

Abstract
Positron emission tomography (PET) has growing importance as a molecular imaging technique for clinical research and drug development. Methods for producing PET radiotracers utilizing cyclotron-produced [18F]fluoride ion (t1/2 = 109.7 min) without the need for complete removal of irradiated target [18O]water and addition of cryptand are keenly sought for practical convenience and efficiency. Several structurally diverse diaryliodonium tosylates, XArI+Ar′Y TsO− (X = H or p-MeO), were investigated in a microfluidic apparatus for their reactivity towards radiofluorination with high specific activity (no-carrier-added) [18F]fluoride ion in mixtures of DMF and irradiated target [18O]water in the absence of cryptand. Salts bearing a para or ortho electron-withdrawing group Y (e.g., Y = p-CN) reacted rapidly (~ 3 min) to give the expected major [18F]fluoroarene product, [18F]ArY, in useful moderate radiochemical yields even when the solvent had [18O]water content up to 28%. Salts bearing electron-withdrawing groups in meta position (e.g., Y = m-NO2), or an electron-donating substituent (Y = p-OMe), gave low radiochemical yields under the same conditions.
Introduction
Methodology development for producing [18F]fluoroarenes in high specific radioactivity from no-carrier-added (NCA) cyclotron-produced [18F]fluoride ion attracts increasing attention because of the growing importance of positron emission tomography (PET) [1,2] as a molecular imaging technique in clinical research [3,4] and drug development [5,6]. The positron-emitter, fluorine-18 (t1/2 = 109.7 min), is widely used as a radiolabel in PET radiotracers.[7] In such radiotracers, an aryl position is often preferred to an aliphatic position for fluorine-18 because of generally greater resistance to undesirable radiodefluorination in vivo.[8] However, the radiosynthesis of [18F]fluoroarenes has been generally more demanding [7].
The preparation of [18F]fluoroarenes from the reactions of [18F]fluoride ion with diaryliodonium salts (Scheme 1) [9,10] is finding increasing application for preparing labeling synthons [11–13] and PET radiotracers [14–17]. This radiofluorination method, unlike aromatic nucleophilic substitution (SNAr) in non-hypervalent substrates, allows NCA [18F]fluoride ion to be introduced readily into electron-deficient or electron-rich rings, irrespective of ring substituent positions [9,10,18,19]. The aryl ring selectivity of the process can be controlled by choosing a relatively electron-rich ring as one of the ring partners so that radiofluorination occurs preferentially at the other ring.[20–24] Often the selected electron-rich ring is 2-thienyl [19–23] or 4 methoxyphenyl [19]. Alkyl substituents in ortho position tend to direct radiofluorination onto the same ring [18], and the addition of a radical scavenger (e.g., TEMPO) improves some radiofluorination reactions.[15,25] These features suggest that many of these reactions have mechanisms that are distinct from that of SNAr reactions in non-hypervalent substrates.
Scheme 1.
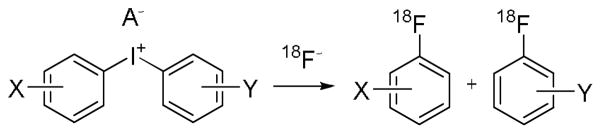
Preparation of NCA [18F]fluoroarenes through the radiofluorination of diaryliodonium salts.
[18F]Fluoride ion is usually produced by proton irradiation of [18O]water according to the high yielding 18O(p,n)18F reaction.[26] Under these circumstances the [18F]fluoride ion is produced at a useful very high NCA specific activity, but is inevitably fully hydrated. Practical [27] and theoretical studies [28–30] indicate that a single fluoride ion in aqueous media may be hydrated with as many as 16 water molecules, and that the hydration energy is exceptionally high at 367 kcal/mol. Theoretically, the nucleophilicity of fluoride ion depends inversely on the number of water molecules of hydration [31], and has been shown experimentally to decrease by almost three orders of magnitude as the number of water molecules increases from zero to six.[27] Traditionally, [18F]fluoride ion is rendered more reactive by removing water, usually by a number of azeotropic evaporations with acetonitrile, and is solubilized in an anhydrous organic solvent by pairing with a large counterion derived from an added base (e.g., Cs2CO3) or base-cryptand mixture (e.g., K 2.2.2-K2CO3).[7] Such procedures for obtaining reactive [18F]fluoride ion in organic solvent are time-consuming and wasteful of radioactivity by decay and especially by surface adsorption. Removal of all water molecules of hydration is difficult and therefore variable reactivity is often observed from the same drying process in day-to-day operation, especially towards poorly reactive substrates. Moreover, if K 2.2.2 cryptand is added to aid [18F]fluoride ion solubilization in organic solvent, this cryptand must be prevented from appearing in the final radiotracer if this is intended to be administered to human subjects.[32] Methods for achieving aryl radiofluorination with [18F]fluoride ion in aqueous or partially aqueous phase and in the absence of cryptand would therefore be attractive for 18F-labeled PET radiotracer production.
In an earlier study of the radiofluorination of simple diaryliodonium salts, we included a low percentage of water (< 0.25% v/v) in the organic reaction solvent (DMF) in order to control the degree of hydration, and therefore the nucleophilicity, of the [18F]fluoride ion.[18] In many cases, the dissolution of the diaryliodonium salt was also enhanced. The low concentration of water was well tolerated in these reactions, which were conducted in the presence of K 2.2.2 cryptand and potassium carbonate. Others have reported remarkable water-tolerance for the radiofluorinations of particular diaryliodonium salts but have not studied this systematically across a wide range of substrates.[33,34,40]
Here, we aimed to study the radiofluorination of diaryliodonium salts in aqueous-organic solvent (DMF), and in the absence of cryptand, in order to assess the possible utility of such conditions. For this study, we selected a range of substituted diaryliodonium tosylates, five of which we have previously evaluated for reactivity towards [18F]fluoride ion in predominantly organic solvent in the presence of cryptand (K 2.2.2) [18,19]. We have previously shown that a microfluidic platform enables sequential radiofluorination reactions to be performed rapidly with very low quantities of reagents under precisely controlled conditions of temperature, time, and reaction stoichiometry.[18,19] Therefore, this reaction platform was used in this study. Our results show that the feasibility to perform radiofluorinations on substituted diaryliodonium tosylates in partially aqueous media depends critically on stereoelectronic factors.
Results and Discussion
Variously substituted diaryliodonium tosylates (2-15; Scheme 2) were selected to explore the influence of substituents on radiofluorination reactions under aqueous-organic conditions in the absence of cryptand. Most of these salts had a 4-methoxyphenyl group attached to the hypervalent iodine atom so that any radiofluorination was preferentially directed to the other aryl ring. The tosylate anion was acceptable in these salts because it was expected to be only weakly competitive with [18F]fluoride ion as a nucleophile. Diphenyliodonium tosylate (1) was also included for study in view of a previous report of its reactivity in water-acetonitrile media.[33] We chose to use water-DMF in our study because we had previously studied the reactivity of some of these salts with [18F]fluoride ion in DMF containing a very low percentage of water (0.25%) in the presence of cryptand (K 2.2.2).
Scheme 2.
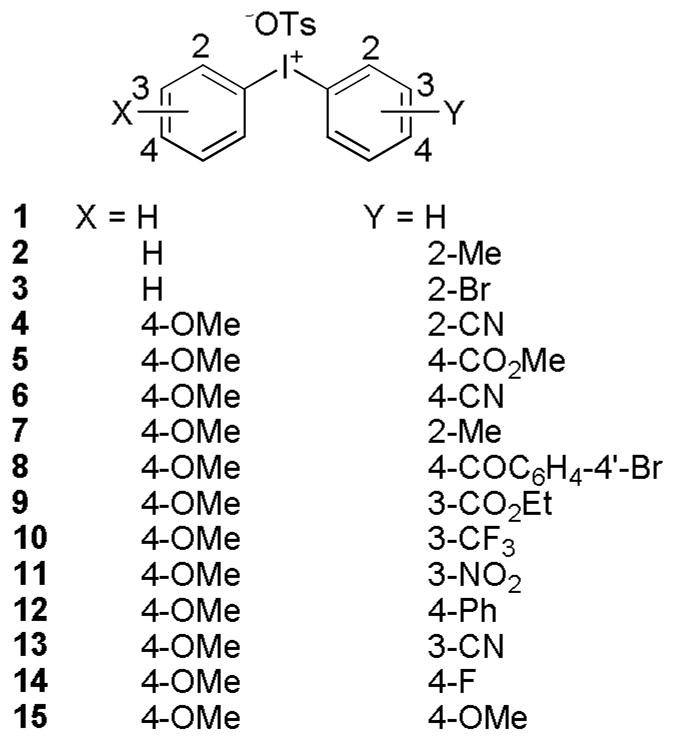
Diaryliodonium tosylates used in this study.
Initially, we aimed to gain insight into the relative reactivity of the selected set of diaryliodonium salts towards NCA [18F]fluoride ion. For this purpose, we used the microfluidic platform to react each salt with NCA [18F]fluoride ion under fixed conditions of salt, K 2.2.2, and K2CO3 concentrations (9.5, 16.6 and 4.5 mM, respectively), solvent composition (DMF-0.25% H2O), temperature (150 °C) and time (181 s), and measured the radiochemical yields (RCYs) of [18F]fluoroarenes with radio-HPLC.
All of the salts gave some [18F]fluoroarene product under these specific conditions, with yields ranging from 12–96% (Table 1). Recoveries of radioactivity from the apparatus were generally high (56–100%; mean ± SD: 87 ± 15%). Diphenyliodonium tosylate (1) gave a moderate yield of [18F]fluorobenzene (49%). For the analogous salt bearing an ortho-methyl substituent (2), overall production of [18F]fluoroarenes was much lower, with [18F]2-fluorotoluene obtained in 2.3-fold preference over [18F]fluorobenzene, in accord with operation of an ‘ortho effect’ [18]. Introduction of an ortho-bromo substituent, as in 3, also suppressed radiofluorination, and gave [18F]2-fluorobromobenzene as the only radioactive product.
Table 1.
Radiochemical yields of [18F]fluoroarenes from the microfluidic NCA radiofluorination of diaryliodonium tosylates (XC6H4I+C6H4Y TsO−; 9.5 mM) in DMF-0.25% H2O containing K 2.2.2-K2CO3 at 150 °C for 181 s.
| XC6H4I+C6H4Y TsO−
|
Radioactivity recoverya (%) | RCY (%)b
|
||||
|---|---|---|---|---|---|---|
| # | X | Y | [18F]FC6H4X | [18F]FC6H4Y | ||
| 1 | H | H | 96 | 49 | ||
| 2 | H | 2-Me | 61 | 7 | 16 | |
| 3 | H | 2-Br | 87 | 0 | 14 | |
| 4 | 4-OMe | 2-CN | 92 | 0 | 96 | |
| 5 | 4-OMe | 4-CO2Me | 100 | 0 | 90 | |
| 6 | 4-OMe | 4-CN | 100 | 0 | 83 | |
| 7 | 4-OMe | 2-Me | 75 | 0 | 66 | |
| 8 | 4-OMe | 4-COC6H4-4′-Br | 80 | 0 | 49 | |
| 9 | 4-OMe | 3-CO2Et | 100 | 0 | 37 | |
| 10 | 4-OMe | 3-CF3 | 100 | 0 | 29 | |
| 11 | 4-OMe | 3-NO2 | 100 | 0 | 17 | |
| 12 | 4-OMe | 4-Ph | 56 | 0 | 16 | |
| 13 | 4-OMe | 3-CN | 73 | 4 | 11 | |
| 14 | 4-OMe | 4-F | 97 | 0 | 13 | |
| 15 | 4-OMe | 4-OMe | 92 | 12 | ||
% of infused radioactivity collected from the microfluidic apparatus at end of reaction.
Radiochemical yield measured by radio-HPLC as an average from two experiments.
All the remaining salts (4–15) had a 4-methoxyphenyl ring. For this group, yields of [18F]fluoroarenes varied from 96% for the 2-cyano salt (4) to 12% for the symmetrical bis(4-methoxyphenyl) salt (15). Among unsymmetrical salts, only the 3-cyano salt (13) gave any appreciable yield of [18F]4-fluoroanisole (4%). Salts bearing a strong electron-withdrawing substituent in ortho (4) or para position (5, 6, and 8) to iodine gave moderate to high yields of the respectively substituted [18F]fluoroarene (49–96%). Salts having a phenyl (12) or 4-fluoro substituent (14), gave 17 and 13% yields, respectively. The symmetrical bis(4-methoxyphenyl) salt (15) also gave a low yield. Overall, these results show the strong influence of the electron-withdrawing power of a para substituent on reaction outcome. Salts having an electron-withdrawing group in meta position to iodine (9–11 and 13) gave low to moderate yields (11–37%) of the respectively substituted [18F]fluoroarene. The salt having a 2-methyl substituent (7) gave a high yield of 2-[18F]fluorotoluene, showing that the steric bulk of the ortho-methyl group dominates over its negative electronic effect in promoting the reaction.
(4-Cyanophenyl) (4′-methoxyphenyl) iodonium tosylate (6) was one of the most reactive salts in the preliminary screen of reactivity towards [18F]fluoride ion in DMF-0.25% H2O. We chose to test the tolerance of the radiofluorination of 6, in the absence of cryptand, to higher concentrations of unremoved [18O]water originating from the cyclotron production of the [18F]fluoride ion. Reactions were possible in water-DMF (DMF, b.p., 154 °C) at temperatures up to 200 °C because the microfluidic reactor operates at a pressure of 300 psi. Therefore, reactions were conducted in the range 100–200°C. When [18O]water content was in the range 14–42% (v/v), 6 did not react at room temperature, but gave the expected [18F]4-fluorobenzonitrile in moderate yields between 100 and 200 °C (Table 2). Yields increased with temperature and declined with water content, although this decline was quite small on doubling water content from 14 to 28% (v/v) at 200 °C. The efficiency of radioactivity recovery from the apparatus was 88–97%. This high recovery shows that the [18F]fluoride ion was well solubilized without need for adding cryptand, due to the substantial concentration of water in the solvent.
Table 2.
Microfluidic NCA radiofluorination of diaryliodonium tosylate 6 in DMF having various water contents.a
| Water content (% v/v) | Radioactivity recoveryb (%) | RCY of [18F]4-FC6H4CNc (%)
|
||
|---|---|---|---|---|
| 100 °C | 150 °C | 200 °C | ||
| 14 | 97 | 35 | 41 | 44 |
| 28 | 96 | 7 | 34 | 39 |
| 42 | 88 | 1 | 15 | 25 |
Reactions were performed with 9.5 mM salt for 181 s.
% of infused radioactivity collected from the microfluidic apparatus at end of reaction.
Radiochemical yield measured by radio-HPLC as an average from two experiments.
We next decided to test all other diaryliodonium tosylates for reactivity at three temperatures over the range 100 to 200 °C in the presence of 28% (v/v) [18O]water (Table 3), because this water content allowed a considerable portion of the batch of irradiated [18O]water, and therefore considerable activities of [18F]fluoride ion, to be used in the reactions. The irradiated [18O]water was not subjected to any drying process and no cryptand was included in reaction mixtures.
Table 3.
Radiofluorination of diaryliodonium tosylates (XC6H4I+C6H4Y TsO−; 9.5 mM) in DMF-28% [18O]water.a
| XC6H4I+ArY TsO−
|
Radioactivity recovery (%)b | RCYc of [18F]FArY (%)
|
||||
|---|---|---|---|---|---|---|
| # | X | Y | 100 (°C) | 150 (°C) | 200 (°C) | |
| 1 | H | H | 93 | 0 | 8 | 20 |
| 2 | H | 2-Me | 83 | 0 | 14d | 32e |
| 3 | H | 2-Br | 98 | 0 | 1 | 3 |
| 4 | 4-OMe | 2-CN | 96 | 12 | 33 | 50 |
| 5 | 4-OMe | 4-CO2Me | 83 | 3 | 33 | 50 |
| 6 | 4-OMe | 4-CN | 96 | 7 | 34 | 39 |
| 7 | 4-OMe | 2-Me | 97 | 0 | 6 | 11 |
| 8 | 4-OMe | 4-COC6H4-4′-Br | 85 | 2 | 25 | 33 |
| 9 | 4-OMe | 3-CO2Et | 85 | 0 | 7 | 10 |
| 10 | 4-OMe | 3-CF3 | 96 | 0 | 2 | 3 |
| 11 | 4-OMe | 3-NO2 | 78 | 1 | 1 | 4 |
| 12 | 4-OMe | 4-Ph | 76 | 0 | 2 | 9 |
| 13 | 4-OMe | 3-CN | 85 | 0 | 2 | 7 |
| 14 | 4-OMe | 4-F | 90 | 0 | 1 | 2 |
| 15 | 4-OMe | 4-OMe | 83 | 0 | 1 | 2 |
Reactions were performed with 9.5 mM salt for 181 s.
% of infused radioactivity collected from the microfluidic apparatus at end of reaction.
Radiochemical yield measured by radio-HPLC as an average from two experiments.
[18F]Fluorobenzene also produced in 9% RCY.
[18F]Fluorobenzene also produced in 19% RCY.
Salts bearing an electron-withdrawing groups in ortho or para position to iodine (4-6 and 8) and also the meta-nitro-substituted salt (11) gave some yield of the expected [18F]fluoroarene at 100 °C (Table 3). All the salts gave some yield at 150 °C and these yields further increased at 200 °C. Radioactivity recoveries were generally very high (mean ± SD: 88 ± 6%). These results were very reproducible. Thus, for example, reactions of 1 and 6 with different batches of cyclotron-produced [18F]fluoride ion gave completely matching yields.
Diphenyliodonium tosylate (1) gave 20% RCY of [18F]fluorobenzene at 200 °C (Table 3). This result is in line with a report from another laboratory of 32% RCY from the radiofluorination of diphenyliodonium triflate in acetonitrile-25% (v/v) water mixture in a batch reaction at 100 °C for the much longer reaction of 15 min.[33] The 2-methyl analog (2) gave a moderate yield of the expected [18F]2-fluorotoluene (32%), accompanied by a lower yield of [18F]fluorobenzene (19%). The ratio of these two products was appreciably lower (1.56 at 150 °C; 1.68 at 200 °C) than in the experiment with a very low water concentration (2.3) (Table 1). Nevertheless, these results show that the ‘ortho effect’ [18] was still in effect under these more aqueous conditions. In our earlier report on the radiofluorination of diaryliodonium salts in DMF-0.25% water, we obtained evidence that the product ratio complied with the Curtin-Hammett principle [35] in being stable over the reaction course [18], thereby indicating that the product ratio depends only on the difference between the free energies of the two transition states. These transition states were postulated to form by initial interaction of fluoride ion with the positively charged iodine center [18]. The lower product ratios may reflect an influence of solvent on the relative free energies of the two transition states, although we did not test whether the Curtin-Hammett principle also applies under these more strongly aqueous conditions.
Notably, salts having a strong electron–withdrawing substituent in either ortho or para position (4-6 and 8) gave usefully moderate yields (33–50%) of the expected [18F]fluoroarenes at 200 °C. In particular, an electron-withdrawing group in ortho or para position to the hypervalent iodine atom promoted formation of the correspondingly substituted [18F]fluoroarene in moderate yield, whereas the symmetrical salt 11 having an electron-donating 4-methoxy group on each ring, gave an extremely low yield of [18F]4-fluoroanisole. In this respect, these reactions strongly resemble SNAr reactions of fluoride ion with non-hypervalent substrates, which also generally require strong electron-withdrawing groups in ortho or para position [36,37]. Nonetheless, the SNAr reactions on non-hypervalent substrates typically need to be conducted in polar aprotic solvents (e.g., DMSO) at high temperature in the presence of a cryptand or another large cation to be useful, unless a rigorously anhydrous (‘naked’) source of fluoride ion is used [37].
Figure 1 shows the relationships of the yields of substituted [18F]fluororenes for reactions in DMF-0.25% H2O (v/v) (Table 1) and for reactions in DMF-28% H2O (v/v) (Table 3) to the Hammett σP value of the para-substituent [38] in the (4-methoxyphenyl)(aryl)iodonium substrate. RCYs generally increased with σP value. Although, these relationships are far from linear and are closer to ‘S-shape’, they imply that the electronic effects of para-substituents are important with regard to reaction outcomes. The two relationships are remarkably congruent with a strong rise between the σP values for p-F and p-CO2Me substituents.
Figure 1.

Relationships between yields of [18F]fluoroarene and Hammett σP constant for reactions of para-substituted (4-methoxyphenyl)(aryl)iodonium tosylates with [18F]fluoride ion in DMF having low (0.25% v/v) and high (28% v/v) water contents.
Each of the tested meta-substituted salts (9-11 and 13) gave low yields of the expected [18F]fluoroarenes (Table 3). Salt 9 having the most bulky substituent (m-CO2Et) gave the highest radiochemical yield of substituted [18F]fluoroarene among this set. For these salts, yields did not increase with Hammett σM value, and a mix of weak electronic and steric effects is likely in effect (Figure 2).
Figure 2.

Relationships between the yields of [18F]fluoroarene and Hammett σM constant for reactions of meta-substituted (4-methoxyphenyl)(aryl)iodonium tosylates with [18F]fluoride ion in DMF having low (0.25% v/v) and high (28% v/v) water contents.
Our results attest to the generally high reactivity of diphenyliodonium salts towards NCA [18F]fluoride ion, even when this ion is weakly nucleophilic because of extensive hydration. We previously determined the Arrhenius activation energy for the reaction of [18F]fluoride ion with (2-methylphenyl)(phenyl)iodonium chloride in DMF-0.25% H2O medium and obtained a value of 18.3 kcal/mol [18]. Microhydration of chloride ion by as few as five water molecules has been calculated to increase the activation energy for reaction with chloromethane in chloroform by 10 kcal/mol [39]. A similar influence of microhydration of fluoride ion on its nucleophilicity may be reasonably expected, as evidenced by the effect of hydration of fluoride ion on rates of reaction with 4-chlorobenzonitrile [31]. The ability to produce NCA [18F]fluoroarenes rapidly from the reaction of [18F]fluoride ion with diaryliodonium salts in substantially aqueous medium is therefore remarkable. Here our observations suggest that reactions do become more difficult as the proportion of water in the reaction solvent is increased (Table 2), but that the increases in activation barriers are not so great as to preclude reactions (Table 3).
Experimental
Methods
NCA [18F]fluoride ion was obtained through the 18O(p,n)18F nuclear reaction by irradiating [18O]water (95 atom %, 2.0 mL) for 90–120 min with a proton beam (16.5 MeV; 20 μA) produced from a PETrace cyclotron (GE Medical Systems; Milwaukee, WI). Radioactivity was measured with a calibrated dose ionization chamber (Atomlab 300; Biodex Medical Systems Inc., Shirley, NY). Radiochemistry was performed in a NanoTek microfluidic apparatus (Advion, Ithaca, NY) housed within a lead-shielded hot-cell. See ESI for the configuration of the apparatus. Details of the operation of the apparatus are described in our previous publications [18,19]. Crude radioactive reaction mixtures were analyzed on a HPLC apparatus comprising a solvent module (System Gold 126; Beckman Coulter, CA) coupled with a UV absorbance detector (Model 168 or Model 166; Beckman Coulter) and a radioactivity detector (PMT, Flow-count; Bioscan, Washington, DC). Diaryliodonium salts used in this study were prepared as described in ESI.
Radiochemistry
Screen of dirayliodonium tosylates for reactivity towards [18F]fluoride ion
Aqueous NCA [18F]fluoride ion solution (100–200 μL, 1.85–3.7 GBq) was transferred to a glass V-vial (5 mL) that had been loaded with a solution of K2CO3/K 2.2.2 (100 μL stock solution of 0.5 mg K2CO3 and 5.0 mg K 2.2.2 in 9:1 MeCN and H2O mixture) and MeCN (600 μL), and placed in a heating well inside a lead-shielded hot-cell. Water was removed by azeotropical distillation with acetonitrile under N2 gas flow (200 mL/min) at 110 °C. The addition of MeCN (650 μL) and heating cycle were repeated three more times. The generated anhydrous NCA [18F]fluoride ion-K+-K 2.2.2 reagent was redissolved in DMF containg 0.5% H2O (450 μL). This solution was then loaded into storage loop 1 of the microfluidic apparatus. A solution of the test diaryliodonium tosylate in anhydrous DMF (19 mM; 275 μL) was loaded into storage loop 2. A quantity of each solution (20 μL) was infused simultaneously at 5 μL/min into the 4 m-length coiled silica glass tube micro-reactor (31.4 μL) held at 150 °C. The effluent from the micro-reactor was quenched by dilution with H2O-MeCN (1: 1 v/v, 0.5 mL).
Evaluation of the reactivity of a second iodonium salt with the same batch of [18F]fluoride ion was performed identically after cleaning storage loop-2 with anhydrous DMF and loaded the loop with the new test iodonium salt solution.
Radiofluorination of diaryliodonium tosylates in DMF-[18O]water mixtures
A mixture (275 μL) of irradiated [18O]water (200 μL; 98 atom %) containing [18F]fluoride ion (1.85–3.7 GBq), DMF (200 μL) and aqueous K2CO3 solution (0.023 M, 50 μL) was loaded into storage loop 1 of the microfluidic apparatus. A solution of the test diaryliodonium tosylate in DMF (19 mM; 275 μL) was loaded into storage loop 2. A quantity of each solution (20 μL) was each infused simultaneously at 5 μL/min into the 4 m-length coiled silica glass tube micro-reactor (31.4 μL) held at a set temperature. The effluent from the micro-reactor was quenched by dilution with H2O-MeCN (1: 1 v/v, 0.5 mL) at room temperature.
Evaluation of the reactivity of a second iodonium salt with the same batch of [18F]fluoride ion was performed identically after cleaning storage loop-2 with anhydrous DMF and loaded the loop with the new test iodonium salt solution.
Analytical HPLC of reaction mixtures
Aliquots of quenched reaction mixtures (20–30 μL) were analyzed with reversed phase radio-HPLC on a Luna C18 column (250 × 4.6 mm i.d., 10 μm; Phenomenex; Torrance, CA) eluted at 1.75 mL/min with a mixture of MeCN and H2O. The MeCN content of the mobile phase was initially 60% v/v. At 1 min after analyte injection this percentage was increased linearly over 7 min to 90% and held at this value until the end of analysis. [18F]Fluoroarenes were identified from their comobility with the respective non-radioactive fluoro-compounds, which eluted with the following retention times: [18F]4-fluorobenzonitrile (3.1 min); [18F]2-fluorobenzonitrile (3.3 min); [18F]3-fluoronitrobenzene (3.7 min); [18F]4-fluoroanisole (4.1 min); [18F]1,4-difluorobenzene (4.2 min); [18F]methyl 4-fluorobenzoate (4.0 min); [18F]fluorobenzene (4.2 min); [18F]ethyl 3-fluorobenzoate (5.2 min); [18F]3-fluorobenzonitrile (5.4 min); [18F]2-fluorotoluene (5.4 min); [18F]2-bromo-fluorobenzene (5.5 min); [18F]1-fluoro-3-(trifluoromethyl)benzene (5.8 min); [18F]4-fluorobiphenyl (7.3 min) and [18F]4-bromo-4′-fluorobenzophenone (7.4 min). The radiochemical yield (RCY) for each identified [18F]fluoroarene was calculated without decay-correction as the percentage of the total radioactivity peak area represented by the [18F]fluoroarene in the recorded radiochromatogram. In selected cases, yields were confirmed by collecting and measuring each radioactive fraction in a calibrated ionization chamber. The percentage recovery of radioactivity from the apparatus was estimated from the amount of radioactivity infused into the micro-reactor and the total radioactivity in the micro-reactor effluent (measured in the calibrated ionization chamber).
In order to assess the effect of irradiated [18O]water content in the reaction solvent on RCYs of [18F]fluoroarenes the percentage of irradiated target water in the solution loaded in storage loop 1 was varied, while keeping the final concentration of K2CO3 at 2.6 mM. Reactions were then performed and analyzed as described above.
Conclusions
The outcomes for reactions of diaryliodonium salts with [18F]fluoride ion in DMF-28% [18O]water in the absence of cryptand were strongly influenced by stereoelectronic effects, particularly an electronic effect for para substituted salts and a steric effect for ortho substituted salts. Our findings that [18F]fluoroarenes having an alkyl ortho substituent or ortho or para electron-withdrawing group, may be produced without need to remove irradiated [18O]water or to add cryptand may be attractive in some radiotracer production settings, particularly as this method saves time, avoids any need for automated drying of cyclotron-produced [18F]fluoride ion, and avoids substantial loss of radioactivity through adsorption to hardware surfaces.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health (NIMH). The authors are grateful to the NIH Clinical Center PET Department (Chief, Dr. Peter Herscovitch) for the cyclotron production of fluorine-18.
Footnotes
Electronic Supplementary Information (ESI) available: Syntheses and characterizations of diaryliodonium salts. See DOI: 10.1039/b000000x/
References
- 1.Phelps ME. Proc Natl Acad Sci, USA. 2000;97:9226–9233. doi: 10.1073/pnas.97.16.9226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ametamey SM, Honer M, Schubiger PA. Chem Rev. 2008;108:1501–1516. doi: 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]
- 3.Gibson RE, Burns HD, Hamill TG, Eng WS, Francis BE, Ryan C. Curr Radiopharm Des. 2000;6:973–989. doi: 10.2174/1381612003399987. [DOI] [PubMed] [Google Scholar]
- 4.Lee CM, Farde L. Trends Pharm Sci. 2006;27:310–316. doi: 10.1016/j.tips.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 5.Matthews PM, Rabiner I, Gunn R. Curr Opin Pharm. 2011;11:501–507. doi: 10.1016/j.coph.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 6.Miller JM, Kumar D, Mann JJ, Parsey RV. Curr Radiopharm. 2008;1:12–16. [Google Scholar]
- 7.Cai LS, Lu SY, Pike VW. Eur J Org Chem. 2008:2853–2873. [Google Scholar]
- 8.Pike VW. Trends Pharm Sci. 2009;30:431–440. doi: 10.1016/j.tips.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pike VW, Aigbirhio FI. J Chem Soc, Chem Commun. 1995;21:2215–2216. [Google Scholar]
- 10.Shah A, Pike VW, Widdowson DA. J Chem Soc, Perkin Trans. 1998;1:2043–2046. [Google Scholar]
- 11.Basuli F, Wu H, Li C, Shi Z, Sulima A, Griffiths GL. J Label Compd Radiopharm. 2011;54:633–636. [Google Scholar]
- 12.Chun J-H, Pike VW. Eur J Org Chem. 2012:4541–4547. doi: 10.1002/ejoc.201200695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chun JH, Pike VW. Chem Commun. 2012;48:9921–9923. doi: 10.1039/c2cc35005j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang MR, Kumata K, Suzuki K. Tetrahedron Lett. 2007;48:8632–8635. [Google Scholar]
- 15.Telu S, Chun JH, Siméon FG, Lu S, Pike VW. Org Biomol Chem. 2011;9:6629–6638. doi: 10.1039/c1ob05555k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moon BS, Kil HS, Park JH, Kim JS, Park J, Chi DY, Lee BC, Kim SE. Org Biomol Chem. 2011;9:8346–8355. doi: 10.1039/c1ob06277h. [DOI] [PubMed] [Google Scholar]
- 17.Lee BC, Dence CS, Zhao H, Parent EE, Welch MJ, Katznellenbogen JA. Nucl Med Biol. 2009;36:147–153. doi: 10.1016/j.nucmedbio.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chun JH, Lu S, Lee YS, Pike VW. J Org Chem. 2010;75:3332–3338. doi: 10.1021/jo100361d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chun JH, Lu S, Pike VW. Eur J Org Chem. 2011:4439–4447. doi: 10.1002/ejoc.201100382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin-Santamaría S, Carroll MA, Carroll CM, Carter CD, Pike VW, Rzepa HS, Widdowson DA. Chem Commun. 2000:649–650. [Google Scholar]
- 21.Ross L, Ermert J, Hocke C, Coenen HH. J Am Chem Soc. 2007;129:8018–8025. doi: 10.1021/ja066850h. [DOI] [PubMed] [Google Scholar]
- 22.Carroll MA, Jones C, Tang SL. J Label Compds Radiopharm. 2007;50:450–451. [Google Scholar]
- 23.Reed CD, Launay GG, Carroll MA. J Fluorine Chem. 2012;143:231–237. [Google Scholar]
- 24.Graskemper JW, Wang BJ, Qin LL, Neumann KD, DiMagno SG. Org Lett. 2011;13:3158–3161. doi: 10.1021/ol201080c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carroll MA, Nairne J, Smith G, Widdowson DA. J Fluorine Chem. 2007;128:127–132. [Google Scholar]
- 26.Guillaume M, Luxen A, Nebeling B, Argentini M, Clark JC, Pike VW. Appl Radiat Isot. 1991;42:749–762. [Google Scholar]
- 27.Landini D, Maia A, Rampoldi A. J Org Chem. 1989;54:328–332. [Google Scholar]
- 28.Zhan CG, Dixon DA. J Phys Chem A. 2004;108:2020–2029. [Google Scholar]
- 29.Kemp DD, Gordon MS. J Phys Chem A. 2005;109:7688–7699. doi: 10.1021/jp058086b. [DOI] [PubMed] [Google Scholar]
- 30.Trumm M, Martínez YOG, Réal F, Masella M, Vallet B, Schimmelpfennig B. J Chem Phys. 2012;136:044509. doi: 10.1063/1.3678294. [DOI] [PubMed] [Google Scholar]
- 31.Pliego JR, Jr, Piló-Veloso D. Phys Chem Chem Phys. 2008;10:1118–1124. doi: 10.1039/b716159j. [DOI] [PubMed] [Google Scholar]
- 32.Alexoff DL, Fowler JS, Gatley SJ. Appl Radiat Isot. 1991;42:1189–1193. doi: 10.1016/0883-2889(91)90195-7. [DOI] [PubMed] [Google Scholar]
- 33.Wadsworth HJ, Devenish T. PCT WO2005/097713. 2005:A1.
- 34.Pascali G, Del Carlo S, Saccomanni G, Manera C, Salvadori PA. Q J Nucl Med Mol Imaging. 2012;56(Suppl 1):12. [Google Scholar]
- 35.Seeman JI. Chem Rev. 1983;83:83–134. [Google Scholar]
- 36.Angelini G, Speranza M, Wolf AP, Shiue CY, Fowler JS, Watanabe M. J Label Compd Radiopharm. 1984;21:1223–1225. [Google Scholar]
- 37.Sun H, Di Magno SG. Angew Chem Int Ed. 2006;45:2720–2725. doi: 10.1002/anie.200504555. [DOI] [PubMed] [Google Scholar]
- 38.Hansch C, Leo A, Taft RW. Chem Rev. 1991;91:165–195. [Google Scholar]
- 39.Nelson KV, Benjamin I. J Chem Phys. 2009;130:194502. doi: 10.1063/1.3138902. [DOI] [PubMed] [Google Scholar]
- 40.Charlton M, Carroll MA. J Label Compd Radiopharm. 2013;56:57. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.