

Abstract
Transmission by fleabite is a relatively recent evolutionary adaptation of Yersinia pestis, the bacterial agent of bubonic plague. To produce a transmissible infection, Y. pestis grows as an attached biofilm in the foregut of the flea vector. Biofilm formation both in the flea foregut and in vitro is dependent on an extracellular matrix (ECM) synthesized by the Yersinia hms gene products. The hms genes are similar to the pga and ica genes of Escherichia coli and Staphylococcus epidermidis, respectively, that act to synthesize a poly-β-1,6-N-acetyl-d-glucosamine ECM required for biofilm formation. As with extracellular polysaccharide production in many other bacteria, synthesis of the Hms-dependent ECM is controlled by intracellular levels of cyclic-di-GMP. Yersinia pseudotuberculosis, the food- and water-borne enteric pathogen from which Y. pestis evolved recently, possesses identical hms genes and can form biofilm in vitro but not in the flea. The genetic changes in Y. pestis that resulted in adapting biofilm-forming capability to the flea gut environment, a critical step in the evolution of vector-borne transmission, have yet to be identified. During a flea bite, Y. pestis is regurgitated into the dermis in a unique biofilm phenotype, and this has implications for the initial interaction with the mammalian innate immune response.
1 The Evolution of Arthropod-Borne Transmission of Yersinia pestis
1.1 Introduction
The genus Yersinia comprises eleven species in the family Enterobacteriaceae of the Gammaproteobacteria. Three of them, Y. pestis, Y. pseudotuberculosis, and Yersinia enterocolitica, are important pathogens of humans and other mammals; one, Yersinia ruckeri, is the agent of red mouth disease in rainbow trout and the others are non-pathogens that live in water and soil (Bottone et al. 2005). Y. pestis, the causative agent of plague, differs conspicuously from its fellow Yersinia species. It is much more invasive and virulent than Y. pseudotuberculosis or Y. enterocolitica, which cause relatively benign enteric diseases transmitted via contaminated food and water. With a lethal dose to susceptible mammals from an intradermal inoculation site of less than ten cells, Y. pestis is one of the most virulent of all microbes and plague one of the most feared diseases of human history (Perry and Fetherston 1997). A second, no less remarkable difference is that Y. pestis, uniquely among the enteric group of Gram-negative bacteria, has evolved an arthropod-borne route of transmission. Y. pestis is primarily a parasite of rodents that is transmitted by fleas. Permanent enzootic foci exist throughout the world, and plague transmission cycles involve many species of wild rodents and their fleas, making the ecology and epizootiology of plague quite complex (Gage and Kosoy 2005; Pollitzer 1954).
1.2 Y. pestis, a Recently Emerged Clone of Y. pseudotuberculosis
Given the radical differences in ecology and natural history between Y. pestis and Y. pseudotuberculosis, it is surprising that they are in fact very closely related subspecies (Bercovier et al. 1980; Ibrahim et al. 1993). Population genetics analyses indicate that fully virulent Y. pestis strains worldwide constitute a clonal group with eight genomic branches separated by minor sequence differences (Achtman et al. 2004). Based on these analyses, it was estimated that Y. pestis diverged from its Y. pseudotuberculosis ancestor only within the last 10,000–20,000 years (Achtman et al. 1999, 2004). Further comparative genomics analyses confirmed a high degree of genomic identity between the two species (Chain et al. 2004; Hinchliffe et al. 2003; Zhou et al. 2004). The close phylogenetic relationship of Y. pseudotuberculosis and Y. pestis implies that the change from a comparatively benign food- and water-borne enteric pathogen to a highly virulent, arthropod-borne systemic pathogen occurred quite recently in evolutionary terms and is based on relatively few genetic differences (Hinnebusch 1997; Lorange et al. 2005).
1.3 Arthropod-Borne Transmission Factors of Y. pestis
The most obvious genetic difference between Y. pestis and Y. pseudotuberculosis is the presence of two Y. pestis-unique plasmids (Ferber and Brubaker 1981), presumably acquired sequentially by the Y. pestis-progenitor strain via horizontal transfer (Carniel 2003). Each of the two plasmids contains a gene important for transmission by fleas. The 100-kb pFra plasmid encodes Yersinia murine toxin (Ymt), a phospholipase D that greatly enhances survival of Y. pestis in the flea midgut (Hinnebusch et al. 2002b). The 9.5-kb pPla plasmid encodes the Y. pestis plasminogen activator (Pla) (McDonough and Falkow 1989; Sodeinde and Goguen 1988). Although Pla is not required to produce a transmissible infection in the flea or to cause the low-incidence primary septicemic form of plague following flea-bite transmission, it is required for systemic dissemination and the development of bubonic plague following intradermal injection of Y. pestis by fleabite or by needle (Brubaker et al. 1965; Hinnebusch et al. 1998; Sebbane et al. 2006; Sodeinde et al. 1992).
As will become evident in the following sections, additional genetic differences between Y. pestis and Y. pseudotuberculosis that are pertinent to the evolution of arthropod-borne transmission remain to be discovered. Although their chromosomes are nearly identical, Y. pestis contains 32 chromosomal genes that so far have not been found in any Y. pseudotuberculosis isolate; conversely, 317 genes on the chromosome of Y. pseudotuberculosis strain IP32953 were not detected in a panel of Y. pestis isolates (Chain et al. 2004). In addition, Y. pestis contains many pseudogenes that are intact in Y. pseudotuberculosis, most of them metabolic genes that are presumably not needed for a mammal-flea, eukaryotic host-associated life cycle (Chain et al. 2004; Parkhill et al. 2001). It is possible that instances of specific gene loss or change of function as well as gene gain contributed to the evolution of arthropod-borne transmission.
2 Plague Transmission Models
Transmission of Y. pestis by fleas can occur by at least three different mechanisms, which may be more or less important among the many different species of flea vectors and at different epizootiologic stages. The first is simple mechanical transmission, which can occur if a flea feeds on a new host shortly after taking a blood meal from a highly septicemic host. Mechanical transmission is akin to dirty-needle transmission, in which the inoculum derives from a residue on the flea mouthparts that remains from a prior infected blood meal (Burroughs 1947). Biological transmission, in contrast, is dependent on bacterial multiplication in the flea midgut and subsequent regurgitation into the bite site. The general mechanism of biological transmission was described by the English medical entomologist A. W. Bacot in two classic papers (Bacot and Martin 1914; Bacot 1915). Bacot observed that Y. pestis grew in the form of large aggregates in the midgut of infected fleas and that in some fleas bacterial aggregates also developed in the lumen of the proventriculus, a valve connecting the esophagus and midgut that opens and closes rhythmically during blood feeding. Colonization of the proventriculus was found to be critical for efficient transmission. As the Y. pestis aggregates grew in the proventriculus, they interfered with its valvular function, permitting regurgitation of blood, carrying bacteria from the midgut or the proventriculus along with it, back into the bite site. In some fleas, consolidation of the Y. pestis aggregate filled the entire lumen of the proventriculus and completely blocked the passage of blood (Fig. 1). However, complete blockage is not essential for efficient transmission; Bacot thought that incompletely blocked fleas might actually be better transmitters (Bacot 1915). This is often overlooked due to the emphasis on blocked fleas, but is an important component of the classic Bacot model because complete proventricular blockage may not develop regularly in all flea vector species (Bacot 1915; Burroughs 1947; Pollitzer 1954). An alternative type of regurgitative transmission that may not involve bacterial interference of proventricular function was recently described for the squirrel flea Oropsylla montana, which likely contributes to the vector competence of fleas in which proventricular blockage is infrequently observed (Eisen et al. 2006; Webb et al. 2006). Whether this newly described mechanism depends on biofilm formation or is closer to simple mechanical transmission remains to be determined.
Fig. 1.
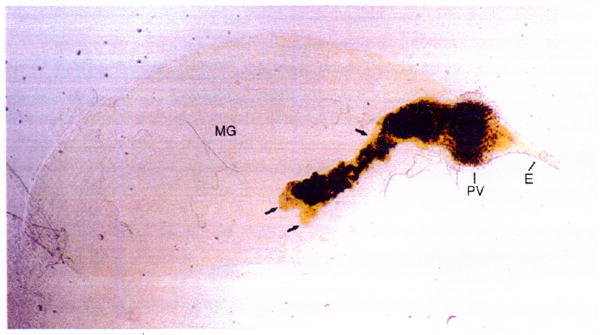
Y. pestis biofilm in the flea. Digestive tract dissected from an X. cheopis flea blocked with a dense biofilm consisting of dark masses of Y. pestis embedded in a paler, viscous ECM (arrows). The contiguous biofilm fills the proventriculus (PV) and extends posteriorly into the lumen of the midgut (MG). E, esophagus
2.1 The Coagulase Model of Proventricular Infection
For many years, bacterial aggregation in the flea midgut and proventricular blockage was attributed to a coagulase activity of the pPla-encoded Y. pestis plasminogen activator (Pla). Although Pla acts to rapidly degrade fibrin clots at 37°C by activating plasminogen, at lower temperatures typical of the flea gut environment an opposite clot-forming or coagulase activity was observed. According to the coagulase model (Cavanaugh 1971), which is still often cited in textbooks, Y. pestis is enveloped and multiplies within a fibrin clot formed from the flea’s blood meal by the coagulase activity of Pla. Proventricular colonization and blockage was hypothesized to result from bacteria-laden clots lodging among the proventricular spines. This hypothesis was compelling because it could also explain the decrease in proventricular blockage and flea-borne transmission during hot weather (Cavanaugh and Marshall 1972). The fibrin-dissolving activity of Pla that predominates at higher temperatures was invoked to explain that phenomenon. (Cavanaugh 1971). McDonough et al. (1993) subsequently reported that a Y. pestis pla mutant caused less mortality to fleas than the isogenic Pla+ parent strain, and suggested that this might be due to a decreased ability to cause blockage. However, the mortality was measured at 4 days after infection, before blockage would be expected to occur.
Several lines of investigation contradict the coagulase model, however. The coagulase activity of Pla is weak and has been observed only with rabbit plasma, and not with rodent or human plasma (Beesley et al. 1967; Jawetz and Meyer 1944; Sodeinde et al. 1992). Pla-negative Y. pestis strains are able to block the rat flea Xenopsylla cheopis as well as wild type strains, with the same inverse relationship between blockage rate and ambient temperature, refuting both predictions of the coagulase model (Hinnebusch et al. 1998). The matrix of the Y. pestis aggregates that form in the flea digestive tract is also unaffected by treatment with proteases, including plasmin (Jarrett et al. 2004). More recent work indicates that instead of a role in producing a transmissible infection in the flea vector, the true biological role of Pla occurs after transmission. Plasminogen activation and other proteolytic activities of Pla are required for the development of bubonic plague, because they facilitate dissemination of Y. pestis from the skin and lymphatic system (Brubaker et al. 1965; Korhonen et al. 2004; Sebbane et al. 2006; Sodeinde et al. 1992).
2.2 The Biofilm Model of Proventricular Infection
The current model for the autoaggregative growth phenotype first observed by Bacot is that Y. pestis grows as a biofilm in the flea digestive tract (Jarrett et al. 2004). According to this model, the decrease in temperature experienced by the bacteria upon leaving the warm-blooded mammal triggers Y. pestis to synthesize a stable extracellular biofilm matrix. This extracellular matrix (ECM) surrounds the dense microcolony of Y. pestis as they grow and also potentiates bacterial aggregation on the surface of the proventricular spines that leads to transmission. The same general strategy is used by two other arthropod-borne microbial pathogens that are transmitted regurgitatively. The phytopathogen Xylella fastidiosa forms a biofilm of polarly attached cells in the foregut of its vectors, which are sap-feeding insects in the leafhopper family (Newman et al. 2004; Purcell et al. 1979); and transmission of Leishmania depends on blockage of the anterior midgut of the sandfly vector by parasites embedded in a polysaccharide-containing secretory gel. (Rogers et al. 2002; Stierhof et al. 1999). The body of evidence that has accumulated in support of the biofilm model of plague transmission is presented in the following sections.
3 Genetic and Molecular Mechanisms of Yersinia Biofilm ECM Synthesis
3.1 The Y. pestis Pigmentation Phenotype, Hms Locus, and Biofilm ECM
One of the first of the many temperature-dependent phenotypes described for Y. pestis was the formation of darkly pigmented colonies after growth at 28°C or less, but not after growth at 37°C, on agar plates containing hemin or the structurally analogous dye Congo red (Jackson and Burrows 1956; Surgalla and Beesley 1969). This phenotype was termed pigmentation, and is due to avid binding of the chromogens to the outer surface of the bacterial cells (Perry et al. 1993). It was also noted that the pigmentation phenotype correlated with cohesive colonies that came off the agar surface in densely packed masses and with pellicle formation on the surface of glass culture vessels (Jackson and Burrows 1956; Surgalla and Beesley 1969). In retrospect, these findings were the first evidence of Yersinia biofilm formation- pellicle growth and Congo red binding to a polysaccharide ECM are now recognized as typical traits of many bacterial biofilms (Heilmann and Götz 1998; Weiner et al. 1999).
Perry et al. (1990) identified a Y. pestis chromosomal region required for pigmentation, termed the hemin storage (hms) locus; and a four-gene operon in this locus, hmsHFRS, was later implicated (Lillard et al. 1997; Pendrak and Perry 1993) (Table 1). Amino acid sequence and domain similarities between the Hms proteins and the Pga and lea proteins of E. coli and staphylococci that synthesize a poly-β-1,6-N-acetyl-d-glucosamine ECM required for biofilm formation suggested a link between pigmentation and biofilm phenotypes; and complementation of non-pigmented Y. pestis hmsR and hmsS mutants with the respective E. coli pga homologs restores the pigmentation phenotype (Darby et al. 2002; Heilmann et al. 1996; Jones et al. 1999; Lillard et al. 1997; Mack et al. 1996; Wang et al. 2004).
Table 1.
Yersinia hms gene products and their predicted functions
| Gene | Location of proteina | Identified protein domainsb | Predicted functionb | Similar genes in other bacteriab |
|---|---|---|---|---|
| Hms locus: | ||||
| hmsH | OM | - | ECM synthesis | pgaA (E. coli) |
| hmsF | OM | polysaccharide deacetylase, COG1649 | ECM synthesis | pgaB (E. coli) icaB (Staph) |
| hmsR | IM | glycosyl transferase | ECM synthesis | pgaC (E. coli) icaA (Staph) |
| hmsS | IM | - | ECM synthesis | pgaD (E. coli) |
| Unlinked gene: | ||||
| hmsT | IM | GGDEF | Diguanylate cyclase | |
| hmsP | IM | GGDEF, EAL | Cyclic-di-GMP phosphodiesterase | yhjK (E. coli) |
OM, outer membrane; IM, inner membrane
See text for references
The ECM of S. epidermidis and Staphylococcus aureus, called PIA (polysaccharide intercellular adhesin), enables biofilm formation on catheters and other indwelling medical devices, making these bacteria one of the most common causes of nosocomial infections (Vadyvaloo and Otto 2005). A model for PIA biosynthesis by the ica (intercellular adhesin) gene products has been developed, based on several genetic and biochemical studies (reviewed in Götz 2002). The glycosyl transferase activity of IcaA, in conjunction with IcaD, first forms intracellular oligomers of 10–20 β-1,6-linked N-acetylglucosamine residues (Gerke et al. 1998). The oligomers are then further polymerized and transported through the cell membrane via IcaC, where the polymer is partially deacylated by the polysaccharide deacetylase activity of the cell-surface protein IcaB (Vuong et al. 2004a). Given the membrane localization and similar domains of the Ica, Pga, and Hms gene products, ECM biosynthesis in E. coli and Yersinia species probably proceeds by an analogous mechanism. Notably, all four of the Y. pestis hmsHFRS gene products are required for pigmentation and biofilm phenotype, and site-directed mutagenesis of the periplasmic, deacetylase, and glycosyl transferase domains of HmsH, F, and R, respectively, severely diminished Congo red binding and in vitro biofilm formation (Forman et al. 2006).
The ECM structures of staphylococcal and E. coli biofilms are biochemically similar, if not identical; both are composed of poly-β-1,6-N-acetylglucosamine (Mack et al. 1996; Wang et al. 2004). The Yersinia ECM structure has not been determined yet. However, Hms-dependent extracellular material has been observed by electron microscopy techniques specifically designed to preserve relatively labile polysaccharide (Fig. 2), and Y. pestis cells expressing the hms genes cross-react with anti-PIA antibody (Erickson and Hinnebusch, unpublished data). Furthermore, dispersin B, a β-hexosaminidase from Actinobacillus actinomycetem-comitans that disrupts biofilms in that organism (Kaplan et al. 2003), degrades poly-β-1,6-N-acetylglucosamine and prevents biofilm formation of S. epider-midis, E. coli, Y. pestis, and other bacteria containing Ica, Pga, and Hms homologs (Itoh et al. 2005). Whereas dispersin B treatment also dispersed preformed E. coli and S. epidermidis biofilms, however, preformed Y. pestis biofilms were unaffected (Itoh et al. 2005). This suggests that the Y. pestis ECM is inaccessible to the enzyme or is further modified during maturation of the biofilm, or that other non-ECM components act to stabilize the Y. pestis biofilm.
Fig. 2.
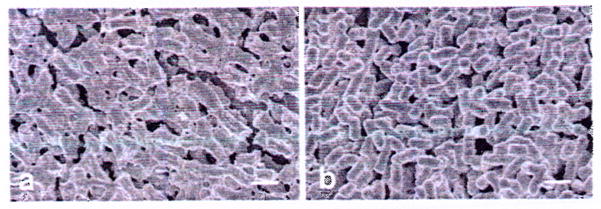
ECM surrounding Hms+ (a) but not Hms− (b) Y. pestis grown on agar plates at 21 °C. Scale bar, 1 μm
Thus, although it remains to be verified biochemically, there is considerable evidence to support the hypothesis that the Yersinia HmsHFRS proteins synthesize a polysaccharide ECM that is structurally related to the Pga- and Ica-dependent ECMs of E. coli and staphylococcal biofilms. As in other bacteria, the Hms-dependent ECM is required for Congo red binding (pigmentation), the binding of other ligands with polysaccharide affinity such as calcofluor (Kirillina et al. 2004) and certain lectins (Tan and Darby 2004), as well as for Yersinia biofilm formation (Darby et al. 2002; Forman et al. 2006; Jarrett et al. 2004; Joshua et al. 2003).
3.2 Role of the Hms Proteins in Producing In Vitro and In Vivo Biofilms
An essential role for the hmsHFRS genes in Yersinia biofilm formation has been amply demonstrated using a variety of experimental systems. The hms locus is required for Y. pestis and Y. pseudotuberculosis biofilm growth in 96-well polystyrene microtiter plates and on the surface of glass continuous-flow (flowcell) chambers (Fig. 3) (Erickson et al. 2006; Forman et al. 2006; Jarrett et al. 2004; Kirillina et al. 2004). Key evidence for the biofilm model of plague transmission came from in vivo studies using the rat flea X. cheopis as an infection model, in which the Y. pestis hms genes were shown to be required to produce a transmissible infection in the flea proventriculus (Hinnebusch et al. 1996; Jarrett et al. 2004). The first explicit statement of the plague biofilm transmission hypothesis, however, was based on the ability of Y. pestis and Y. pseudotuberculosis to form Hms-dependent biofilm on the surface of Caenorhabditis elegans nematodes, a model that may incorporate aspects of both in vitro and in vivo biofilm formation (Darby et al. 2002; Joshua et al. 2003). In this model, Yersinia biofilm aggregates on the mouthparts and blocks the feeding of the nematodes as they crawl across a preformed lawn of ECM-producing bacteria. The in vitro and C. elegans models have proven to be useful surrogates to identify genes in addition to hms that are important for flea-borne transmission (Darby et al. 2005); however, in vitro, C. elegans and flea biofilm phenotypes do not always correlate (Erickson et al. 2006).
Fig. 3.
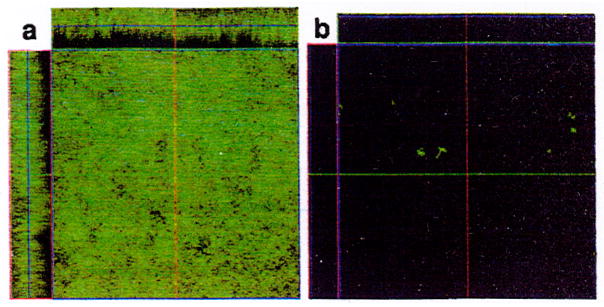
Biofilm produced on the glass surface of a flow cell after 48 h at 21°C by Hms+ (a) but not Hms− (b) Y. pestis
3.3 Regulation of Hms-Dependent Biofilm Formation
Proximal regulation of Hms-dependent ECM synthesis in Y. pestis appears to depend primarily on intracellular levels of the bacterial second messenger cyclic-di-GMP. hmsT and hmsP, two recently identified chromosomal genes that are unlinked to the hmsHFRS locus and to each other, encode a GGDEF-domain protein with c-di-GMP synthesizing diguanylate cyclase activity and an EAL-domain protein with phosphodiesterase activity, respectively (Bobrov et al. 2005; Hare and McDonough 1999; Jones et al. 1999; Kirillina et al. 2004; Simm et al. 2005). HmsT and HmsP are hypothesized to control Hms-dependent ECM biosynthesis via their opposing c-di-GMP forming and degrading activities. In keeping with this regulatory scheme, copy number of hmsT is directly related to c-di-GMP levels and biofilm thickness in Y. pestis; conversely, deletion of hmsT or disruption of its GGDEF-encoding region result in poor biofilm formation (Kirillina et al. 2004; Simm et al. 2005). As also predicted by the model, elimination of HmsP or its EAL domain result in thicker biofilm (Kirillina et al. 2004). Accordingly, the Yersinia Hms system joins a list of systems in several other Gram-negative bacteria, including Salmonella, Gluconacetobacter, Rhizobium, Pseudomonas, and Vibrio, in which GGDEF- and EAL-protein control of c-di-GMP concentration is involved in the regulation of extracellular polysaccharide and biofilm ECM synthesis (D’Argenio and Miller 2004; Römling and Amikam 2006).
GGDEF and EAL domain proteins are two of the largest superfamilies in eubacteria, with multiple members present in most species (Römling and Amikam 2006). This redundancy suggests that c-di-GMP flux may be influenced by a complex composite of different GGDEF and EAL family member pairs that come into play under different environmental conditions. For example, different GGDEF-domain proteins have been demonstrated to participate in a hierarchical fashion in control of the rdar biofilm phenotype in Salmonella (Kader et al. 2006). In addition to hmsT and hmsP, Y. pestis contains seven additional genes that are predicted to encode GGDEF- and EAL-domain proteins (Parkhill et al. 2001) (Table 2). Their role, if any, in Yersinia Hms or biofilm regulation is unknown. Interestingly, however, one (YPO3988) is highly similar to E. coli yhjH, which inhibits biofilm formation in Pseudomonas putida (Gjermansen et al. 2006). Another (YPO0998) is highly similar to yhdA, which regulates the Csr small regulatory RNA system known to control the E. coli Pga-dependent biofilm ECM (Jackson et al. 2002; Wang et al. 2005).
Table 2.
Predicted GGDEF- and EAL-domain proteins in Yersinia pestis CO92
| GGDEF domain | EAL domain | Both GGDEF and EAL domains | |||
|---|---|---|---|---|---|
| Gene no. | Gene name (homolog) | Gene no. | Gene name (homolog) | Gene no. | Gene name (homolog) |
| YPO0425 | hmsT | YPO1274a | rtn | YPO3996a | hmsP (yhjK) |
| YPO0449a | - | YPO2779 | - | YPO3664a | (yhdA) |
| YPO1752b | - | YPO3988 | (yhjH) | YPO0998a | - |
Predicted membrane protein
Pseudogene in CO92 but intact in Y. pestis KIM
Many questions remain about the regulation of the Hms system in Yersinia. The low-temperature dependence of the Hms phenotype known since the original description (Jackson and Burrows 1956) can be attributed to degradation of HmsT, H, and R at 37°C (Perry et al. 2004), but details of this posttranslational regulation are unknown. The molecular mechanism of c-di-GMP enhancement of biosynthetic Hms protein activity has not been determined. The hmsT promoter region contains a binding site for the iron-response regulator protein Fur, suggesting a link between the Hms and low iron response systems that has yet to be fully explored (Jones et al. 1999; Staggs et al. 1994). Answers to these and other questions will provide important insight into the biology and evolution of the Yersinia. For example, strains of Y. pestis and Y. pseudotuberculosis that have identical hmsHFRS, hmsT, and hmsP genes differ in their biofilm phenotype in different in vitro conditions (Chain et al. 2004; Darby et al. 2002; Deng et al. 2002; Joshua et al. 2003; Parkhill et al. 2001). Of greatest biological significance, Y. pseudotuberculosis can infect the flea midgut but never forms a biofilm in that environment (Erickson et al. 2006). Conversely, Y. pseudotuberculosis typically forms thicker biofilms in the C. elegans model system than does Y. pestis (Tan and Darby 2004).
4 Other Factors Implicated in Yersinia Biofilm Formation
4.1 Bacterial Factors
The biofilm growth state is recognized as a complex developmental cycle, involving initial adherence, ECM production, maturation, and eventual dispersion (Stoodley et al. 2002). Initial attachment to a surface can rely on relatively weak and nonspecific physicochemical and electrostatic interactions that are influenced by characteristics of the bacterial outer membrane, the surrounding medium, and the substrate (Beloin et al. 2005). The ECM itself may also promote attachment.
Lipopolysaccharide (LPS), the major component of the Gram-negative outer membrane, has been shown to affect Yersinia biofilm. Mutation of waaA, yrbH, and gmhA, which encode enzymes required for the addition of the LPS inner core sugars Kdo (3-deoxy-D-manno-octulosonic acid) and heptose to lipid A, all result in decreased biofilm in Y. pestis (Darby et al. 2005; Tan and Darby 2004, 2006). Of these, only the heptose-less Y. pestis gmhA mutant has been evaluated in the flea, where it was severely deficient in ability to produce proventricular blockage even though it colonized the flea gut normally (Darby et al. 2005). Deficient biofilm formation of the Y. pestis waa and yrbH mutant strains (which produce LPS consisting solely of lipid A) on C. elegans may be related to their reduced growth rate (Tan and Darby 2005, 2006); alternatively, LPS inner core alteration could conceivably affect initial attachment, or export or stability of the ECM on the outer surface. An unidentified separate activity of YrbH, in addition to its known role in KDO biosynthesis, was also implicated in biofilm formation (Tan and Darby 2006). Y pestis, unlike Y. pseudotuberculosis, produces a rough form of LPS lacking O-polysaccharide at temperatures at which the Hms-dependent ECM is made. Loss of O-polysaccharide does not markedly affect the in vitro biofilm forming ability of Y. pseudotuberculosis, however (Erickson et al. 2006).
In many bacteria, specific cell-surface adhesins such as certain types of fimbriae or outer membrane proteins are important for initial attachment to the substrate on which a biofilm forms (reviewed in Beloin et al. 2005). The Y. pestis genome contains at least ten genetic loci predicted to encode surface fimbriae or adhesins (Parkhill et al. 2001). Two of them, responsible for the F1 protein fibrillar capsule and the pH6 antigen, have known roles in the pathogenesis of plague (Brubaker 1991; Lindler et al. 1990), but functions have not been attributed to the others.
The complex bacterial physiology involved in biofilm development and maturation is indicated by the number of environmental sensing and gene regulatory systems that have been implicated in these processes (reviewed in Beloin et al. 2005; Stoodley et al. 2002). Among them are specific bacterial two-component regulatory systems as well as global regulators of central metabolism. The role of bacterial quorum sensing, a means of intercellular signaling within a community, in biofilm maturation has attracted particular attention (Davies et al. 1998; Parsek and Greenberg 2005; Vuong et al. 2003; Zhu and Mekalanos 2003). Interestingly, a connection between quorum sensing and c-di-GMP regulatory systems has been proposed (Camilli and Bassler 2006). The environmental sensing and subsequent redirection of gene expression pathways that lead to biofilm formation in Yersinia remain to be discovered. We have found that a Y pestis strain deleted of all three known quorum-sensing systems is able to form a normal biofilm in the flea (Jarrett et al. 2004). Nevertheless, it is possible that Y. pestis quorum-sensing signaling is required for the final step in the biofilm developmental cycle, dispersal of individual cells from the biofilm, which might affect regurgitative transmission from a pro ventricular biofilm.
Patel et al. recently discovered that the polyamines spermidine and putrescine are important for ECM production and biofilm formation in Y. pestis, and suggested possible roles for these cationic molecules in Hms-related signaling pathways or as biosynthetic intermediates or components of ECM (Patel et al. 2006). In Agrobacterium tumefasciens and Vibrio cholerae, homologs of polyamine transport (Pot) membrane proteins are important for surface attachment (Karatan et al. 2005; Matthysse et al. 1996). Although the Y. pestis polyamine transport genes are highly induced in the flea, mutational loss of this system did not affect biofilm formation in the flea, ruling out an essential role for these proteins in adherence, but the polyamine biosynthesis genes of this pot mutant were intact (Vadyvaloo et al. 2007).
4.2 Environmental Factors
Besides the many bacterial factors that have been implicated, the biophysical properties of the substrate and surrounding medium can also greatly influence bacterial biofilm formation (Heydorn et al. 2000; Prouty and Gunn 2003). For example, C. elegans mutants with an altered surface cuticle do not accumulate Yersinia biofilm (Höflich et al. 2004; Joshua et al. 2003). Thus, initiation and development of the biofilm growth phenotype can be multifactorial and environment-specific. Accordingly, for reasons that have yet to be defined but that are likely to be complex, the biofilm growth phenotypes of Y. pestis and Y pseudotuberculosis in different in vitro and in vivo environments do not always correlate, and Y. pseudotuberculosis never forms biofilm in the digestive tract of X. cheopis fleas (Erickson et al. 2006).
5 Characteristics of the Y. pestis Transmissible Biofilm Produced in the Flea
During septicemic plague, Y pestis occurs in peripheral blood as individual cells. Immediately after being ingested into the midgut of an X. cheopis flea, however, it begins to form multicellular aggregates with evidence of an ECM. Usually, these aggregates are free-floating in the lumen of the midgut, unattached to a substrate. Y. pestis does not adhere to or invade the midgut epithelium, putting it at risk of being eliminated in the feces because fleas feed and defecate frequently. In fact, up to half of X. cheopis rapidly clear themselves of infection in this way even after feeding on blood containing more than 108 Y. pestis per milliliter (Engelthaler et al. 2000; Lorange et al. 2005; Pollitzer 1954). Thus, the formation of multicellular aggregates that are too large to pass in the feces may be important to produce a stable infection. For efficient transmission, however, adherence and consolidation of a biofilm to the spines in the proventriculus is required. This usually occurs secondary to the formation of midgut aggregates, although biofilm can occur simultaneously at both sites (Pollitzer 1954). Complete blockage does not usually occur until at least 1–2 weeks after an infectious blood meal, and proventricular colonization does not occur in all fleas with stable midgut infections.
Hms-negative Y. pestis strains are able to stably infect the flea midgut in the form of multicellular aggregates, but never colonize the proventriculus (Hinnebusch et al. 1996; Jarrett et al. 2004). This indicates that other bacterial factors besides the ECM are involved in the autoaggregative growth phenotype in the midgut, but that the ECM is essential for attachment and development of a biofilm on the surface of the proventricular spines. The outer coating of the spines is similar or identical to cuticle, the hard, hydrophobic, acellular material that makes up the flea exoskeleton. The Hms-dependent ECM of a Y. pestis biofilm may promote aggregation on that surface, in the same way that the biochemically similar Ica-dependent ECM of staphylococcal biofilms promotes aggregation to the hydrophobic, abiotic surface of catheters and other indwelling medical devices (Rupp et al. 1999).
In vivo biofilms of Y. pestis in the flea differ in some respects from in vitro biofilms. They are a dark brown color, most likely due to adsorption of hemin derived from red blood cell digestion in the flea midgut; in other words, they exhibit the pigmentation phenotype (Jarrett et al. 2004). The ECM of Y. pestis biofilm in the flea gut also appears to be heterogenous and more complex than is observed in vitro. Notably, it has a viscous appearance (Fig. 1) and, unlike in vitro Y. pestis biofilm, avidly takes up lipid stains (Jarrett et al. 2004). This suggests that the in vivo ECM consists not only of bacterial exopolysaccharide, but also exogenous material incorporated from the flea gut environment, such as lipid derived from the flea blood meal.
6 Implications of the Biofilm Transmission Phenotype at the Flea-Y. pestis-Host Interface
The ability to form an in vivo biofilm is now recognized as an integral part of the infection and disease process of many microbial pathogens (reviewed in Costerton et al. 1999; Parsek and Singh 2003). For Y. pestis, an in vivo biofilm type of infection occurs in the invertebrate vector, where it is important for efficient transmission, and not in the vertebrate host. The Y. pestis Hms-dependent ECM is not synthesized at mammalian body temperature and is not required for bubonic plague pathogenesis (Lillard et al. 1999). However, Y pestis enters the mammal directly from an infectious biofilm in the flea, and this has implications for the initial interaction with the mammalian innate immune system at the dermal flea bite site.
The ECM that surrounds bacteria in a biofilm has been shown in some cases to protect against phagocytosis and/or intracellular killing, cationic antimicrobial peptides, and antibiotics (Costerton et al. 1999; Jesaitis et al. 2003; Vuong et al. 2004b). It is likely that the infectious inoculum delivered by a flea consists of small clumps of Y. pestis that are associated with a complex ECM (Fig. 4) (Jarrett et al. 2004; Lorange et al. 2005). The Y. pestis virulence factors known to protect against innate immunity, such as the antiphagocytic F1 capsule and the Type III secretion system that delivers cytotoxic Yop proteins into innate immune cells, are not synthesized in the flea. Thus, it is possible that the ECM may protect Y. pestis during the initial encounter with innate immune effector cells immediately after transmission, before the synthesis of specific virulence factors is induced. In addition, if the biofilm ECM made in the flea gut contains lipids derived from mammalian blood, these self components may mask bacterial antigens and avert initial recognition by the immune system. In an initial experiment, we found that both Hms+ and Hms− Y. pestis recovered from flea guts were significantly more resistant to uptake by human polymorphonuclear leukocytes than the same bacteria grown in culture (Jarrett et al. 2004).
Fig. 4.
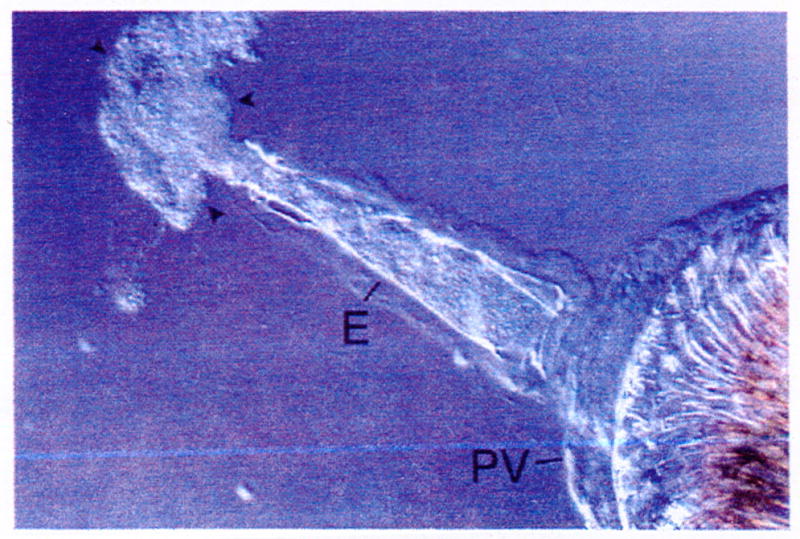
Y. pestis biofilm (arrowheads) expelled through the esophagus (E) by application of a cover slip to the digestive tract dissected from a blocked X. cheopis flea. The array of spines that line the interior of the proventriculus (PV) is visible
Y. pestis containing conjugative plasmids that encode high-level resistance to multiple antibiotics have been isolated from human patients in Madagascar (Galimand et al. 1997; Guiyoule et al. 2001). The source of these newly acquired plasmids is unknown, but conjugative plasmid transfer among bacteria can occur at high frequency within a dense, multicellular biofilm (Hausner and Wuertz 1999; Molin and Tolker-Nielsen 2003). High-frequency conjugative transfer to Y pestis within a mixed biofilm in the flea gut has been demonstrated, suggesting that gene transfer from commensal microbial flora in the flea gut that become incorporated into a Y. pestis biofilm may be a source of multiple-drug resistant strains (Hinnebusch et al. 2002a).
7 Summary
The genetic changes in Y. pestis since it diverged from Y. pseudotuberculosis that enabled biofilm development and growth in the digestive tract of the flea was clearly of fundamental importance in the evolution of the new arthropod-borne route of transmission. Because the hms genes and the ability to produce environmental biofilm appear to predate this recent divergence, probably relatively few changes were needed; perhaps involving only fine-tuning of environmental sensing and regulatory pathways that induce the Hms system (Erickson et al. 2006). Bacterial biofilm development is multifactorial, however, and much remains to be learned about it in the genus Yersinia. Adaptation of biofilm-forming potential to the flea gut environment, along with other discrete changes such as acquisition of two new plasmids, made possible the abrupt (in evolutionary terms) change to fleaborne transmission. Application of the biofilm developmental state to enable arthropod-borne transmission represents a novel function that illustrates the utility of multicellular, adherent growth.
Acknowledgments
This work was supported by the Intramural Research Program of the NIH, NIAID.
References
- Achtman M, Zurth K, Morelli G, Torrea G, Guiyoule A, Carniel E. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc Natl Acad Sci U S A. 1999;96:14043–14048. doi: 10.1073/pnas.96.24.14043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achtman M, Morelli G, Zhu P, Wirth T, Diehl I, Kusecek B, Vogler AJ, Wagner DM, Allender CJ, Easterday WR, Chenal-Francisque V, Worsham P, Thomson NR, Parkhill J, Lindler LE, Carniel E, Keim P. Microevolution and history of the plague bacillus, Yersinia pestis. Proc Natl Acad Sci U S A. 2004;101:17837–17842. doi: 10.1073/pnas.0408026101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacot AW, Martin CJ. Observations on the mechanism of the transmission of plague by fleas. J Hygiene (Plague Suppl 3) 1914;13:423–439. [PMC free article] [PubMed] [Google Scholar]
- Bacot AW. Further notes on the mechanism of the transmission of plague by fleas. J Hygiene (Plague Suppl 4) 1915;14:774–776. [PMC free article] [PubMed] [Google Scholar]
- Beesley ED, Brubaker RR, Janssen WA, Surgalla MJ. Pesticins. III. Expression of coagulase and mechanism of fibrinolysis. J Bacteriol. 1967;94:19–26. doi: 10.1128/jb.94.1.19-26.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloin C, Da Re S, Ghigo J-M. Colonization of abiotic surfaces. In: Böck A, Curtis R III, Kaper JB, Neidhardt FC, Nyström K, Rudd E, Squires CL, editors. EcoSal - Escherichia coli and Salmonella: cellular and molecular biology. ASM Press; Washington DC: 2005. [Google Scholar]
- Bercovier H, Mollaret HH, Alonso JM, Brault J, Fanning GR, Steigerwalt A, Brenner DJ. Intra- and interspecies relatedness of Yersinia pestis by DNA hybridization and its relationship to Yersinia pseudotuberculosis. Curr Microbiol. 1980;4:225–229. [Google Scholar]
- Bobrov AG, Kirillina O, Perry RD. The phosphodiesterase activity of the HmsP EAL domain is required for negative regulation of biofilm formation in Yersinia pestis. FEMS Microbiol Lett. 2005;247:123–130. doi: 10.1016/j.femsle.2005.04.036. [DOI] [PubMed] [Google Scholar]
- Bottone EJ, Bercovier H, Mollaret HH. Genus XLI. Yersinia. In: Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s manual of systematic bacteriology. 2. Springer; Berlin New York Heidelberg: 2005. [Google Scholar]
- Brubaker RR, Beesley ED, Surgalla MJ. Pasteurella pestis: role of pesticin I and iron in experimental plague. Science. 1965;149:422–424. doi: 10.1126/science.149.3682.422. [DOI] [PubMed] [Google Scholar]
- Brubaker RR. Factors promoting acute and chronic diseases caused by yersiniae. Clin Microbiol Rev. 1991;4:309–324. doi: 10.1128/cmr.4.3.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs AL. Sylvatic plague studies. The vector efficiency of nine species of fleas compared with Xenopsylla cheopis. J Hygiene. 1947;45:371–396. doi: 10.1017/s0022172400014042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilli A, Bassler BL. Bacterial small-molecule signaling pathways. Science. 2006;311:1113–1116. doi: 10.1126/science.1121357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carniel E. Evolution of pathogenic Yersinia, some lights in the dark. In: Skurnik M, Bengoechea JA, Granfors K, editors. The genus Yersinia: entering the functional genomic era. Kluwer Academic; New York: 2003. pp. 3–11. [Google Scholar]
- Cavanaugh DC. Specific effect of temperature upon transmission of the plague bacillus by the oriental rat flea, Xenopsylla cheopis. Am J Trop Med Hyg. 1971;20:264–273. doi: 10.4269/ajtmh.1971.20.264. [DOI] [PubMed] [Google Scholar]
- Cavanaugh DC, Marshall JD. The influence of climate on the seasonal prevalence of plague in the Republic of Vietnam. J Wildl Dis. 1972;8:85–94. doi: 10.7589/0090-3558-8.1.85. [DOI] [PubMed] [Google Scholar]
- Chain PSG, Carniel E, Larimer FW, Lamerdin J, Stoutland PO, Regala WM, Georgescu AM, Vergez LM, Land ML, Motin VL, Brubaker RR, Fowler J, Hinnebusch J, Marceau M, Medigue C, Simonet M, Chenal-Francisque V, Souza B, Dacheaux D, Elliot JM, Derbise A, Hauser LJ, Garcia E. Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis. Proc Natl Acad Sci USA. 2004;101:13826–13831. doi: 10.1073/pnas.0404012101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- D’Argenio DA, Miller SI. Cyclic di-GMP as a bacterial second messenger. Microbiology. 2004;150:2497–2502. doi: 10.1099/mic.0.27099-0. [DOI] [PubMed] [Google Scholar]
- Darby C, Hsu JW, Ghori N, Falkow S. Caenorhabditis elegans: plague bacteria biofilm blocks food intake. Nature. 2002;417:243–244. doi: 10.1038/417243a. [DOI] [PubMed] [Google Scholar]
- Darby C, Ananth SL, Tan L, Hinnebusch BJ. Identification of gmhA, a Yersinia pestis gene required for flea blockage, by using a Caenorhabditis elegans biofilm system. Infect Immun. 2005;73:7236–7242. doi: 10.1128/IAI.73.11.7236-7242.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies DG, Parsek MR, Pearson JP, Iglewski BH, Costerton JW, Greenberg EP. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science. 1998;280:295–298. doi: 10.1126/science.280.5361.295. [DOI] [PubMed] [Google Scholar]
- Deng W, Burland V, Plunkett G, Boutin A, Mayhew GF, Liss P, Perna NT, Rose DJ, Mau B, Zhou S, Schwartz DC, Fetherston JD, Lindler LE, Brubaker RR, Plano GV, Straley SC, McDonough KA, Nilles ML, Matson JS, Blattner FR, Perry RD. Genome sequence of Yersinia pestis KIM. J Bacteriol. 2002;184:4601–4611. doi: 10.1128/JB.184.16.4601-4611.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen RJ, Bearden SW, Wilder AP, Montenieri JA, Antolin MF, Gage KL. Early-phase transmission of Yersinia pestis by unblocked fleas as a mechanism explaining rapidly spreading plague epizootics. Proc Natl Acad Sci U S A. 2006;103:15380–15385. doi: 10.1073/pnas.0606831103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelthaler DM, Hinnebusch BJ, Rittner CM, Gage KL. Quantitative competitive PCR as a technique for exploring flea-Yersina pestis dynamics. Am J Trop Med Hyg. 2000;62:552–560. doi: 10.4269/ajtmh.2000.62.552. [DOI] [PubMed] [Google Scholar]
- Erickson DL, Jarrett CO, Wren BW, Hinnebusch BJ. Serotype differences and lack of bio-film formation characterize Yersinia pseudotuberculosis infection of the Xenopsylla cheopis flea vector of Yersinia pestis. J Bacteriol. 2006;188:1113–1119. doi: 10.1128/JB.188.3.1113-1119.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferber DM, Brubaker RR. Plasmids in Yersinia pestis. Infect Immun. 1981;31:839–841. doi: 10.1128/iai.31.2.839-841.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman S, Bobrov AG, Kirillina O, Craig SK, Abney J, Fetherston JD, Perry RD. Identification of critical amino acid residues in the plague biofilm Hms proteins. Microbiology. 2006;152:3399–3410. doi: 10.1099/mic.0.29224-0. [DOI] [PubMed] [Google Scholar]
- Gage KL, Kosoy MY. Natural history of plague: perspectives from more than a century of research. Annu Rev Entomol. 2005;50:505–528. doi: 10.1146/annurev.ento.50.071803.130337. [DOI] [PubMed] [Google Scholar]
- Galimand M, Guiyoule A, Gerbaud G, Rasoamanana B, Chanteau S, Carniel E, Courvalin P. Multidrug resistance in Yersinia pestis mediated by a transferable plasmid. N Engl J Med. 1997;337:677–680. doi: 10.1056/NEJM199709043371004. [DOI] [PubMed] [Google Scholar]
- Gerke C, Kraft A, Sussmuth R, Schweitzer O, Götz F. Characterization of the N-acetylglucosaminyltransferase activity involved in the biosynthesis of the Staphylococcus epidermidis polysaccharide intercellular adhesin. J Biol Chem. 1998;273:18586–18593. doi: 10.1074/jbc.273.29.18586. [DOI] [PubMed] [Google Scholar]
- Gjermansen M, Ragas P, Tolker-Nielsen T. Proteins with GGDEF and EAL domains regulate Pseudomonas putida biofilm formation and dispersal. FEMS Microbiol Lett. 2006;265:215–224. doi: 10.1111/j.1574-6968.2006.00493.x. [DOI] [PubMed] [Google Scholar]
- Guiyoule A, Gerbaud G, Buchrieser C, Galimand M, Rahalison L, Chanteau S, Courvalin P, Carniel E. Transferable plasmid-mediated resistance to streptomycin in a clinical isolate of Yersinia pestis. Emerg Infect Dis. 2001;7:43–48. doi: 10.3201/eid0701.010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz F. Staphylococcus and biofilms. Mol Microbiol. 2002;43:1367–1378. doi: 10.1046/j.1365-2958.2002.02827.x. [DOI] [PubMed] [Google Scholar]
- Hare JM, McDonough KA. High-frequency RecA-dependent and -independent mechanisms of Congo red binding mutations in Yersinia pestis. J Bacteriol. 1999;181:4896–4904. doi: 10.1128/jb.181.16.4896-4904.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausner M, Wuertz S. High rates of conjugation in bacterial biofilms as determined by quantitative in situ analysis. Appl Environ Microbiol. 1999;65:3710–3713. doi: 10.1128/aem.65.8.3710-3713.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilmann C, Schweitzer O, Gerke C, Vanittanakom N, Mack D, Götz F. Molecular basis of intercellular adhesion in the biofilm-forming Staphylococcus epidermidis. Mol Microbiol. 1996;20:1083–1091. doi: 10.1111/j.1365-2958.1996.tb02548.x. [DOI] [PubMed] [Google Scholar]
- Heilmann C, Götz F. Further characterization of Staphylococcus epidermidis transposon mutants deficient in primary attachment or intercellular adhesion. Zentralbl Bakteriol. 1998;287:69–83. doi: 10.1016/s0934-8840(98)80149-7. [DOI] [PubMed] [Google Scholar]
- Heydorn A, Ersbøll BK, Hentzer M, Parsek MR, Givskov M, Molin S. Experimental reproducibility in flow-chamber biofilms. Microbiology. 2000;146:2409–2415. doi: 10.1099/00221287-146-10-2409. [DOI] [PubMed] [Google Scholar]
- Hinchliffe SJ, Isherwood KE, Stabler RA, Prentice MB, Rakin A, Nichols RA, Oyston PC, Hinds J, Titball RW, Wren BW. Application of DNA microarrays to study the evolutionary genomics of Yersinia pestis and Yersinia pseudotuberculosis. Genome Res. 2003;13:2018–2029. doi: 10.1101/gr.1507303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch BJ, Perry RD, Schwan TG. Role of the Yersinia pestis hemin storage (hms) locus in the transmission of plague by fleas. Science. 1996;273:367–370. doi: 10.1126/science.273.5273.367. [DOI] [PubMed] [Google Scholar]
- Hinnebusch BJ. Bubonic plague: a molecular genetic case history of the emergence of an infectious disease. J Mol Med. 1997;75:645–652. doi: 10.1007/s001090050148. [DOI] [PubMed] [Google Scholar]
- Hinnebusch BJ, Fischer ER, Schwan TG. Evaluation of the role of the Yersinia pestis plasminogen activator and other plasmid-encoded factors in temperature-dependent blockage of the flea. J Inf Dis. 1998;178:1406–1415. doi: 10.1086/314456. [DOI] [PubMed] [Google Scholar]
- Hinnebusch BJ, Rosso M-L, Schwan TG, Carniel E. High-frequency conjugative transfer of antibiotic resistance genes to Yersinia pestis in the flea midgut. Mol Microbiol. 2002a;46:349–354. doi: 10.1046/j.1365-2958.2002.03159.x. [DOI] [PubMed] [Google Scholar]
- Hinnebusch BJ, Rudolph AE, Cherepanov P, Dixon JE, Schwan TG, Forsberg Å. Role of Yersinia murine toxin in survival of Yersinia pestis in the midgut of the flea vector. Science. 2002b;296:733–735. doi: 10.1126/science.1069972. [DOI] [PubMed] [Google Scholar]
- Höflich J, Berninsone P, Göbel C, Gravato-Nobre MJ, Libby BJ, Darby C, Politz SM, Hodgkin J, Hirschberg CB, Baumeister R. Loss of srf-3-encoded nucleotide sugar transporter activity in Caenorhabditis elegans alters surface antigenicity and prevents bacterial adherence. J Biol Chem. 2004;279:30440–30448. doi: 10.1074/jbc.M402429200. [DOI] [PubMed] [Google Scholar]
- Ibrahim A, Goebel BM, Liesack W, Griffiths M, Stackebrandt E. The phylogeny of the genus Yersinia based on 16S rDNA sequences. FEMS Microbiol Lett. 1993;114:173–177. doi: 10.1111/j.1574-6968.1993.tb06569.x. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Wang X, Hinnebusch BJ, Preston JF, Romeo T. Depolymerization of β-1,6-N-acetyl-D-glucosamine disrupts the integrity of diverse bacterial biofilms. J Bacteriol. 2005;187:382–387. doi: 10.1128/JB.187.1.382-387.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DW, Suzuki K, Oakford L, Simecka JW, Hart ME, Romeo T. Biofilm formation and dispersal under the influence of the global regulator CsrA of Escherichia coli. J Bacteriol. 2002;184:290–301. doi: 10.1128/JB.184.1.290-301.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson S, Burrows TW. The pigmentation of Pasteurella pestis on a defined medium containing haemin. Br J Exp Pathol. 1956;37:570–576. [PMC free article] [PubMed] [Google Scholar]
- Jarrett CO, Deak E, Isherwood KE, Oyston PC, Fischer ER, Whitney AR, Kobayashi SD, DeLeo FR, Hinnebusch BJ. Transmission of Yersinia pestis from an infectious biofilm in the flea vector. J Inf Dis. 2004;190:783–792. doi: 10.1086/422695. [DOI] [PubMed] [Google Scholar]
- Jawetz E, Meyer KF. Studies on plague immunity in experimental animals. II. Some factors of the immunity mechanism in bubonic plague. J Immunol. 1944;49:15–29. [Google Scholar]
- Jesaitis AJ, Franklin MJ, Berglund D, Sasaki M, Lord CI, Bleazard JB, Duffy JE, Beyenal H, Lewandowski Z. Compromised host defense on Pseudomonas aeruginosa biofilms: characterization of neutrophil and biofilm interactions. J Immunol. 2003;171:4329–4339. doi: 10.4049/jimmunol.171.8.4329. [DOI] [PubMed] [Google Scholar]
- Jones HA, Lillard JW, Perry RD. HmsT, a protein essential for expression of the haemin storage (Hms+) phenotype of Yersinia pestis. Microbiology. 1999;145:2117–2128. doi: 10.1099/13500872-145-8-2117. [DOI] [PubMed] [Google Scholar]
- Joshua GWP, Karlyshev AV, Smith MP, Isherwood KE, Titball RW, Wren BW. A Caenorhabditis elegans model of Yersinia infection: biofilm formation on a biotic surface. Microbiology. 2003;149:3221–3229. doi: 10.1099/mic.0.26475-0. [DOI] [PubMed] [Google Scholar]
- Kader A, Simm R, Gerstel U, Morr M, Römling U. Hierarchical involvement of various GGDEF domain proteins in rdar morphotype development of Salmonella enterica serovar Typhimurium. Mol Microbiol. 2006;60:602–616. doi: 10.1111/j.1365-2958.2006.05123.x. [DOI] [PubMed] [Google Scholar]
- Kaplan JB, Ragunath C, Ramasubbu N, Fine DH. Detachment of Actinobacillus actinomycetemcomitans biofilm cells by an endogenous beta-hexosaminidase activity. J Bacteriol. 2003;185:4693–4698. doi: 10.1128/JB.185.16.4693-4698.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatan E, Duncan TR, Watnick PI. NspS, a predicted polyamine sensor, mediates activation of Vibrio cholerae biofilm formation by norspermidine. J Bacteriol. 2005;187:7434–7443. doi: 10.1128/JB.187.21.7434-7443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirillina O, Fetherston JD, Bobrov AG, Abney J, Perry RD. HmsP, a putative phosphodiesterase, and HmsT, a putative diguanylate cyclase, control Hms-dependent biofilm formation in Yersinia pestis. Mol Microbiol. 2004;54:75–88. doi: 10.1111/j.1365-2958.2004.04253.x. [DOI] [PubMed] [Google Scholar]
- Korhonen TK, Kukkonen M, Virkola R, Lang H, Suomalainen M, Kyllönen P, Lähteenmäki K. The plasminogen activator Pla of Yersinia pestis: localized proteolysis and systemic spread. In: Carniel E, Hinnebusch BJ, editors. Yersinia Molecular and Cellular Biology. Horizon Bioscience; Norfolk, UK: 2004. pp. 349–362. [Google Scholar]
- Lillard JW, Fetherston JD, Pedersen L, Pendrak ML, Perry RD. Sequence and genetic analysis of the hemin storage (hms) system of Yersinia pestis. Gene. 1997;193:13–21. doi: 10.1016/s0378-1119(97)00071-1. [DOI] [PubMed] [Google Scholar]
- Lillard JW, Bearden SW, Fetherston JD, Perry RD. The haemin storage (Hms+) phenotype of Yersinia pestis is not essential for the pathogenesis of bubonic plague in mammals. Microbiology. 1999;145:197–209. doi: 10.1099/13500872-145-1-197. [DOI] [PubMed] [Google Scholar]
- Lindler LE, Klempner MS, Straley SC. Yersinia pestis pH 6 antigen: genetic, biochemical, and virulence characterization of a protein involved in the pathogenesis of bubonic plague. Infect Immun. 1990;58:2569–2577. doi: 10.1128/iai.58.8.2569-2577.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorange EA, Race BL, Sebbane F, Hinnebusch BJ. Poor vector competence of fleas and the evolution of hypervirulence in Yersinia pestis. J Inf Dis. 2005;191:1907–1912. doi: 10.1086/429931. [DOI] [PubMed] [Google Scholar]
- Mack D, Fischer W, Krokotsch A, Leopold K, Hartmann R, Egge H, Laufs R. The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear beta-l,6-linked glucosaminoglycan: purification and structural analysis. J Bacteriol. 1996;178:175–183. doi: 10.1128/jb.178.1.175-183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthysse AG, Yarnall HA, Young N. Requirement for genes with homology to ABC transport systems for attachment and virulence of Agrobacterium tumefaciens. J Bacteriol. 1996;178:5302–5308. doi: 10.1128/jb.178.17.5302-5308.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough KA, Falkow S. A Yersinia pestis-specific DNA fragment encodes temperature-dependent coagulase and fibrinolysin-associated phenotypes. Mol Microbiol. 1989;3:767–775. doi: 10.1111/j.1365-2958.1989.tb00225.x. [DOI] [PubMed] [Google Scholar]
- McDonough KA, Barnes AM, Quan TJ, Montenieri J, Falkow S. Mutation in the pla gene of Yersinia pestis alters the course of the plague bacillus-flea (Siphonaptera: Ceratophyllidae) interaction. J Med Entomol. 1993;30:772–780. doi: 10.1093/jmedent/30.4.772. [DOI] [PubMed] [Google Scholar]
- Molin S, Tolker-Nielsen T. Gene transfer occurs with enhanced efficiency in biofilms and induces enhanced stabilisation of the biofilm structure. Curr Opin Biotechnol. 2003;14:255–261. doi: 10.1016/s0958-1669(03)00036-3. [DOI] [PubMed] [Google Scholar]
- Newman KL, Almeida RP, Purcell AH, Lindow SE. Cell-cell signaling controls Xylella fastidiosa interactions with both insects and plants. Proc Natl Acad Sci U S A. 2004;101:1737–1742. doi: 10.1073/pnas.0308399100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhill J, Wren BW, Thomson NR, Titball RW, Holden MTG, Prentice MB, Sebhaihia M, James KD, Churcher C, Mungall KL, Baker S, Basham D, Bentley SD, Brooks K, Cerdeño-Tárraga AM, Chillingworth T, Cronin A, Davies RM, Davis P, Dougan G, Feltwell T, Hamlin N, Holroyd S, Jagels K, Karlyshev AV, Leather S, Moule S, Oyston PCF, Quail M, Rutherford K, Simmonds M, Skelton J, Stevens K, Whitehead S, Barrell BG. Genome sequence of Yersinia pestis, the causative agent of plague. Nature. 2001;413:523–527. doi: 10.1038/35097083. [DOI] [PubMed] [Google Scholar]
- Parsek MR, Singh PK. Bacterial biofilms: an emerging link to disease pathogenesis. Ann Rev Micriobiol. 2003;57:677–701. doi: 10.1146/annurev.micro.57.030502.090720. [DOI] [PubMed] [Google Scholar]
- Parsek MR, Greenberg EP. Sociomicrobiology: the connections between quorum sensing and biofilms. Trends Microbiol. 2005;13:27–33. doi: 10.1016/j.tim.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Patel CN, Wortham BW, Lines JL, Fetherston JD, Perry RD, Oliveira MA. Polyamines are essential for the formation of plague biofilm. J Bacteriol. 2006;188:2355–2363. doi: 10.1128/JB.188.7.2355-2363.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendrak ML, Perry RD. Proteins essential for expression of the Hms+ phenotype of Yersinia pestis. Mol Microbiol. 1993;8:857–864. doi: 10.1111/j.1365-2958.1993.tb01632.x. [DOI] [PubMed] [Google Scholar]
- Perry RD, Pendrak ML, Schuetze P. Identification and cloning of a hemin storage locus involved in the pigmentation phenotype of Yersinia pestis. J Bacteriol. 1990;172:5929–5937. doi: 10.1128/jb.172.10.5929-5937.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RD, Lucier TS, Sikkema DJ, Brubaker RR. Storage reservoirs of hemin and inorganic iron in Yersinia pestis. Infect Immun. 1993;61:32–39. doi: 10.1128/iai.61.1.32-39.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RD, Fetherston JD. Yersinia pestis - etiologic agent of plague. Clin Microbiol Rev. 1997;10:35–66. doi: 10.1128/cmr.10.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RD, Bobrov AG, Kirillina O, Jones HA, Pedersen L, Abney J, Fetherston JD. Temperature regulation of the hemin storage (Hms+) phenotype of Yersinia pestis is posttranscriptional. J Bacteriol. 2004;186:1638–1647. doi: 10.1128/JB.186.6.1638-1647.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollitzer R. Plague. World Health Organization; Geneva: 1954. [Google Scholar]
- Prouty AM, Gunn JS. Comparative analysis of Salmonella enterica serovar Typhimurium biofilm formation on gallstones and on glass. Infect Immun. 2003;71:7154–7158. doi: 10.1128/IAI.71.12.7154-7158.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell AH, Finlay AH, McLean DL. Pierce’s disease bacterium: mechanism of transmission by leafhopper vectors. Science. 1979;206:839–841. doi: 10.1126/science.206.4420.839. [DOI] [PubMed] [Google Scholar]
- Rogers ME, Chance ML, Bates PA. The role of promastigote secretory gel in the origin and transmission of the infective stage of Leishmania mexicana by the sandfly Lutzomyia longipalpis. Parasitology. 2002;124:495–507. doi: 10.1017/s0031182002001439. [DOI] [PubMed] [Google Scholar]
- Rupp ME, Ulphani JS, Fey PD, Bartscht K, Mack D. Characterization of the importance of polysaccharide intercellular adhesin/hemagglutinin of Staphylococcus epidermidis in the pathogenesis of biomaterial-based infection in a mouse foreign body infection model. Infect Immun. 1999;67:2627–2632. doi: 10.1128/iai.67.5.2627-2632.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Römling U, Amikam D. Cyclic di-GMP as a second messenger. Curr Opin Microbiol. 2006;9:218–228. doi: 10.1016/j.mib.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Sebbane F, Jarrett CO, Gardner D, Long D, Hinnebusch BJ. Role of the Yersinia pestis plasminogen activator in the incidence of distinct septicemic and bubonic forms of flea-borne plague. Proc Natl Acad Sci U S A. 2006;103:5526–5530. doi: 10.1073/pnas.0509544103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simm R, Fetherston JD, Kader A, Römling U, Perry RD. Phenotypic convergence mediated by GGDEF-domain-containing proteins. J Bacteriol. 2005;187:6816–6823. doi: 10.1128/JB.187.19.6816-6823.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodeinde OA, Goguen JD. Genetic analysis of the 9.5-kilobase virulence plasmid of Yersinia pestis. Infect Immun. 1988;56:2743–2748. doi: 10.1128/iai.56.10.2743-2748.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodeinde OA, Subrahmanyam YV, Stark K, Quan T, Bao Y, Goguen JD. A surface protease and the invasive character of plague. Science. 1992;258:1004–1007. doi: 10.1126/science.1439793. [DOI] [PubMed] [Google Scholar]
- Staggs TM, Fetherston JD, Perry RD. Pleiotropic effects of a Yersinia pestis fur mutation. J Bacteriol. 1994;176:7614–7624. doi: 10.1128/jb.176.24.7614-7624.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stierhof YD, Bates PA, Jacobson RL, Rogers ME, Schlein Y, Handman E, Ilg T. Filamentous proteophosphoglycan secreted by Leishmania promastigotes forms gel-like three-dimensional networks that obstruct the digestive tract of infected sandfly vectors. Eur J Cell Biol. 1999;78:675–689. doi: 10.1016/S0171-9335(99)80036-3. [DOI] [PubMed] [Google Scholar]
- Stoodley P, Sauer K, Davies DG, Costerton JW. Biofilms as complex differentiated communities. Ann Rev Microbiol. 2002;56:187–209. doi: 10.1146/annurev.micro.56.012302.160705. [DOI] [PubMed] [Google Scholar]
- Surgalla MJ, Beesley ED. Congo red agar plating medium for detecting pigmentation in Pasteurella pestis. Appl Microbiol. 1969;18:834–837. doi: 10.1128/am.18.5.834-837.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L, Darby C. A movable surface: formation of Yersinia sp. biofilms on motile Caenorhabditis elegans. J Bacteriol. 2004;186:5087–5592. doi: 10.1128/JB.186.15.5087-5092.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L, Darby C. Yersinia. pestis is viable with endotoxin composed of only lipid A. J Bacteriol. 2005;187:6599–6600. doi: 10.1128/JB.187.18.6599-6600.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L, Darby C. Yersinia pestis YrbH is a multifunctional protein required for both 3-deoxy-D-manno-oct-2-ulosonic acid biosynthesis and biofilm formation. Mol Microbiol. 2006;61:861–870. doi: 10.1111/j.1365-2958.2006.05265.x. [DOI] [PubMed] [Google Scholar]
- Vadyvaloo V, Otto M. Molecular genetics of Staphylococcus epidermidis biofilms on indwelling medical devices. Int J Art Org. 2005;28:1069–1078. doi: 10.1177/039139880502801104. [DOI] [PubMed] [Google Scholar]
- Vadyvaloo V, Jarrett CO, Sturdevant DE, Sebbane F, Hinnebusch BJ. Analysis of Yersinia pestis gene expression in the flea vector. Adv Exp Med Biol. 2007;603:192–200. doi: 10.1007/978-0-387-72124-8_16. [DOI] [PubMed] [Google Scholar]
- Vuong C, Gerke C, Somerville GA, Fischer ER, Otto M. Quorum-sensing control of biofilm factors in Staphylococcus epidermidis. J Infect Dis. 2003;188:706–718. doi: 10.1086/377239. [DOI] [PubMed] [Google Scholar]
- Vuong C, Kocianova S, Voyich JM, Yao Y, Fischer ER, DeLeo FR, Otto M. A crucial role for exopolysaccharide modification in bacterial biofilm formation, immune evasion, and virulence. J Biol Chem. 2004a;279:54881–54886. doi: 10.1074/jbc.M411374200. [DOI] [PubMed] [Google Scholar]
- Vuong C, Voyich JM, Fischer ER, Braughton KR, Whitney AR, DeLeo FR, Otto M. Polysaccharide intercellular adhesin (PIA) protects Staphylococcus epidermidis against major components of the human innate immune system. Cell Microbiol. 2004b;6:269–275. doi: 10.1046/j.1462-5822.2004.00367.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Preston JF, Romeo T. The pgaABCD locus of Escherichia coli promotes the synthesis of a polysaccharide adhesin required for biofilm formation. J Bacteriol. 2004;186:2442–2449. doi: 10.1128/JB.186.9.2724-2734.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Dubey AK, Suzuki K, Baker CS, Babitzke P, Romeo T. CsrA post-transcriptionally represses pgaABCD, responsible for synthesis of a biofilm polysaccharide adhesin of Escherichia coli. Mol Microbiol. 2005;56:1648–1663. doi: 10.1111/j.1365-2958.2005.04648.x. [DOI] [PubMed] [Google Scholar]
- Webb CT, Brooks CP, Gage KL, Antolin MF. Classic flea-borne transmission does not drive plague epizootics in prairie dogs. Proc Natl Acad Sci U S A. 2006;103:6236–6241. doi: 10.1073/pnas.0510090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner R, Seagren E, Arnosti C, Quintero E. Bacterial survival in biofilms: probes for exopolysaccharide and its hydrolysis, and measurements of intra- and interphase mass fluxes. Methods Enzymol. 1999;310:403–426. doi: 10.1016/s0076-6879(99)10032-6. [DOI] [PubMed] [Google Scholar]
- Zhou D, Han Y, Song Y, Huang P, Yang R. Comparative and evolutionary genomics of Yersinia pestis. Microbes Infect. 2004;6:1226–1234. doi: 10.1016/j.micinf.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Zhu J, Mekalanos JJ. Quorum sensing-dependent biofilms enhance colonization in Vibrio cholerae. Dev Cell. 2003;5:647–656. doi: 10.1016/s1534-5807(03)00295-8. [DOI] [PubMed] [Google Scholar]