

Abstract
IL-17- and IFN-γ-secreting T cells play an important role in autoimmune responses in multiple sclerosis and the model system experimental autoimmune encephalomyelitis (EAE). Dendritic cells (DCs) in the periphery and microglia in the CNS are responsible for cytokine polarization and expansion of this T cell subset. Our results indicate that in vivo administration of a signal transduction inhibitor that targets DCs to mice with EAE led to a decrease in CNS infiltration of pathogenic Ag-specific T cells. Since this approach does not target T cells directly, we assessed the effects on the APCs that are involved in generating the T cell responses. Since in EAE and multiple sclerosis, both microglia and peripheral DCs are likely to contribute to disease, we utilized a bone marrow chimera system to distinguish between these two populations. These studies show that peripheral DCs are the primary target but that microglia are also modestly affected by CEP-701, as numbers and activation states of the cells in the CNS are decreased after therapy. Our results also showed a decrease in secretion of TNF-α, IL-6, and IL-23 by DCs as well as a decrease in expression of costimulatory molecules. We further determined that levels of phospho-Stat1, Stat3, Stat5, and NF-κB, which are signaling molecules that have been implicated in these pathways, were decreased. Thus, use of this class of signal transduction inhibitors may represent a novel method to treat autoimmunity by dampening the autoreactive polarizing condition driven by DCs.
The pathogenesis of many autoimmune diseases is dependent on activation of CD4+ T cells. While most current therapies for autoimmune disorders have targeted T cells, which are the direct mediators of autoimmune diseases, the contribution of dendritic cells (DCs)3 to autoimmune disease has been more extensively examined in recent years. We have focused on inhibiting the DCs as a method to decrease the autoreactive response. Our previous studies and those of others showed that signaling through FLT3 helps to maintain survival in DCs, and that inhibition of DCs with the kinase inhibitor CEP-701 led to the improvement of established disease in a mouse model for multiple sclerosis (MS), experimental autoimmune encephalomyelitis (EAE). In the present report, we have extended these findings to investigate both peripheral DCs and CNS-resident microglia as targets for this therapy and to identify more detailed mechanisms of action.
Both peripheral DCs and CNS-resident microglia are likely to participate in the presentation of myelin proteins to T cells. The exact mechanism by which autoimmunity is generated is not known, and while ultimately the pathology of both MS and EAE resides in the CNS, Ag presentation to T cells may occur in the periphery, with subsequent trafficking of activated T cells to the CNS (1). There is also evidence that T cells can be activated within the CNS (2). Thus, it is likely that during some phases of disease, both APCs in the periphery and CNS-resident APCs contribute to T cell activation, as further suggested by studies showing the activation of T cells by peripheral DCs as well as trafficking of those DCs into the brain (3). Resident microglia have been shown to be important for Ag presentation and additionally as mediators of epitope spreading (4-7). Further evidence for an important role of microglial activation in the pathogenesis of EAE was shown in a recent study in which a chemical inhibitor of microglial activation led to an improvement in EAE (8). Thus, targeting both of these populations of APCs could be potentially beneficial.
DCs provide an attractive target both because of their potential to activate and polarize autoreactive T cells and because of their secretion of cytokines that help to recruit other cells to the site. Additionally, in some cases, DCs and microglia may have direct pathogenic effects themselves. Thus, we evaluated the effects of inhibiting signal transduction on both the stimulatory and cytokine profiles of DCs. Specifically, major functions of DCs in activating autoreactive responses include stimulation of T cells, and through the skewing of immune responses. In this light, one particular cytokine that has come into focus recently for its possible role in autoimmune disease is IL-23. IL-23 was first reported in 2000 as a cytokine that shared the p40 subunit of IL-12 but had some significantly different properties (9). The secretion of IL-23 and the generation and expansion of Th17 cells have been shown to be contributory to several autoimmune disease models, including diabetes (10), arthritis (11), and EAE/MS (12-15). Studies have indicated that secretion of a number of cytokines is likely to be important in the generation and expansion of Th17 cells; while IL-23 plays a key role in expansion of Th17 cells, IL-6, IL-21, and TGFβ are thought to be critical for lineage differentiation (16-21).
CEP-701 was originally developed as a FLT3 inhibitor because of its relative selectivity for this pathway (22). We initially evaluated the use of CEP-701 in DCs because of the well-known role of FLT3 in DC development. For example, in vivo administration of FLT3 ligand (FL) generates large numbers of functional DCs (23, 24), and mice lacking either FL (25) or receptor (26) have significant deficiencies in DC populations. Our previous studies showed that mature DCs express active FLT3 and that treatment of DCs with CEP-701 inhibited its phosphorylation (27). As further evidence of the importance of this molecule in generating autoimmune disease, another study showed that treatment of mice with a FL-Ig led to a worsening of EAE (28). We show in the present report that mice deficient in FL have a delayed onset of disease. Based on these and additional reports demonstrating the importance of FLT3 in DC development and function, our hypothesis is that at least a part of the mechanism of disease amelioration is due to inhibition of FLT3 in DCs. However, as with most drugs, there is not absolute specificity, and other pathways, including Jak2 (29), may possibly be targets as well.
Many cytokines that are important for DC function depend on signaling through NF-κB (30), Stat1, Stat3, and Stat5 (31). NF-κB has been shown in multiple systems to have an impact on both survival and cytokine secretion in DCs (32-34), including regulating expression of both IL-6 (35), which has been shown to be a proinflammatory cytokine in the context of EAE/MS (3), and IL-23 (30). Thus, we also analyzed the effects of CEP-701 on downstream signaling molecules and the subsequent impact on cytokine expression.
Materials and Methods
Mice
Mice were purchased from the National Cancer Institute (except where noted otherwise) and maintained in the Johns Hopkins Oncology Animal Facility. All procedures were conducted under approved protocols, and mice were euthanized at appropriate signs of distress for the studies involving EAE. The 2D2 mice were a gift from Dr. V. Kuchroo (Harvard Medical School) and maintained in the Johns Hopkins University animal facility. FL-deficient mice (25) were bred and maintained in the Johns Hopkins University animal facility. Bone marrow-derived dendritic cell (BMDCs) were generated by flushing bone marrow from the femurs and tibias, and the cells were propagated by following a modified protocol (36). Day 8 and day 10 DCs were used for the experiments as noted. ELISA kits were purchased from Pierce (for TNF and IL-6) and eBiosciences (IL-23) and used according to the manufacturer’s recommendations.
2D2 CD4+ isolation
Spleens and lymph nodes were harvested from 8-wk-old naive 2D2 female mice. A single-cell suspension was prepared, RBCs were lysed in hypotonic buffer (ACK lysis buffer; BioSource International/BioFluids), and CD4+ cells were selected with a MACS CD4 negative selection kit (Miltenyi Biotec) and LS columns (Miltenyi Biotec) per the manufacturer’s instructions. After selection, cells were counted and the viability was determined by trypan blue exclusion (>99%), while the purity based on Vα3.2, Vβ11, and CD4 (>92%) was assessed by FACS (BD FACSCalibur; BD Biosciences). Immediately following isolation, cells were injected i.v. into mice.
Western analysis
Western analysis was conducted as previously described (27) for experiments utilizing whole cell lysates. Nuclear extracts were isolated using NE-PER extraction reagents from Thermo Scientific. Abs were purchased from Cell Signaling Technologies.
Perfusion and immunostaining
Mice were processed with some modifications, according to methods described in Curtin et al. (37). Mice were anesthetized with 1 ml of Avertin (Sigma-Aldrich) and perfused through the left cardiac ventricle with 30 ml of oxygenated Tyrode’s solution (Sigma-Aldrich) plus 10 U/ml heparin (Sigma-Aldrich). The forebrain and cerebellum were dissected and spinal cords flushed out with HBSS (Invitrogen) by hydrostatic pressure. CNS tissue was passed through a cell strainer and pelleted and digested in HBSS-based enzyme solution of DNAseI (0.1 mg/ml) (Roche) and Liberase Blendzyme 2 (0.03 mg/ml) (Roche) for 15 min at 37°C with intermediate shaking. Ten milliliters and 5 ml of enzyme solution were used for brain and cord, respectively. Enzyme digestion was followed by blocking solution (10% FCS, 10 mM EDTA in HBSS) for 5 min with 40 and 20 ml per brain and cord, respectively. The tissue was pelleted and resuspended in 5 ml of 30% isotonic Percoll (GE Healthcare) (diluted with 10x HBSS (Invitrogen) and distilled water). The tissue was underlaid with 5 ml of 70% isotonic Percoll. Mononuclear cells were isolated from the 30/70 interphase after gradient centrifugation. Collected cells were washed in 7× volume of FACS buffer (2 mM EDTA, 5% FCS in PBS), pelleted, and counted.
Cells were resuspended in FACS buffer and stained with anti-mouse CD16/CD32 (BD Biosciences) for 5 min and afterward with different combinations of Abs CD45.1-FITC, CD45.2-PE, CD45.1-allophycocyanin (eBioscience), CD11b-PECy7, CD11b-PerCP-Cy5.5, CD11c-allophycocyanin, CD86-PE, CD4-PerCP, Vα3.2-FITC, and Vβ11-PE (BD Biosciences) for 20 min at 4°C. For intracellular cytokine staining, cells were isolated as described and stimulated in culture medium containing PMA (50 ng/ml; Sigma-Aldrich), ionomycin (1 μg/ml; Sigma-Aldrich), and monensin (GolgiStop, 4 μl/6 ml; BD Biosciences) at 37°C in a humidified 5% CO2 atmosphere for 4 h. After CD16/CD32 blocking and staining of surface markers (Vα3.2, Vβ11, and CD4), cells were fixed and permeabilized using Cytofix/Cytoperm and Perm/Wash buffer from BD Biosciences according to the manufacturer’s instructions. The cells were stained with IL-17-allophycocyanin (BioLegend), IFN-γ-PerCP-Cy5.5 (eBioscience), and corresponding isotype controls. Cells were incubated (1:100) at 4°C for 20 min and washed twice in Perm/Wash before analysis. All analysis was done using four-color BD FACSCalibur (BD Biosciences).
EAE induction
EAE was induced by injecting the mice s.c. (into the flanks) with 200 μl of an emulsion (1:1 PBS/CFA, made by syringe extrusion) containing 100 μg of myelin oligodendrocyte glycoprotein (MOG)35–55 peptide (MEVG WYRSPFSRVVHLYRNGK) (Invitrogen) in PBS and 400 μg of Mycobacterium tuberculosis extract H37Ra (Difco) in CFA (Sigma-Aldrich). Additionally, the mice received 200 ng of pertussis toxin (List Biological Laboratories) i.v. after immunization and 48 h later. In some experiments, 5 × 106 2D2 CD4+ T cells were adoptively transferred i.v. before immunization. When needed, mice were injected with either vehicle control or CEP-701 (Cephalon) (20 mg/kg, twice a day, 12 h apart) for 8 days before tissue harvest. Clinical signs of EAE were assessed by a blinded observer based on the following scale: 0, no abnormalities noted; 0.5, loss of tail tonicity; 1, loss of tail reflex; 2, affected gait; 2.5, hind limb paresis; 3, full hind limb paralysis; 3.5 front limb paresis; 4, front and hind limb paralysis; 5, moribund/death.
Statistical analysis
All statistics shown were conducted using the GraphPad Prism program. Unless otherwise indicated, t tests were conducted to determine statistical significance.
Results
Signaling through FLT3 contributes to EAE pathogenesis
To delineate the role of FLT3 in EAE, we compared the course of disease in wild-type mice and mice deficient in FL. Fig. 1 shows that mice lacking FL (denoted FLT3L−/−) developed a later course, with a significant delay in progression of disease. An analysis of the area under the curve shows that there is a significant difference in disease severity until a plateau at approximately day 30 (p = 0.016), at which time the difference becomes not significant, although there was a trend toward less severe disease, indicating that signaling through FLT3 contributes to the pathology of EAE.
FIGURE 1.
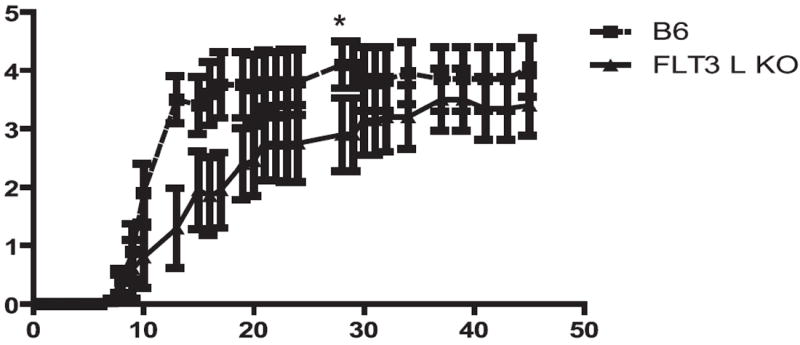
Mice deficient in FL have a delayed onset of EAE. EAE was induced in age-matched wild-type or FL−/−) mice. No therapy was given to either group; mice were scored daily. Shown is the course of disease, with the averages and SEM for each group (10 mice/group); this result is representative of three independent experiments. Difference in disease course was significant up to day 30 by area under the curve analysis.
In vivo administration of CEP-701 decreases numbers of Th17 and Th1 2D2 cells in the CNS
Our previous studies showed that treatment of mice with CEP-701 significantly improved the clinical course of established disease in mice inflicted with EAE (27). The present studies were designed to assess the mechanisms behind the observed therapeutic effect. Because it is ultimately activated T cells that cause disease, we first utilized a model system in which we could directly assess changes in the numbers and characteristics of Ag-specific pathogenic T cells that infiltrated into the CNS. For these studies, we tracked T cells that are specific for MOG (obtained from the 2D2 transgenic mice) (38) in control and treated mice and analyzed their numbers and phenotypes after treatment. In these experiments, we first adoptively transferred 2D2 T cells into host C57BL/6 mice, which were then immunized with MOG35–55 and treated with either the vehicle control or CEP-701. Eight days later, mice were perfused, and brains and spinal cords were harvested. Cells were analyzed for TCR markers identifying the 2D2 T cells and for intracellular production of IL-17 and IFN-γ. Our results show that treatment with CEP-701 decreased both the infiltration of 2D2 cells into the CNS as well as their secretion of these two critical cytokines (Fig. 2). This decrease in influx and/or reactivation of Th17 cells may account for a significant part of the observed beneficial effect of the therapy.
FIGURE 2.
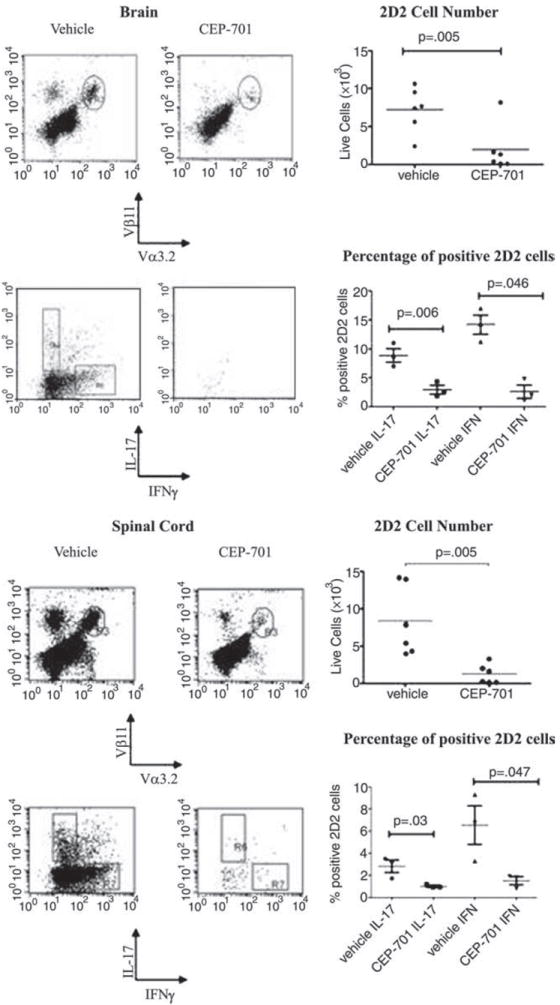
Treatment of mice with CEP-701 decreases trafficking into CNS and IL-17 and IFN-γ expression of pathogenic 2D2 cells. Purified CD4+ 2D2 cells were injected into C57BL/6 mice, which were then immunized with MOG35–55. Mice were treated for 8 days with either vehicle control or CEP-701. Mice were then perfused, brains and spinal cords harvested, and single-cell suspensions were prepared for FACS analysis. Cells were analyzed for Vα3.2/Vβ11/IFN-γ and IL-17. Representative FACS plots are shown as well as all data points. Each point shown on the graph represents three pooled mice from two different experiments.
CEP-701 therapy in vivo diminishes the numbers and activation states of both peripheral DCs and microglia in the CNS
Our previous studies had shown that this therapy targeted DCs and that downstream effects on T cell responses were likely due to the effects on the DCs and not to direct effects on the T cells themselves (27). We thus sought to assess the impact of CEP-701 therapy on the DCs in the periphery that migrated into the CNS as well as on the resident microglia. Although pathogenic T cells exert their effects in the CNS, their activation is dependent on their interactions with APCs, whether by DCs in the periphery or by microglia in the CNS. Peripheral DCs migrate into the brains in EAE, so to be able to distinguish these infiltrating cells from resident microglia, we utilized a bone marrow chimera system in which we could differentiate the origin of APCs that were found in the brain at a given point during the course of disease. To differentiate these cell types, we first transplanted C57BL/6 (CD45.2) mice with bone marrow from the congenic CD45.1 B6.SJL donor mice and allowed them sufficient time to fully engraft. Thus, the peripheral blood and bone marrow of these mice express CD45.1 on their hematopoietically derived cells, but the microglia retain expression of CD45.2. Following engraftment, mice were immunized with MOG35–55 and treated with either vehicle control or CEP-701. The mice were treated for 8 days and then they were perfused and their brains and spinal cords were analyzed to determine the origin and phenotype of the APCs present. After immunization, there was a high level of trafficking of peripherally derived DCs into the brains, as expected. Treatment of mice with CEP-701 led to a decrease in numbers in the CNS of both peripherally derived DCs and resident microglia (which also express FLT3, data not shown), although to a much lesser extent than do peripheral DCs at this time point. This suggests that at least part of the mechanism of action may be through decreasing the numbers of both of these types of APCs that are found in the brain during EAE. Additionally, there was a consistent decrease in the percentage of CD86high cells in both peripherally derived DCs and in microglia, indicating a decrease in the activation state of both infiltrated and resident APCs (Figs. 3 and 4).
FIGURE 3.
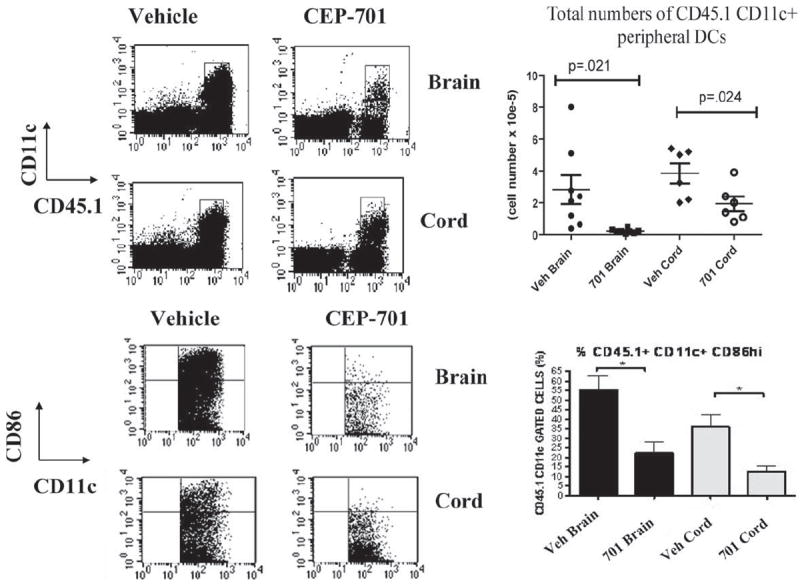
Treatment of mice with CEP-701 decreases trafficking of peripheral DCs into the brain and their activation state. Bone marrow chimeras (CD45.1→CD45.2) were established such that peripherally derived DCs (CD45.1) could be distinguished from CNS-resident microglia (CD45.2). EAE was induced and mice were treated for 8 days with either vehicle control or CEP-701 (20 mg/kg, twice daily). Mice were perfused and brains and spinal cords were harvested and prepared for FACS analysis. Cells were analyzed for expression of CD11c, CD45.1, CD45.2, and CD86. Shown are representative plots from individual mice and graphs of all mice. Each point shown on the graph represents three pooled mice from two different experiments.
FIGURE 4.
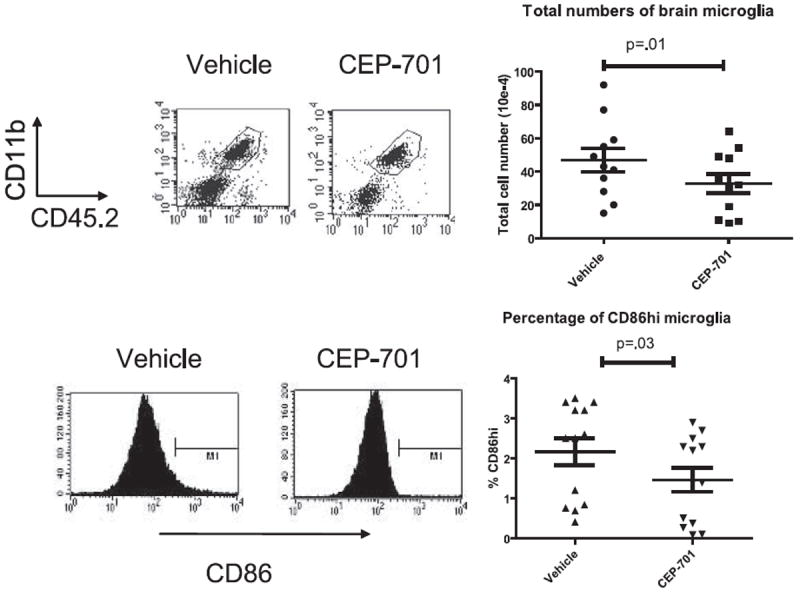
CEP-701 decreases the numbers and activation state of brain-resident microglia. EAE was induced in the CD45.1→CD45.2 transplanted mice, which were then treated for 8 days with either vehicle control or CEP-701. Mice were then perfused and brains were harvested and prepared for FACS analysis. Cells were analyzed for expression of CD11c, CD45.1, CD45.2, and CD86. Shown are representative plots from individual mice and graphs of all mice. Each point shown on the graph represents three pooled mice from two different experiments.
Signal transduction inhibition decreases the secretion of IL-23, IL-6, and TNF-α and the expression of costimulatory molecules by DCs
To further dissect the mechanism by which FLT3 inhibition has efficacy in this autoimmune disease model, we also tested whether other cytokines known to contribute to an inflammatory/autoimmune state are decreased in cultured DCs by treatment with CEP-701. For these studies, BMDCs were generated in culture and then treated with CEP-701. Supernatants were harvested for the analysis of cytokine secretion and FACS for analysis of costimulatory molecule expression. As shown in Fig. 5a, decreases in IL-23 TNF and IL-6 were observed by ELISA at time points that precede apoptosis (which occurs at >24 h after exposure).
FIGURE 5.
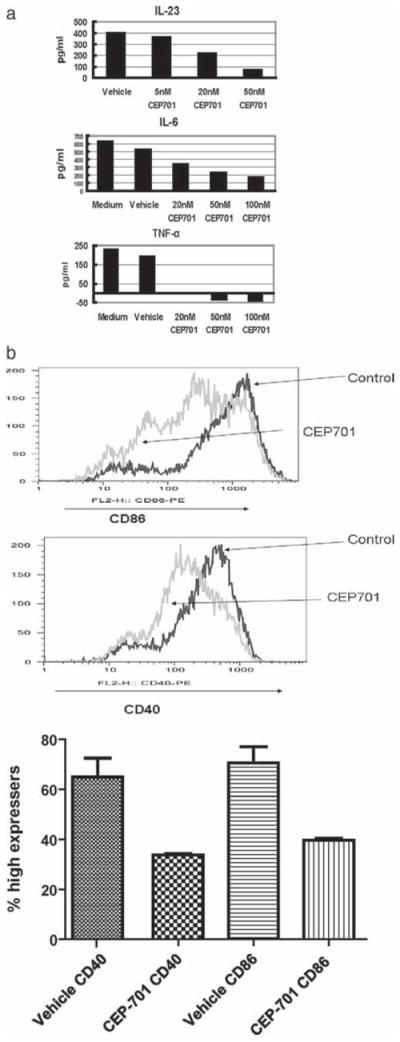
a, CEP-701 decreases production of IL-23, IL-6, and TNF by DCs. BMDCs were treated with no inhibitor (vehicle, labeled V) or the concentrations of CEP-701 shown on the graph for ~12 h before analysis. Supernatants were harvested and cytokine quantification was conducted by ELISA. The results of these studies are shown in each graph; a representative result is shown for each cytokine and is representative of three to five experiments for each. b, CEP-701 decreases costimulatory molecule expression in annexin V-negative DCs. BMDCs were treated for 16 h and then labeled with Abs to CD11c, annexin V, and either CD86 (top) or CD40 (bottom). Shown are histograms of these two molecules, gated on CD11c+annexin V− cells, and a graph with statistical analysis for three different experiments.
DC-mediated stimulation of T cells occurs through the expression of costimulatory molecules by DCs that directly interact with cognate molecules on T cells. Therefore, we also measured levels of expression of CD86 and CD40 after treatment. Cells were treated as above and analyzed for expression of annexin V and costimulatory molecules. As shown in Fig. 5b, both CD86 and CD40 were significantly decreased in viable cells (gated on annexin V-negative cells) after exposure to drug. Thus, the effects of CEP-701 in ameliorating EAE may be mediated in part not only by induction of DC apoptosis but also through a decrease in DC function as well.
Decrease of NF-κB, Stat1, Stat3, and Stat5 in DCs after treatment
We hypothesized that one mechanism of action of signal transduction blockade in DCs leading to the observed changes in cytokine secretion and costimulatory expression was through the disruption of downstream signaling pathways. We thus assessed effects of CEP-701 treatment of DCs on signaling pathways known to be important components in the production of cytokines. For these studies, DCs were generated as above and exposed to vehicle or inhibitor for the indicated times and then analyzed by Western analysis. Treatment of DCs with CEP-701 led to a decrease in NF-κB activation (Fig. 6a), which could help to explain the decrease in inflammatory cytokine expression that we observed in vivo.
FIGURE 6.
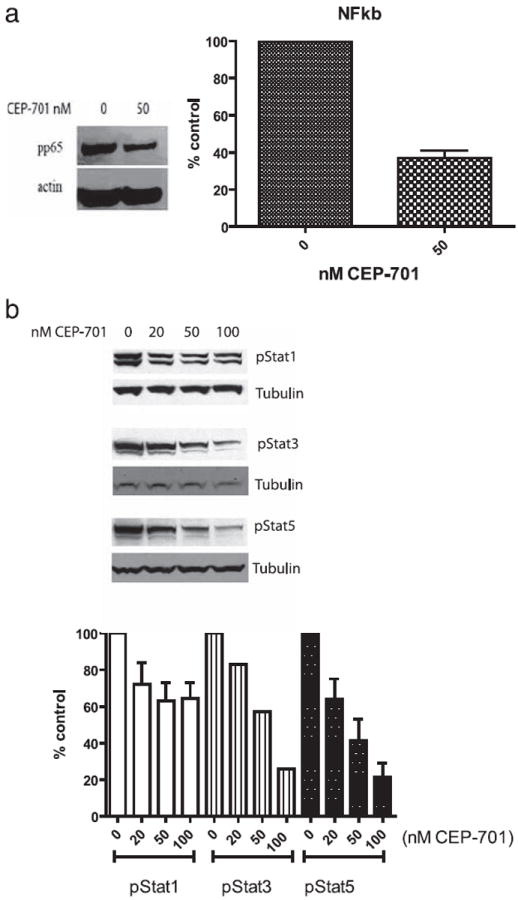
CEP-701 targets specific signaling pathways necessary for DC function. BMDCs were treated with no CEP-701 (vehicle) or 50 nM CEP-701 for 1 h to assess changes in signaling molecules. Cells were lysed and either nuclear extracts (for NF-κB) (a) or whole cell lysates (for pStat1, pStat3, and pStat5) (b) were prepared. Shown are representative Western analyses for these molecules and densitometry analysis.
We also assessed the impact of treatment on the phosphorylation/activation state of Stat1, Stat3, and Stat5 (Fig. 6b). The results show a decrease in Stat1 (albeit to a much lesser degree), Stat3, and Stat5 activation after inhibition, indicating the possibility that these molecules may also play a part in generating a therapeutic effect.
Discussion
Our studies suggest that using a signal transduction inhibitor to target pathways in APCs may be a novel method to limit the trafficking of peripheral DCs into the brain and concomitant pathogenic T cell responses. Th17 cells have been shown to be required for EAE, at least in some settings and stages of disease (39-41). We used the MOG-specific transgenic 2D2 T cells to assess the infiltration of Ag-specific Th17 cells into the CNS and showed that there was a significant decrease in the numbers of these cells following treatment. While IL-17 has recently come into focus as being a critical cytokine in EAE, there is evidence that Th1 cells also play a role in disease at certain time points (42), and activation of these cells may constitute an independent event (43). Our results that IFN-γ-secreting 2D2 cells were also decreased suggest the potential for this therapy to inhibit the influx of Th1 cells into the CNS as well. Our previous studies showed that T cells are not direct targets for this class of inhibitors (27), and thus blocking this pathway in DCs may provide a novel means to diminish autoimmunity by inhibiting the activation of autoreactive T cells. CEP-701/lestaurtinib was originally developed for its selectivity against FLT3; however, it has been shown to inhibit Jak2 as well (29). To support our hypothesis that FLT3 plays a role in the pathogenesis of EAE, we compared the course of disease in wild-type mice and those deficient for FL. The mice deficient for FL had a significant delay in the onset and progression of disease, indicating that FLT3 contributes to the severity of disease. Thus, we hypothesize that while other targets may also be involved, FLT3 signaling is an important pathway in this disease process.
We assessed the effects of treatment of mice with CEP-701 on both peripherally derived DCs and CNS derived-microglia in EAE. Although their relative contributions to the activation and reactivation of pathogenic T cells have not been clearly demonstrated, numerous studies have shown that both cell types likely play a role in disease. In EAE, mice are immunized peripherally, thus initiating the response outside the CNS, and many studies have shown that infiltration of peripherally derived CD11c+ DCs mediate disease and are necessary for lesion formation in EAE. It was recently shown that migration of monocyte-derived DCs in the inflamed brain leads to stimulation of pathogenic Th17 responses as a result of blood-brain barrier-secreted factors (13). However, microglia are likely to be involved in the reactivation of T cells and possibly in the initial onset of disease (44). Activation of microglia has been shown to be necessary for the onset of disease in some settings (45). Additionally, one recent study evaluated mice relatively early in the course of disease and identified areas of microglial inflammation, with magnetic resonance imaging results paralleling those of MS in humans (46). Furthermore, resident microglia may also present Ag and thus contribute to ongoing disease. Recent studies showed that Th1 and Th17 cells preferentially migrate across the blood-brain barrier, and that expansion of Th17 cells occurs locally (47). Our studies show that there was a dramatic decrease in both the numbers and the activation states of peripherally derived DCs and a more subtle effect on microglia at a relatively early time point. These findings may account for the decrease in activation and migration of activated pathogenic T cells observed. While in the early stages of disease, it is likely that the influx of peripheral DCs is primarily responsible for pathology in this model system, it is possible that activated CNS-resident microglia may play a greater role in later stages of the process and in human disease, which may be due either to directly targeting microglia or alternatively could be a secondary result from the earlier events.
Cytokine secretion by DCs is necessary for activation as well as for polarization of T cells. Because of the significance of IL-23 and IL-6 (48) in the generation and/or expansion of Th17 cells, we analyzed these cytokines as well as TNF-α, which perpetuates the inflammatory process through recruitment of cells and also has the potential for direct toxicity. IL-6 has been associated with a number of autoimmune conditions, including rheumatoid arthritis, systemic lupus erythematosus, MS, transverse myelitis (49), and others. IL-6 is proposed to work both by generating the recruitment of inflammatory cells and by contributing to the differentiation of Th17 cells as mentioned above. As T cells are not directly affected by CEP-701 (27), the change in secretion of both IL-23 and IL-6 could have a significant impact on the reduction of Th17 cells in this setting.
We next sought to assess whether CEP-701 treatment of DCs would affect pathways that are potentially important for DC function, and thus we assessed changes in activation of some of these molecules that are known to participate in DC survival and/or function. We first analyzed NF-κB activation since many of the inflammatory and activation-inducing cytokines are mediated by NF-κB (50-52). NF-κB has been shown in multiple systems to have an impact on both survival and cytokine secretion in DCs (32-34), including mediating expression of IL-6 (35) and IL-23, in that mice deficient in the c-Rel component of NF-κB had significantly diminished p19 production, indicating the importance of this pathway in IL-23 generation (30). Our results show that CEP-701 inhibited the activation of NF-κB, suggesting that may this pathway be a mediator involved in the suppression of DC activation.
We also analyzed Stat1 and Stat5, which have been identified as transcription factors that were up-regulated after stimulation of DCs with GM-CSF, indicating that they may play a role in activation of DCs (53). The importance of Stat5 was also shown in mice deficient in this molecule, which showed defects in growth and proliferation of monocytes (54). Furthermore, relevant to both activity and survival, during the final maturation stages of myeloid differentiation, Stat5 was shown to have an antiapoptotic effect (55). Stat3 has been shown to play an indispensable role in mediating effects of FLT3 on development of DCs in that mice deficient for Stat3 had significantly lower numbers of DCs and did not respond to FL stimulation (56). Furthermore, forced expression of FLT3 in the megakaryocyte/erythroid progenitor population led to an activation of Stat3 (57), providing additional evidence for the linkage of these two pathways. Since FLT3 is a target of CEP-701, we evaluated levels of phospho-Stat3 and Stat5, which were significantly decreased after treatment, while pStat1 was less affected. This finding suggests that there is specificity to the pathways targeted by CEP-701 and that general induction of apoptosis may not be entirely responsible for the observed effects and that the effects of inhibiting signaling may be mediated by specific downstream pathway molecules. The exact pathway by which these inhibitors have this effect is not known. Previous studies have indicated that the inhibition of Stat3 and Stat5 phosphorylation by CEP-701 is likely an indirect effect of inhibiting FLT3 kinase activity, as FLT3 is not known to directly phosphorylate the Stats (58). Also, no tyrosine kinase inhibitors studied to date are specific for a single kinase, so some fraction of the reduced phosphorylation observed could be due to CEP-701 inhibiting another kinase pathway that also leads to Stat phosphorylation.
Results of our studies thus indicate that CEP-701 treatment of mice with EAE led to a decrease in the influx of pathogenic Th17 cells and APCs capable of reactivating them in the brain. Taken together, these findings suggest that targeting signaling pathways in DCs may be a novel method to treat autoimmune disease by decreasing these downstream pathogenic responses.
Footnotes
This work was supported by National Cancer Institute Grant RO1-CA11989 (to K.A.W.), National Institutes of Health Grant NS041435 and National Multiple Sclerosis Society Grant TR-3760-A3 (to P.A.C.), and National Cancer Institute Grant RO1-CA70970 and the Kyle Haydock Professorship (to D.S.).
Abbreviations used in this paper: DC, dendritic cell; BMDC, bone marrow-derived dendritic cell; EAE, experimental autoimmune encephalomyelitis; FL, FLT3 ligand; MOG, myelin oligodendrocyte glycoprotein; MS, multiple sclerosis.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.de Vos A, van Meurs FM, Brok HP, Boven LA, Hintzen RQ, van der Valk P, Ravid R, Rensing S, Boon L, ’t Hart BA, Laman JD. Transfer of central nervous system autoantigens and presentation in secondary lymphoid organs. J Immunol. 2002;169:5415–5423. doi: 10.4049/jimmunol.169.10.5415. [DOI] [PubMed] [Google Scholar]
- 2.Juedes AE, Ruddle NH. Resident and infiltrating central nervous system APCs regulate the emergence and resolution of experimental autoimmune encephalomyelitis. J Immunol. 2001;166:5168–5175. doi: 10.4049/jimmunol.166.8.5168. [DOI] [PubMed] [Google Scholar]
- 3.Miller SD, McMahon EJ, Schreiner B, Bailey SL. Antigen presentation in the CNS by myeloid dendritic cells drives progression of relapsing experimental autoimmune encephalomyelitis. Ann NY Acad Sci. 2007;1103:179–191. doi: 10.1196/annals.1394.023. [DOI] [PubMed] [Google Scholar]
- 4.McRae BL, Vanderlugt CL, Dal Canto MC, Miller SD. Functional evidence for epitope spreading in the relapsing pathology of experimental autoimmune encephalomyelitis. J Exp Med. 1995;182:75–85. doi: 10.1084/jem.182.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vanderlugt CL, Begolka WS, Neville KL, Katz-Levy Y, Howard LM, Eagar TN, Bluestone JA, Miller SD. The functional significance of epitope spreading and its regulation by co-stimulatory molecules. Immunol Rev. 1998;164:63–72. doi: 10.1111/j.1600-065x.1998.tb01208.x. [DOI] [PubMed] [Google Scholar]
- 6.McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. 2005;11:335–339. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- 7.Tuohy VK, Yu M, Weinstock-Guttman B, Kinkel RP. Diversity and plasticity of self recognition during the development of multiple sclerosis. J Clin Invest. 1997;99:1682–1690. doi: 10.1172/JCI119331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo X, Nakamura K, Kohyama K, Harada C, Behanna HA, Watterson DM, Matsumoto Y, Harada T. Inhibition of glial cell activation ameliorates the severity of experimental autoimmune encephalomyelitis. Neurosci Res. 2007;59:457–466. doi: 10.1016/j.neures.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 9.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 10.Mensah-Brown EP, Shahin A, Al-Shamisi M, Wei X, Lukic ML. IL-23 leads to diabetes induction after subdiabetogenic treatment with multiple low doses of streptozotocin. Eur J Immunol. 2006;36:216–223. doi: 10.1002/eji.200535325. [DOI] [PubMed] [Google Scholar]
- 11.Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 12.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 13.Ifergan I, Kebir H, Bernard M, Wosik K, Dodelet-Devillers A, Cayrol R, Arbour N, Prat A. The blood-brain barrier induces differentiation of migrating monocytes into Th17-polarizing dendritic cells. Brain. 2008;131:785–799. doi: 10.1093/brain/awm295. [DOI] [PubMed] [Google Scholar]
- 14.Becher B, Durell BG, Noelle RJ. IL-23 produced by CNS-resident cells controls T cell encephalitogenicity during the effector phase of experimental autoimmune encephalomyelitis. J Clin Invest. 2003;112:1186–1191. doi: 10.1172/JCI19079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ivanov II, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Semin Immunol. 2007;19:409–417. doi: 10.1016/j.smim.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent pro- and anti-inflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 19.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-β induces development of the TH17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 20.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Zhou L, I, Ivanov I, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs TH17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 22.Levis M, Allebach J, Tse KF, Zheng R, Baldwin BR, Smith BD, Jones-Bolin S, Ruggeri B, Dionne C, Small D. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood. 2002;99:3885–3891. doi: 10.1182/blood.v99.11.3885. [DOI] [PubMed] [Google Scholar]
- 23.Borges L, Miller RE, Jones J, Ariail K, Whitmore J, Fanslow W, Lynch DH. Synergistic action of fms-like tyrosine kinase 3 ligand and CD40 ligand in the induction of dendritic cells and generation of antitumor immunity in vivo. J Immunol. 1999;163:1289–1297. [PubMed] [Google Scholar]
- 24.Lynch DH, Andreasen A, Maraskovsky E, Whitmore J, Miller RE, Schuh JC. Flt3 ligand induces tumor regression and antitumor immune responses in vivo. Nat Med. 1997;3:625–631. doi: 10.1038/nm0697-625. [DOI] [PubMed] [Google Scholar]
- 25.McKenna HJ, Stocking KL, Miller RE, Brasel K, De Smedt T, Maraskovsky E, Maliszewski CR, Lynch DH, Smith J, Pulendran B, et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood. 2000;95:3489–3497. [PubMed] [Google Scholar]
- 26.Waskow C, Liu K, Darrasse-Jeze G, Guermonprez P, Ginhoux F, Merad M, Shengelia T, Yao K, Nussenzweig M. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat Immunol. 2008;9:676–683. doi: 10.1038/ni.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whartenby KA, Calabresi PA, McCadden E, Nguyen B, Kardian D, Wang T, Mosse C, Pardoll DM, Small D. Inhibition of FLT3 signaling targets DCs to ameliorate autoimmune disease. Proc Natl Acad Sci USA. 2005;102:16741–16746. doi: 10.1073/pnas.0506088102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ, Becher B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. 2005;11:328–334. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- 29.Hexner EO, Serdikoff C, Jan M, Swider CR, Robinson C, Yang S, Angeles T, Emerson SG, Carroll M, Ruggeri B, Dobrzanski P. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood. 2008;111:5663–5671. doi: 10.1182/blood-2007-04-083402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carmody RJ, Ruan Q, Liou HC, Chen YH. Essential roles of c-Rel in TLR-induced IL-23 p19 gene expression in dendritic cells. J Immunol. 2007;178:186–191. doi: 10.4049/jimmunol.178.1.186. [DOI] [PubMed] [Google Scholar]
- 31.Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, Pflanz S, Zhang R, Singh KP, Vega F, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rβ1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–5708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- 32.Kriehuber E, Bauer W, Charbonnier AS, Winter D, Amatschek S, Tamandl D, Schweifer N, Stingl G, Maurer D. Balance between NF-κB and JNK/AP-1 activity controls dendritic cell life and death. Blood. 2005;106:175–183. doi: 10.1182/blood-2004-08-3072. [DOI] [PubMed] [Google Scholar]
- 33.Onishi H, Kuroki H, Matsumoto K, Baba E, Sasaki N, Kuga H, Tanaka M, Katano M, Morisaki T. Monocyte-derived dendritic cells that capture dead tumor cells secrete IL-12 and TNF-α through IL-12/TNF-α/NF-κB autocrine loop. Cancer Immunol Immunother. 2004;53:1093–1100. doi: 10.1007/s00262-004-0568-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rescigno M, Martino M, Sutherland CL, Gold MR, Ricciardi-Castagnoli P. Dendritic cell survival and maturation are regulated by different signaling pathways. J Exp Med. 1998;188:2175–2180. doi: 10.1084/jem.188.11.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mann J, Oakley F, Johnson PW, Mann DA. CD40 induces interleukin-6 gene transcription in dendritic cells: regulation by TRAF2, AP-1, NF-κB, and CBF1. J Biol Chem. 2002;277:17125–17138. doi: 10.1074/jbc.M109250200. [DOI] [PubMed] [Google Scholar]
- 36.Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 37.Curtin JF, King GD, Barcia C, Liu C, Hubert FX, Guillonneau C, Josien R, Anegon I, Lowenstein PR, Castro MG. Fms-like tyrosine kinase 3 ligand recruits plasmacytoid dendritic cells to the brain. J Immunol. 2006;176:3566–3577. doi: 10.4049/jimmunol.176.6.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med. 2003;197:1073–1081. doi: 10.1084/jem.20021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, Blumenschein WM, McClanahan T, Brombacher F, Hurst SD, et al. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med. 2007;204:161–170. doi: 10.1084/jem.20061738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thakker P, Leach MW, Kuang W, Benoit SE, Leonard JP, Marusic S. IL-23 is critical in the induction but not in the effector phase of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:2589–2598. doi: 10.4049/jimmunol.178.4.2589. [DOI] [PubMed] [Google Scholar]
- 41.Kroenke MA, Segal BM. Th17 and Th1 responses directed against the immunizing epitope, as opposed to secondary epitopes, dominate the autoimmune repertoire during relapses of experimental autoimmune encephalomyelitis. J Neurosci Res. 2007;85:1685–1693. doi: 10.1002/jnr.21291. [DOI] [PubMed] [Google Scholar]
- 42.Gocke AR, Cravens PD, Ben LH, Hussain RZ, Northrop SC, Racke MK, Lovett-Racke AE. T-bet regulates the fate of Th1 and Th17 lymphocytes in autoimmunity. J Immunol. 2007;178:1341–1348. doi: 10.4049/jimmunol.178.3.1341. [DOI] [PubMed] [Google Scholar]
- 43.Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12-and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535–1541. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Serafini B, Columba-Cabezas S, Di Rosa F, Aloisi F. Intracerebral recruitment and maturation of dendritic cells in the onset and progression of experimental autoimmune encephalomyelitis. Am J Pathol. 2000;157:1991–2002. doi: 10.1016/S0002-9440(10)64838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ponomarev ED, Shriver LP, Maresz K, Dittel BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J Neurosci Res. 2005;81:374–389. doi: 10.1002/jnr.20488. [DOI] [PubMed] [Google Scholar]
- 46.Nessler S, Boretius S, Stadelmann C, Bittner A, Merkler D, Hartung HP, Michaelis T, Bruck W, Frahm J, Sommer N, Hemmer B. Early MRI changes in a mouse model of multiple sclerosis are predictive of severe inflammatory tissue damage. Brain. 2007;130:2186–2198. doi: 10.1093/brain/awm105. [DOI] [PubMed] [Google Scholar]
- 47.Ifergan I, Kebir H, Bernard M, Wosik K, Dodelet-Devillers A, Cayrol R, Arbour N, Prat A. The blood brain barrier induces differentiation of migrating monocytes into Th17-polarizing dendritic cells. Brain. 2008;131:785–799. doi: 10.1093/brain/awm295. [DOI] [PubMed] [Google Scholar]
- 48.Kimura A, Naka T, Kishimoto T. IL-6-dependent and -independent pathways in the development of interleukin 17-producing T helper cells. Proc Natl Acad Sci USA. 2007;104:12099–12104. doi: 10.1073/pnas.0705268104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaplin AI, Deshpande DM, Scott E, Krishnan C, Carmen JS, Shats I, Martinez T, Drummond J, Dike S, Pletnikov M, et al. IL-6 induces regionally selective spinal cord injury in patients with the neuroinflammatory disorder transverse myelitis. J Clin Invest. 2005;115:2731–2741. doi: 10.1172/JCI25141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Utsugi M, Dobashi K, Ishizuka T, Kawata T, Hisada T, Shimizu Y, Ono A, Mori M. Rac1 negatively regulates lipopolysaccharide-induced IL-23 p19 expression in human macrophages and dendritic cells and NF-κB p65 trans activation plays a novel role. J Immunol. 2006;177:4550–4557. doi: 10.4049/jimmunol.177.7.4550. [DOI] [PubMed] [Google Scholar]
- 51.Ashton AW, Ware GM, Kaul DK, Ware JA. Inhibition of tumor necrosis factor α-mediated NFκB activation and leukocyte adhesion, with enhanced endothelial apoptosis, by G protein-linked receptor (TP) ligands. J Biol Chem. 2003;278:11858–11866. doi: 10.1074/jbc.M210766200. [DOI] [PubMed] [Google Scholar]
- 52.Keller ET, Chang C, Ershler WB. Inhibition of NFκB activity through maintenance of IκBα levels contributes to dihydrotestosterone-mediated repression of the interleukin-6 promoter. J Biol Chem. 1996;271:26267–26275. doi: 10.1074/jbc.271.42.26267. [DOI] [PubMed] [Google Scholar]
- 53.Welte T, Koch F, Schuler G, Lechner J, Doppler W, Heufler C. Granulocyte-macrophage colony-stimulating factor induces a unique set of STAT factors in murine dendritic cells. Eur J Immunol. 1997;27:2737–2740. doi: 10.1002/eji.1830271038. [DOI] [PubMed] [Google Scholar]
- 54.Feldman GM, Rosenthal LA, Liu X, Hayes MP, Wynshaw-Boris A, Leonard WJ, Hennighausen L, Finbloom DS. STAT5A-deficient mice demonstrate a defect in granulocyte-macrophage colony-stimulating factor-induced proliferation and gene expression. Blood. 1997;90:1768–1776. [PubMed] [Google Scholar]
- 55.Kieslinger M, Woldman I, Moriggl R, Hofmann J, Marine JC, Ihle JN, Beug H, Decker T. Antiapoptotic activity of Stat5 required during terminal stages of myeloid differentiation. Genes Dev. 2000;14:232–244. [PMC free article] [PubMed] [Google Scholar]
- 56.Laouar Y, Welte T, Fu XY, Flavell RA. STAT3 is required for Flt3L-dependent dendritic cell differentiation. Immunity. 2003;19:903–912. doi: 10.1016/s1074-7613(03)00332-7. [DOI] [PubMed] [Google Scholar]
- 57.Onai N, Obata-Onai A, Tussiwand R, Lanzavecchia A, Manz MG. Activation of the Flt3 signal transduction cascade rescues and enhances type I interferon-producing and dendritic cell development. J Exp Med. 2006;203:227–238. doi: 10.1084/jem.20051645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tse KF, Mukherjee G, Small D. Constitutive activation of FLT3 stimulates multiple intracellular signal transducers and results in transformation. Leukemia. 2000;14:1766–1776. doi: 10.1038/sj.leu.2401905. [DOI] [PubMed] [Google Scholar]