

Abstract
The α-tocopherol transfer protein (α-TTP) is a liver protein that transfers α-tocopherol (vitamin E) to very-low-density lipoproteins (VLDLs). These VLDLs are then circulated throughout the body to maintain blood α-tocopherol levels. Mutations to the α-TTP gene are associated with ataxia with vitamin E deficiency (AVED), a disease characterized by peripheral nerve degeneration. In this study, molecular dynamics simulations of the E141K and R59W disease-associated mutants were performed. The mutants displayed disruptions in and around the ligand-binding pocket. Structural analysis and ligand docking to the mutant structures predicted a decreased affinity for α-tocopherol. To determine the detailed mechanism of the mutation-related changes, we developed a new tool called ContactWalker that analyzes contact differences between mutant and wild-type proteins and highlights pathways of altered contacts within the mutant proteins. Taken together, our findings are in agreement with experiment and suggest structural explanations for the reduced ability of the mutants to bind and carry α-tocopherol.
Keywords: α-tocopherol, α-tocopherol transfer protein, antioxidant, AutoDock Vina, AVED, binding affinity, contact network, depth-first search, docking, MD, molecular-dynamics simulations, Vitamin E, VLDL
INTRODUCTION
α-Tocopherol transfer protein (α-TTP) transfers α-tocopherol, a strong antioxidant and a form of vitamin E, to very-low-density lipoproteins (VLDLs). These lipoproteins, located in the liver, are then circulated throughout the body to maintain blood α-tocopherol levels. Vitamin E occurs in multiple forms including α-, β-, γ-, and δ-tocopherol. Each form has a hydrophobic tail with three chiral centers, and α-TTP preferentially binds RRR-α-tocopherol (Figure 1a).1
Figure 1.
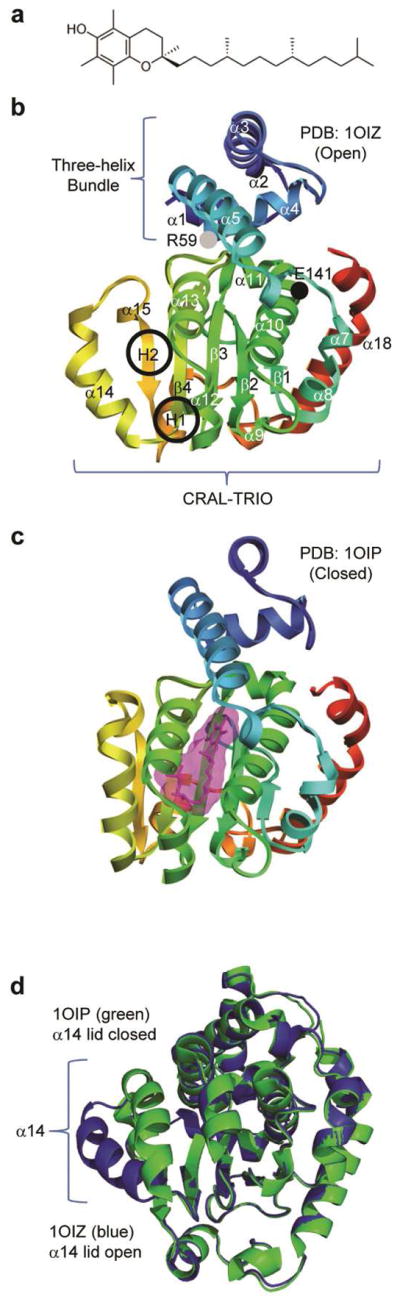
Molecular structures of vitamin E and the different forms of α-TTP. (a) RRR-α-tocopherol (vitamin E). (b) In the unbound structure (PDB code: 1OIZ), a ‘lid’ formed by the α14 region is open, allowing entrance to a hydrophobic binding pocket. The CRAL-TRIO domain is indicated in brackets. The black and gray circles indicate the E141K and R59W mutation regions, respectively. The two hinge regions are circled and labeled as H1 and H2. (c) In the bound conformation (PDB code: 1OIP), the lid is closed, enclosing the binding pocket. (d) 1OIZ (blue) and 1OIP (green) overlaid and aligned by Cα RMSD. The Cα RMSD between the two structures excluding α14 was 0.5 Å
Patients with mutations in the α-TTP gene have a reduced ability to transfer α-tocopherol to VLDLs and, as a result, are unable to maintain blood α-tocopherol levels. This condition, known as ataxia with vitamin E deficiency (AVED), is associated with peripheral nerve degeneration. Although the link between α-tocopherol deficiency and AVED-associated nerve damage is not fully understood, it is likely that α-tocopherol protects tissues from oxidative stress. Increasing dietary consumption of vitamin E can be an effective treatment for AVED.2
α-TTP belongs to the SEC14-like family of proteins and is composed of two domains: an N-terminal three-helix bundle and a C-terminal CRAL-TRIO domain (Figure 1b). The latter domain is used for transporting hydrophobic molecules through hydrophilic environments; other SEC14-like proteins include human supernatant protein factor and Cellular Retinaldehyde-Binding Protein (CRALBP). The CRAL-TRIO domain has a ligand binding pocket, entry to which is controlled by a swinging ‘lid’. Investigations of SEC14 and CRALBP indicate that the lid is ‘hinged’ by two regions of residues: residues 242–244 and 263–265 for CRALBP3 and residues 212–213 and 239–240,242 for Sec14.4 Based on sequence alignment of these proteins3, these regions fall in the vicinity of residues 194–201 and 220–224 for α-TTP. These regions are referred to as hinge 1 (H1) and hinge 2 (H2), respectively (Figure 1b). In α-TTP, the lid is formed by residues 198–2211, the central structure of which is the helix α14.
Meier et al. crystallized α-TTP in both the lid-open (PDB code: 1OIZ) (Figure 1b) and lid-closed (PDB code: 1OIP) (Figure 1c) forms.1 When α-TTP is not bound to α-tocopherol, the lid is open and the ligand binding pocket is accessible. When α-TTP is bound to α-tocopherol, the lid is closed and the binding pocket is separated from the external environment. The open and closed forms are overlaid in Figure 1d.
In this study we investigated two mutants of α-TTP: one replaces a Glu with a Lys at residue 141 (E141K) and the other replaces an Arg with a Trp at residue 59 (R59W). Both mutants alter highly conserved residues5 and are associated with severe clinical effects.1,6,7 Although experimental binding, ligand transfer, and urea denaturation data have been reported for these mutants7, experimentally determined structures of these mutants are not available. Consequently, we have used all-atom molecular dynamics (MD) simulations to investigate the structural effects of these mutations. This protein has not been the subject of previous MD simulations to our knowledge, but Ryan et al4 and Schaff et al8 have performed MD simulations of related Sec14 and Sfh1 proteins. In addition to standard structural analyses of the αTTP simulations, docking calculations were also performed to assess the impact of mutations on the protein’s ability to bind RRR-α-tocopherol. Finally, to characterize how effects are propagated between the mutation site and the binding site, we created a new tool called ContactWalker. Analysis of these pathways provides a molecular description of the mutation-associated structural changes resulting from the E141K and R59W mutations.
METHODS
Structural Models
The 1.88 Å resolution crystal structure of α-TTP was obtained from the Protein Data Bank9 (PDB) (http://www.pdb.org) (PDB code 1OIZ)1 and was used for the starting structure for the wild type and mutant simulations. The unbound, apo structure was used so we could investigate mutation-induced changes in the intrinsic structural and dynamic behavior of the protein to avoid ligand-induced structural bias. Starting structures for both mutants were constructed by making the appropriate amino acid substitutions to the wild-type protein. The energies of the resulting structures were minimized for 100 steps in vacuo using the ENCAD simulation package10 and the Levitt et al. force field.11 A different 1.95 Å resolution crystal structure of α-TTP (PDB code 1OIP)1 crystallized with bound α-tocopherol and the lid ‘closed’ was used as a ligand docking baseline. The model of RRR-α-tocopherol used for docking studies was extracted from the 1OIP crystal structure.
Molecular dynamics simulations and analysis
MD simulations were performed using the in lucem molecular mechanics (ilmm) software package12 using a previously published protocol and potential energy function.11,13,14,15 Starting structures were prepared for MD using 1000 steps of steepest descent minimization and then solvated with flexible F3C water molecules14 in a periodic box with walls located at least 10 Å from all protein atoms. The solvent density was set to the experimentally determined value for water at 37°C, 0.993 g/mL.16 The solvent energy was minimized for at least 500 steps before the solvent was heated again for 1 ps. The solvent energy was then minimized for an additional 500 steps and followed by an energy minimization of the entire system for 500 steps. Atomic velocities were assigned from a Maxwellian distribution at low temperature and then brought to the target temperature of 37°C (310 K). Thereafter we used the NVE microcanonical ensemble, where the box volume, number of particles, and total energy are fixed. A force-shifted nonbonded cutoff range of 10 Å was used for nonbonded interactions17 and the interaction list was updated every 2 steps. Simulation steps were 2 fs and the structures were saved every 1 ps. The temperature was set to 37°C and the simulations were performed at neutral pH (Asp and Glu negatively charged, Arg and Lys positively charged, and His neutral). Three independent simulations of wild type and each mutant were performed for at least 51 ns. These simulations were performed as part of the SNP (single-nucleotide polymorphism) thrust of our Dynameomics project,18,19 an ongoing project to determine the native state dynamics and unfolding pathways of all known protein folds. α-TTP’s 3-helix bundle N-terminal domain is represented in fold rank 89 and the C-terminal CRAL-TRIO domain is represented in fold rank 1251 in the 2009 release set of our Consensus Domain Dictionary20,21 (CDD). Protein images were created using Pymol22 and UCSF Chimera.23
Inter-residue Contacts
Inter-residue contact occupancies were calculated at 1 ps granularity using heavy-atom contacts from time steps greater than 25 ns. Contact distance thresholds were 5.4 Å for carbon-carbon contacts and 4.6 Å for all other heavy-atom contacts; residues were considered to be in contact if at least one inter-atomic contact was within the appropriate threshold. Contacts between neighboring residues were ignored. The “residue occupancy difference” was calculated as the difference in occupancy between each wild-type residue and the corresponding mutant residue. As a result, negative values indicated greater prevalence in the mutant (mutant stabilization) and positive values reflected greater prevalence in wild type (mutant destabilization).
ContactWalker
Because of the dynamic nature of proteins, inter-atomic contact distances change over time and even stable contacts oscillate around some mean distance. As a result, searching for structurally important contact changes between wild type and mutant proteins can be challenging. To address this challenge, we built ContactWalker, a tool that measures the difference in inter-atomic contact occupancies between wild type and mutant proteins.
ContactWalker calculates contact occupancy differences between a set of one or more wild-type simulations and a set of one or more mutant simulations. It uses the minimum demonstrated occupancy change between the wild type and mutant simulations to avoid over-estimating the significance of the change in occupancy. For example, the R54:Q145 contact had occupancy values of 24%, 53%, and 61% for the three wild-type simulations and 100%, 100%, and 98% for the three E141K simulations. The minimum demonstrated occupancy change between wild type and the E141K mutant was −37% (61%–98%). Using the minimum demonstrated occupancy change provided the most conservative measure of significant changes in strength of protein contacts.
Contact Networks
A residue whose contacts have been altered, either by stabilization or destabilization, is considered to be a disrupted residue. Preliminary observations found that disrupted residues tended to exist in connected networks rather than in isolation. ContactWalker can be used to visualize these networks, highlighting pathways of disrupted contacts (disruption pathways) and building on discretized, non-interactive network depictions such as those reported by Schmidlin et al24.
To do this, it builds a connected graph to represent all the contact changes between the wild type and mutant proteins. Graph nodes represent residues and graph edges represent nonbonded contacts with edge weights proportional to the change in contact occupancy. Backbone peptide bonds are inserted with an occupancy-change value of zero.
Following construction of the contact graph, ContactWalker identifies the target residues. These are residues among which ContactWalker looks for networks of disrupted contacts. Residues can be manually specified if a region of interest is already known or they can be automatically specified by selecting those residues with changes in contact occupancy that lie outside of some threshold. If the automatic method is used, additional manual targets may be specified as well.
Once the target residues have been identified, the user specifies the search parameters. These include options such as maximum search depth (the maximum number of contacts to search before trying another path), and a minimum edge value to determine if an edge is eligible for search. A cutoff in terms of absolute occupancy may also be specified. Backbone peptide bonds are always available for traversal.
Next, ContactWalker performs the actual search. A depth-first search (DFS) is performed between every pair of target nodes to find pathways of significant occupancy change. A DFS is a graph-traversal algorithm that searches a graph by moving from parent node to child node, often limited to a maximum number of steps, before backing out and trying a new path. In this scenario, this limits the search to a maximum number of inter-residue contacts between significant residues.
In the final step, the output graph is laid out and rendered. The output graph is a subset of the original connectivity graph containing only those edges and nodes that meet the user’s significance criteria. Graph images are rendered using the Graphviz graph layout tool25 and the built-in scalable force directed placement graph layout algorithm.26 The graph layout is determined by the connectivity patterns of significantly-changed edges, not necessarily the proteins’ three-dimensional structure. Approximate structural regions of the protein are highlighted on the graph. Edge colors indicate the degree of occupancy change along a green (mutant stabilized) to orange (mutant destabilized) spectrum and edge size indicates the magnitude of occupancy change. Node color designates secondary structure per the legend. ContactWalker also outputs a Pymol22 script to visualize the contact change data mapped onto the protein structure.
Ligand Docking
To predict the effect of distortions of the binding site on the binding affinity of α-tocopherol, docking calculations of RRR-α-tocopherol were performed on MD structures taken between 25 and 51 ns. Docking energies and conformational pose calculations were made using AutoDock Vina.27 Of the 26000 structures between 26 ns and 51 ns, 260 (1%) were selected using simple random sampling without replacement. Sample mean, 95% confidence interval (CI), and the sample standard deviation were calculated using a two-tailed t-test, α = 0.05, and either 260 or 780 degrees of freedom for individual simulation data or aggregated data, respectively. Docking energies were reported as (sample mean) ± (stdev). Additionally, a baseline docking energy was established by docking RRR-α-tocopherol into the 1OIP crystal structure.
Cα RMSD and Cα RMSF
Cα root-mean-squared deviation (Cα RMSD) values were calculated using only α-carbons and excluded both termini (residues 9–23 and 266–274) and the α14 region (residues 201–213). Cα root-mean-squared fluctuation (Cα RMSF) data were calculated using MD structures beginning at 25 ns, used only α-carbons, and also excluded both termini. The α14 region was included in the Cα RMSF calculations because visual inspection of the trajectories showed that the lid had closed by approximately 10 ns.
α-Tocopherol Headgroup-Coordinating Residues
The α-Tocopherol headgroup-coordinating residues were defined as those residues with at least one atom within 5.4 Å of the headgroup of the vitamin E molecule. Water-coordinating residues were defined as residues with a nitrogen or oxygen atom within 4.6 Å of a water molecule. Calculations were performed using Pymol22 and the 1OIP PDB structure1 of αTTP.
RESULTS AND DISCUSSION
In general, all simulations maintained the overall binding-pocket structure and did not undergo significant unfolding; this is consistent with the urea denaturation experiments reported by Morley et al7 that showed a 0.4 M change in C50 concentrations for the R59W and E141K mutants relative to the WT. The Cα root-mean-squared deviation (Cα RMSD) of all three sets of simulations indicated that the proteins were relatively stable between 2–3 Å Cα RMSD with the exceptions that each mutant had one simulation with Cα RMSD values of 4.0 Å (simulation 1 in each case) (Figure 2). Cα root-mean-squared fluctuation (Cα RMSF) analysis of all three proteins showed a similar trend (Figure 3); each protein demonstrated Cα RMSF values around 1 Å in regions of secondary structure and in each case the simulation with higher Cα RMSD values also showed higher Cα RMSF values, often in the vicinity of α13 and α14.
Figure 2.

Cα RMSD values over time for all three proteins. Values were calculated using α-carbons excluding both termini and α14. (a) Wild type. (b) E141K. (c) R59W.
Figure 3.

Cα RMSF calculated using α-carbons and excluding both termini. (a) Black and gray bands indicate α-helices and β-sheets respectively, blue squares indicate mutation sites, and red bands indicate hinge regions 1 and 2 (labeled H1 and H2). (b) Wild type. (c) E141K mutant. (d) R59W mutant.
α-Tocopherol docking studies resulted in a range of docking energies from −6.5 to −10.2 kcal/mol (Figure 4). The 95% confidence intervals for the mean potential energies were 0.1 kcal/mol and 0.0 kcal/mol for the per-simulation and aggregated docking data, respectively. For reference, the potential energy of docking α-tocopherol into the 1OIP ‘closed’ structure was − 10.8 kcal/mol, and the Cα RMSD between the 1OIZ and 1OIP crystal structures excluding α14 was 0.5 Å. Thus, this control suggests that the method can correctly identify and recover both the experimentally determined structure of the complex and its correct binding interactions. Per-simulation α-tocopherol docking analyses demonstrated that both the wild type and the mutant proteins were capable of a range of docking energies (Figures 4a, b and c). In aggregate, wild type and the E141K mutant had the same mean docking energy (−8.5 ± 0.7 and −8.5 ± 0.6 kcal/mol, respectively) and the R59W mutant performed less favorably (−8.1 ± 0.6 kcal/mol) (Figure 4d). Converting these potential energies to dissociation constants (Kd) resulted in 1000 nM for wild type and the E141K mutant and 1900 nM for the R59W mutant. This is approximately in keeping with the findings of Morley et al.7 who reported 2X and 4X increases in Kd for the E141K and R59W mutants, respectively.
Figure 4.

Histograms of α-tocopherol docking potential energies. AutoDock Vina was used to dock α-tocopherol into 260 randomly-selected structures from each simulation. Mean potential energies and standard deviations are shown in the legends. (a, b, c) Per-simulation histograms of wild type, E141K mutant, and R59W mutant, respectively. (d) Aggregated energies for each protein.
Wild-type Simulations
All three wild-type trajectories stabilized between 2.5–3 Å Cα RMSD (Figure 2a), and simulation 2 displayed more structural fluctuation than the other two (Figures 3b and 5). In all three wild-type simulations, α14 underwent a swinging motion from the lid-open position to the lid-closed position; Ryan et al4 reported a similar conformational change in MD simulations of the related Sec14 protein. The β-sheet wall of the binding pocket remained stable in all simulations while the α-helix wall exhibited more dynamic behavior (Figures 3b and 5). Despite these similarities, the three wild-type simulations each behaved slightly differently; to better characterize the range of behaviors of the wild-type protein, we analyzed each simulation separately.
Figure 5.

Representative wild-type structures rendered as tubes. Tube radius and color reflect Cα RMSD (left column) and Cα RMSF (right column). E82, I83 and K178 are colored per the legend and rendered as spheres. (a) Simulation 1. (b) Simulation 2. (c) Simulation 3. Note the separation of K178 from E82 and I83 in simulation 2.
As shown in Figure 5a, structures from simulation 1 did not deviate far from the minimized crystal structure with the exceptions of the α3–α4 loop, α5, α7, α13, N227, and the α17–α18 loop. Simulation 1 also provided the most favorable docking conformations of the three wild-type simulations with a mean potential energy of −9 ± 0.5 kcal/mol (Figure 4a). Because of its stability and favorable docking performance, wild-type simulation 1 was used as a structural baseline for subsequent contact comparisons.
Simulation 2 was more dynamic than either simulation 1 or simulation 3, particularly near α14 (Figures 3b and 5b), and the mean docking energy (−8.4 ± 0.6 kcal/mol) was between the energies of the other two simulations (Figure 4a). Contact occupancy comparisons of simulations 1 and 2 indicated that the structures in simulation 2 were missing several binding-pocket contacts that were present in simulation 1 including E82:K178 (100% difference) and I83:K178 (66% difference) (Figure 5b). These two contacts tethered α5 to α13 and helped maintain the structure of the α-helix wall of the binding pocket. Both E82 and I83 established alternative stabilizing contacts including 100% occupancy contacts with R134. K178 did not establish significant alternative stabilizing contacts and remained destabilized relative to simulation 1 (1.5 Å Cα RMSF in simulation 2 vs. 0.75 Å Cα RMSF in simulation 1). Structures from simulation 2 showed, relative to simulation 1, a general lack of contacts between α12, α13, and α14 lid and hinge regions. Of the 19 contacts in this region with occupancy differences greater than 50%, six were stabilized and 13 were destabilized. This difference in occupancy is reflected in the increased mobility and fluctuation of α14 in simulation 2 (Figures 3b and 5b). The internal structure of α14 saw a similar destabilization; of the 13 contacts within α14 with occupancy differences greater than 50%, three contacts were stabilized and ten contacts were destabilized.
Simulation 3 had relatively low Cα RMSF values, similar to those of simulation 1 (Figures 3b and 5c), but it also had the least favorable docking energy of the three wild-type simulations (−8.1 ± 0.5 kcal/mol) (Figure 4a). In total, 27 contacts had occupancy differences greater than 99% relative to simulation 1, the majority of which were in the vicinity of the hinge and lid region formed by α12, α13, and α14. Also, like simulation 2, the I83:K178 contact (66% occupancy) was not present in simulation 3. However, unlike simulation 2, the E82:K178 contact was intact, retaining some of the α13 tethering. Furthermore, also unlike simulation 2, K178 formed alternative stabilizing contacts with L183 (93%). Thus, K178 was not completely disconnected from E82 and I83 nor was it left in a destabilized state, resulting in slightly lower Cα RMSF values (1.1 Å vs. 1.5 Å in simulation 2).
E141K Simulations
Overall, the E141K simulations were fairly stable and did not undergo any significant unfolding. As indicated by the Cα RMSD calculations (Figure 2b) simulations 2 and 3 stabilized between 2–2.5 Å while simulation 1 deviated from the starting structure by as much as 4 Å Consistent with the Cα RMSD data, the Cα RMSF data indicated that simulation 1 of the E141K mutant was more dynamic than simulations 2 or 3 (Figures 3c and 6).
Figure 6.

Representative E141K structures rendered as tubes. Tube radius and color reflect Cα RMSD (left column) and Cα RMSF (right column). The K141 mutation site is rendered as magenta spheres. (a) Simulation 1. (b) Simulation 2. (c) Simulation 3.
Comparing contact occupancies of all three E141K simulations against those of the wild-type simulation 1 baseline, we found 15 contacts with a minimum demonstrated occupancy change of greater than 95%. Of these, three were stabilizations of the hinge 2 region (K217:R221, K217:E220, and E216:E220), five were destabilizations that included the lid (F213:K217, F213:E216, L214:K217, P212:E216, and K211:T215), one tethered α13 to α14 (T184:T215), and one untethered α5 from α13 (W76:S186). All ten of these contact changes were also observed in the wild type simulations. However, their presence in the E141K mutant was both more common (all three E141K simulations) and, in the case of hinge 2 region and α13–α14 tethering, larger in magnitude (Table 1).
Table 1.
Occupancy Differences in Contacts Common to Both E141K and Wild Type
| Residue 1 | Residue 2 | WT1 vs. WT2a | WT1 vs. WT3a | WT1 vs. E141K (all)a |
|---|---|---|---|---|
| W76 | S186 | 68% | 97% | 97% |
| K211 | T215 | 96% | 97% | 95% |
| P212 | E216 | 99% | 99% | 98% |
| F213 | E216 | 100% | 100% | 100% |
| F213 | K217 | 100% | 100% | 100% |
| L214 | K217 | 100% | 98% | 100% |
| T184 | T215 | −43% | −49% | −96% |
| K217 | E220b | −24% | −4% | −99% |
| K217 | R221b | −86% | −57% | −99% |
| E216 | E220b | −20% | 0% in both WT1 and WT3) | −100% |
Negative numbers indicate stabilization in the compared structure (underlined).
Hinge 2 region.
To isolate the specific effects of the E141K mutation from the inherent propensities of the protein, we compared all three E141K simulations against all three wild-type simulations. Only nine contacts were observed that had a minimum demonstrated occupancy difference of greater than 20% (Table 2). Of these, four of the nine contacts were in or near hinge 2. Disruption pathways between the mutation site and the hinge 2 region (Figure 7) were found by increasing the sensitivity of ContactWalker (significance threshold: 9%, allowable threshold: 6%, search depth: 2, absolute occupancy cutoff: 0.01). The primary contact change at the mutation site was the R54:K141 contact (92% loss). As indicated in the figure, the disruption pathway included α5, α10, α13, and the hinge 2 region.
Table 2.
E141K Occupancy Change Magnitudes > 20%
| Residue 1 | Residue 2 | Occupancy Difference |
|---|---|---|
| E216 | E220 | −80% |
| K217 | E220 | −75% |
| A10 | D60 | −53% |
| T184 | T215 | −47% |
| R54 | Q145 | −37% |
| L183 | I218 | −37% |
| A10 | F61 | −27% |
| R77 | L137 | 73% |
| R54 | K141 | 92% |
Figure 7.

Contact disruption pathways in the E141K mutant. Contact occupancy differences were calculated between all wild-type simulations and all E141K simulations with a significance threshold of 0.09, an allowable traversal threshold of 0.06, and a search depth of 2. (a) ContactWalker disruption pathways. Line thickness indicates magnitude of occupancy change, green lines indicate mutant-associated stabilization, and orange-hashed lines indicate mutant-associated destabilization. Nodes are colored per the legend. The black line highlights a pathway between the mutation site and the hinge 2 region. (b) 51 ns structure of E141K simulation 1. The black line indicates the same pathway that was labeled in (a). Pathway residues are rendered as sticks and labeled for orientation. The K141 mutation site is rendered as magenta spheres. The final residue of the pathway (E220) is rendered as orange spheres.
Zhang et al reported that membrane binding is dependent on several hydrophobic residues including F165 and F169.28 While these residues did not experience appreciable contact disruption, they did have different average Cα RMSF and Cα RMSD values than WT; the Cα RMSD values were 2.9 and 2.6 Å in WT vs. 1.9 and 1.9 Å in E141K for residues F165 and F169, respectively and the Cα RMSF values for F169 were 1.4 in WT and 1.2 Å in E141K.
Several headgroup-coordinating residues were disrupted: F133, L137, V182, L183, F187, and L198 (Figure 7). Although docking studies with MD structures did not indicate significant disruption to α-tocopherol binding for this mutant, these contact changes may be indicative of binding-pocket disruptions that take place over longer time scales than are currently available with molecular dynamics. Also, the 1OIP crystal structure shows four crystallographic water molecules involved with the α-tocopherol headgroup binding. Three of the disrupted headgroup-coordinating residues, F133, V182, and F187, also help to coordinate these waters. Data reported by Schaaf et al8 suggest that water coordination within the analogous hydrophobic ligand binding pocket of the related Sec14 and Sfh1 proteins may be important to the functioning of those proteins.
In addition to possible binding site disruption, these results indicate that a structural effect of the E141K mutation was to disrupt the hinge 2 region. This is reflective of the in silico findings of Ryan et al that the G266D mutant of the related Sec14 protein disrupts hinge function4, although we did not observe significant disruption to the gating module reported in that same study. The ubiquity of these disruptions within the mutant simulations suggests an explanation for part of the increase in Kd relative to wild type reported by Morley et al7; the destabilization caused by the E141K mutation was enough to disrupt the hinge and lid region of the protein, but not enough to severely disrupt the actual α-tocopherol binding pocket (although, as discussed above, some loss in binding ability may be attributable to pocket disruption). As a result, the E141K in silico docking energies were almost identical to wild type while the in vitro experiments, which were dependent on the wild type-like gate and hinge behavior, demonstrated an increase in Kd.
R59W Simulations
The R59W mutant simulations, like the E141K simulations, did not undergo significant unfolding. Simulations 2 and 3 were relatively stable with Cα RMSD values stabilizing between 2.5 and 3 Å (Figure 2c). Simulation 1, however, was considerably more dynamic with Cα RMSD values as high as 4.5 Å and Cα RMSF values often twice those of the other simulations (Figure 3d).
Comparison of all three R59W simulations against the wild-type simulation 1 baseline revealed four disrupted contacts in the lid region: K211:T215 (83%), P212:E216 (86%), F213:E216 (100%), and F213:K217 (100%) Four contacts that tethered α13 in place were also destabilized: W76:F187 (58%), Y73:P188 (83%), W59:T184 (87%), and W76:S186 (91%).
Contact occupancy comparison between all three wild-type simulations and all three R59W simulations resulted in low minimum demonstrated occupancy change values. Only 18 contacts demonstrated minimum occupancy changes greater than 20%. Of these, only six contacts exhibited occupancy change values greater than 50%. Notably, the W59 mutation site did not see significant occupancy change; in the aggregate, the largest minimum demonstrated occupancy change for a W59 contact was a 6% contact occupancy increase with R54.
R59W simulations 1 and 3, considered on their own, exhibited less favorable α-tocopherol docking energies than simulation 2 (Figures 4 and 8). They were also structurally similar to wild-type simulation 2, displaying a characteristic separation of α13 from α5 and α10 (Figure 5b). Contact comparison of only these two simulations against wild-type simulation 1 revealed additional disruptions to T184:K217 (98% destabilization), L214:K217 (100% destabilization), and K217:R221 (99% stabilization). The T184:K217 and L214:K217 disruptions were also present in the poorer-docking wild-type simulations and in the E141K simulations. Contact analysis also revealed stabilization in the hinge 2 region, similar to the E141K hinge 2 stabilizations.
Figure 8.

Representative R59W structures rendered as tubes. Tube radius and color reflect Cα RMSD (left column) and Cα RMSF (right column). The W59 mutation site is rendered as magenta spheres. (a) Simulation 1. (b) Simulation 2. (c) Simulation 3.
Given these data, we took R59W simulation 1 and 3 conformations to be representative of mutation-induced disruptions. To isolate the structural disruptions associated with decreased docking performance, R59W simulations 1 and 3 were compared against R59W simulation 2; 50 contacts with greater than 90% occupancy change were identified, of which 72% were destabilized (Figure 9). Contacts responsible for holding α13 in place were destabilized, including E82:K178 (100%), E141:F187 (99%), Y73:S186 (100%), N72:D185 (99%), W76:V182 (97%), N72:S186 (96%), and F133:L183 (91%). Of the stabilized contacts, three contacts strengthened the connection between the C-terminus of α13 and the N-terminal three-helix bundle: W59:F187 (93%), L65:S186 (93%), and A58:L189 (98%). Notably, one of these (W59:F187) included the mutated residue. Furthermore, the hinge 1 region contained three significant destabilizing interactions (E199:N227 (91%), P200:H225 (95%), and I197:P200 (94%)), the hinge 2 region contained one significant stabilizing interaction (E216:E220 (94%) as well as others in the near vicinity), and the α14 lid contained one significant destabilization (K211:E216 (97%)). The gating module reported by Ryan et al4 was disrupted only in the H101:S136 contact.
Figure 9.
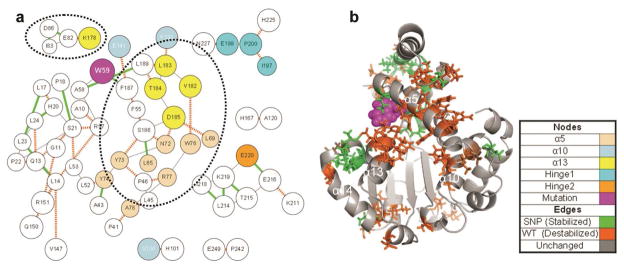
Contact disruption pathways in the R59W mutant. Contact occupancy difference was calculated between simulation 2 and simulations 1 and 3 with a significance threshold of 0.9, an allowable traversal threshold of 0.9, and a search depth of 1. (a) ContactWalker disruption pathways. Line thickness indicates magnitude of occupancy change, green lines indicate stabilization of the mutant, and orange-hashed lines indicate mutant destabilization. Nodes are colored per the legend. The dotted circles highlight destabilized α13 residues. (b) 51 ns structure of R59W simulation 1. Significant nodes are rendered as sticks and colored based on overall stability (green) or instability (orange). Note the prevalence of destabilized residues between α5, α10, and α13 coincident with the expansion of the ligand-binding pocket.
The R59W mutation increased the tendency of the protein to adopt conformations that were unfavorable to ligand docking; relative to the wild-type simulation 1 baseline, the lid and hinge regions were disrupted and α13 became disrupted more often in the R59W simulations than in the wild-type simulations. Furthermore, the in silico docking was less favorable than wild type, reflecting structural changes to the binding pocket in addition to disruptions to the lid and hinge regions. Like the E141K mutation, the R59W mutation disrupted several headgroup-coordinating residues including F133, S136, V182, L183, F187, and L189 (Figure 9). All of these residues except L183 also help to coordinate crystallographic waters in the ligand binding pocket of the 1OIP (closed) crystal structure.
Finally, like the E141K mutant, the R59W mutant did not cause significant contact disruption to F165 and F169, the residues reported by Zhang et al28 to be important to membrane binding. These simulations did, however, show a lower average Cα RMSD relative to the starting structure than did the WT residues (2.9 and 2.6 Å in WT vs. 2.3 and 2.2 Å in R59W for residues F165 and F169, respectively). Also, F165 had a larger average Cα RMSF in the R59W simulations (1.2 Å in WT vs. 1.5 Å in R59W).
CONCLUSIONS
In aggregate, the mutants had very few large occupancy changes relative to wild type, indicating that their observed behaviors were already present in wild type. The primary effect of both mutations was to exacerbate pre-existing tendencies; in the case of the E141K mutant, we observed changes in the hinge 2 region. In the case of the R59W mutant, we observed a widening of the ligand binding pocket. In both cases, the mutant proteins remained folded and relatively stable, indicating that these mutants’ decreased α-tocopherol binding is due to a shift in the population with an increase in conformers with altered binding regions.
Genetic diseases can have severe clinical effects while causing only subtle changes to protein structure. As demonstrated by the E141K and R59W α-TTP mutations, the cause of these disruptions can be masked by the dynamic propensities of the protein. In the case of the E141K mutant, finding that the hinge was disrupted with only minor disruptions to the actual ligand binding pocket highlights the complexity and subtlety of protein dynamics. Occupancy comparison was a useful tool for characterizing these dynamic systems because we were able to establish a wild-type baseline against which we could compare mutation-associated structural changes. We were also able to characterize large-scale structural changes without losing single-contact data resolution. Visualization of these contact changes, together with docking studies and traditional analyses, facilitated the exploration of gigabytes of MD data and provided both quantitative and qualitative accounts of the changes that occurred as a result of mutations to α-TTP. These findings offer a structural description of the E141K and R59W mutants and suggest a molecular basis for their attenuated ability to transport α-tocopherol.
Acknowledgments
We are grateful for computer resources provided through the DOE Office of Biological and Environmental Research as provided by the National Energy Research Scientific Computing Center, which is supported by the Office of Science of the U.S. Department of Energy under contract DE-AC02-05CH11231. Financial support was also provided by the National Institutes of Health (GM50789 to V.D. and TG 3 T15 LM007442-04S1 to P.C.A. and D.B.)
Footnotes
The authors declare no competing financial interests.
References
- 1.Meier R, Tomizaki T, Schulze-Briese C, Baumann U, Stocker A. The molecular basis of vitamin E retention: structure of human α-tocopherol transfer protein. J Mol Biol. 2003;331:725–734. doi: 10.1016/s0022-2836(03)00724-1. [DOI] [PubMed] [Google Scholar]
- 2.Di Donato I, Bianchi S, Federico A. Ataxia with vitamin E deficiency: update of molecular diagnosis. Neurol Sci. 2010;31:511–515. doi: 10.1007/s10072-010-0261-1. [DOI] [PubMed] [Google Scholar]
- 3.Liu T, Jenwitheesuk E, Teller DC, Samudrala R. Structural insights into the cellular retinaldehyde binding protein (CRALBP) Proteins. 2005;61:412–422. doi: 10.1002/prot.20621. [DOI] [PubMed] [Google Scholar]
- 4.Ryan MM, Temple BRS, Phillips SE, Bankaitis VA. Conformational Dynamics of the Major Yeast Phosphatidylinositol Transfer Protein Sec14p: Insight into the Mechanisms of Phospholipid Exchange and Diseases of Sec14p-Like Protein Deficiencies. Mol Biol Cell. 2007;18:1928–1942. doi: 10.1091/mbc.E06-11-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Panagabko C, Morley S, Hernandez M, Cassolato P, Gordon H, Parsons R, Manor D, Atkinson J. Ligand specificity in the CRAL-TRIO protein family. Biochemistry. 2003;42:6467–6474. doi: 10.1021/bi034086v. [DOI] [PubMed] [Google Scholar]
- 6.Cavalier L, Ouahchi K, Kayden HJ, Di Donato S, Reutenauer L, Mandel J-L, Koenig M. Ataxia with Isolated Vitamin E Deficiency: Heterogeneity of Mutations and Phenotypic Variability in a Large Number of Families. Am J Hum Genet. 1998;62:301–310. doi: 10.1086/301699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morley S, Panagabko C, Shineman D, Mani B, Stocker A, Atkinson J, Manor D. Molecular Determinants of Heritable Vitamin E Deficiency. Biochemistry. 2004;43:4143– 4149. doi: 10.1021/bi0363073. [DOI] [PubMed] [Google Scholar]
- 8.Schaaf G, Dynowski M, Mousley CJ, Shah SD, Yuan P, Winklbauer EM, de Campos MKF, Trettin K, Quinones MC, Smirnova TI, Yanagisawa LL, Ortlund EA, Bankaitis VA. Resurrection of a functional phosphatidylinositol transfer protein from a pseudo-Sec14 scaffold by directed evolution. Mol Biol Cell. 2011;22:892–905. doi: 10.1091/mbc.E10-11-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernstein FC, Koetzle TF, Williams GJ, Meyer EF, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. The protein data bank: A computer-based archival file for macromolecular structures. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 10.Levitt M. Molecular Applications Group. Stanford University and Yeda; Palo Alto, CA and Rehovot, Israel: 1990. ENCAD-Energy calculations and dynamics. [Google Scholar]
- 11.Levitt M, Hirshberg M, Sharon R, Daggett V. Potential energy function and parameters for simulations of the molecular dynamics of proteins and nucleic acids in solution. Comput Phys Commun. 1995;91:215–231. [Google Scholar]
- 12.Beck DAC, Alonso DOV, Daggett V. ilmm, in lucem Molecular Mechanics. Seattle, WA: University of Washington; 2000–2013. [Google Scholar]
- 13.Beck DAC, Daggett V. Methods for molecular dynamics simulations of protein folding/unfolding in solution. Methods. 2004;34:112–120. doi: 10.1016/j.ymeth.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 14.Levitt M, Hirshberg M, Sharon R, Laidig KE, Daggett V. Calibration and Testing of a Water Model for Simulation of the Molecular Dynamics of Proteins and Nucleic Acids in Solution. J Phys Chem. 1997;101:5051–5061. [Google Scholar]
- 15.Armen RS, Bernard BM, Day R, Alonso DO, Daggett V. Characterization of a possible amyloidogenic precursor in glutamine-repeat neurodegenerative diseases. P Natl Acad Sci USA. 2005;102:13433–13438. doi: 10.1073/pnas.0502068102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kell GS. Precise representation of volume properties of water at one atmosphere. J Chem Eng Data. 1967;12:66–69. [Google Scholar]
- 17.Beck DA, Armen RS, Daggett V. Cutoff size need not strongly influence molecular dynamics results for solvated polypeptides. Biochemistry. 2005;44:609–616. doi: 10.1021/bi0486381. [DOI] [PubMed] [Google Scholar]
- 18.Beck DAC, Jonsson AL, Schaeffer RD, Scott KA, Day R, Toofanny RD, Alonso DO, Daggett V. Dynameomics: mass annotation of protein dynamics and unfolding in water by high-throughput atomistic molecular dynamics simulations. Protein Eng Des Sel. 2008;21:353–368. doi: 10.1093/protein/gzn011. [DOI] [PubMed] [Google Scholar]
- 19.Van der Kamp MW, Schaeffer RD, Jonsson AL, Scouras AD, Simms AM, Toofanny RD, Benson NC, Anderson PC, Merkley ED, Rysavy S, Bromley D, Beck DAC, Daggett V. Dynameomics: a comprehensive database of protein dynamics. Structure. 2010;18:423–435. doi: 10.1016/j.str.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Day R, Beck DAC, Armen RS, Daggett V. A consensus view of fold space: Combining SCOP, CATH, and the Dali Domain Dictionary. Protein Sci. 2003;12:2150–2160. doi: 10.1110/ps.0306803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaeffer RD, Jonsson AL, Simms AM, Daggett V. Generation of a consensus protein domain dictionary. Bioinformatics. 2011;27:46–54. doi: 10.1093/bioinformatics/btq625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeLano WL. The PyMOL molecular graphics system. 2002 http://www.pymol.org.
- 23.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 24.Schmidlin T, Kennedy BK, Daggett V. Structural changes to monomeric CuZn superoxide dismutase caused by the familial amyotrophic lateral sclerosis-associated mutation A4V. Biophys J. 2009;97:1709–1718. doi: 10.1016/j.bpj.2009.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gansner ER, North SC. An open graph visualization system and its applications to software engineering. Software Pract Exper. 2000;30:1203–1233. [Google Scholar]
- 26.Hu Y. Efficient, high-quality force-directed graph drawing. Mathematica J. 2005;10:37–71. [Google Scholar]
- 27.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang WX, Thakur V, Lomize A, Pogozheva I, Panagabko C, Cecchini M, Baptist M, Morley S, Manor D, Atkinson J. The contribution of surface residues to membrane binding and ligand transfer by the α-tocopherol transfer protein (α-TTP) J Mol Biol. 2010;405:972–988. doi: 10.1016/j.jmb.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]