828):1153–1158. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- Hell SW. Microscopy and its focal switch. Nat Methods. 2009;6(1):24–32. doi: 10.1038/nmeth.1291. [DOI] [PubMed] [Google Scholar]
- Hess ST, Girirajan TPK, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Babcock H, Zhuang X. Breaking the diffraction barrier: super-resolution imaging of cells. Cell. 2010;143:1047–1058. doi: 10.1016/j.cell.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HD, Lord SJ, Iwanaga S, et al. Superresolution imaging of targeted proteins in fixed and living cells using photoactivatable organic fluorophores. J Am Chem Soc. 2010;132(43):15099–15101. doi: 10.1021/ja1044192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord SJ, Lee HD, Moerner WE. Single-molecule spectroscopy and imaging of biomolecules in living cells. Anal Chem. 2010;82(6):2192–2203. doi: 10.1021/ac9024889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerner WE. New directions in single-molecule imaging and analysis. Proc Natl Acad Sci USA. 2007;104:12596–12602. doi: 10.1073/pnas.0610081104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerner WE. Single-molecule optical spectroscopy and imaging: from early steps to recent advances. In: Graslund A, Rigler R, Widengren J, editors. Single Molecule Spectroscopy in Chemistry, Physics and Biology: Nobel Symposium 138 Proceedings. Springer-Verlag; Berlin: 2009. pp. 25–60. [Google Scholar]
- Moerner WE, Fromm DP. Methods of single-molecule fluorescence spectroscopy and microscopy. Rev Sci Instrum. 2003;74:3597–3619. [Google Scholar]
- Ptacin JL, Lee SF, Garner EC, et al. A spindle-like apparatus guides bacterial chromosome segregation. Nat Cell Biol. 2010;12(8):791–798. doi: 10.1038/ncb2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3 (10):793–796. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MA. The Development of Techniques for Three-Dimensional Superresolution Fluorescence Microscopy and Their Application to Biological Systems. Stanford University; Stanford, CA, USA: 2011. [Google Scholar]
- Thompson RE, Larson DR, Webb WW. Precise nanometer localization analysis for individual fluorescent probes. Biophys J. 2002;82:2775–2783. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MA, Biteen JS, Lord SJ, Conley NR, Moerner WE. Molecules and methods for super-resolution imaging. Meth Enzymol. 2010;475:27–59. doi: 10.1016/S0076-6879(10)75002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]