

Abstract
Lentiviral vectors (LVs) are a powerful means of delivering genetic material to many types of cells. Because of safety concerns associated with these HIV-1 derived vectors, producing large quantities of LVs is challenging. In this paper, we report a method for producing high titers of self-inactivating LVs. We retrovirally transduce the tet-off stable producer cell line GPR to generate a cell line, GPRS, which can express all the viral components, including a dendritic cell-specific glycoprotein, SVGmu. Then, we use concatemeric DNA transfection to transfect the LV transfer plasmid encoding a reporter gene GFP in combination with a selectable marker. Several of the resulting clones can produce LV at a titer 10-fold greater than what we achieve with transient transfection. Plus, these viruses efficiently transduce dendritic cells in vitro and generate a strong T cell immune response to our reporter antigen. This method may be a good option for producing strong LV-based vaccines for clinical studies of cancer or infectious diseases.
Keywords: Immunology, Issue 76, Virology, Genetics, Molecular Biology, Cellular Biology, Biochemistry, Chemical Engineering, Bioengineering, Biomedical Engineering, Medicine, Infection, Pharmacology, Lentivirus, Cancer Vaccines, Vaccines, Virus-Like Particle, life sciences, microbiology, bioengineering (general), Lentiviral vector, stable cell line, dendritic cells, vaccine, concatemeric transfection, retrovirus, virus, plasmid, cell culture
Introduction
Many vector systems have been developed for gene delivery. Vectors based on lentiviruses have been among the most commonly studied viral system. These vectors are advantageous because they can efficiently transduce both dividing and non-dividing cells 1, achieve long-term expression due to integration into the host genome, exhibit low natural anti-vector immunity in most human populations 2, and have a low potential for genotoxicity from insertional mutagenesis 3,4.
Production of lentiviruses has always been colored by safety concerns. Lentiviral vectors are generally derived from HIV-1, the etiological agent of AIDS. Transient transfection of individual components of the lentivector (transfer, envelope, and packaging plasmids) is a common and flexible means of delivering genetic material in laboratory settings. However, scaling-up transient transfections for clinical applications is cumbersome and may lead to the development of replication-competent lentivirus 5,6. To overcome these hurdles, several stable packaging and producer cell lines have been developed 6-11. One of these lines, the GPR packaging line 11, has the attractive advantage of being regulated by tetracycline. In this paper, we demonstrate how to adapt this system to produce self-inactivating lentiviral vectors that are specifically targeted toward dendritic cells (DC-LVs) 12.
Dendritic cells (DCs) are the most robust antigen presenting cells of the immune system. They have been the subject of great interest in cancer vaccine development because they directly initiate, program, and regulate tumor-specific immune responses 13. Incorporating a vaccination protocol to include DCs has the potential to elicit a stronger antitumor immune response than peptide or DNA vaccines. Recently, we have developed a lentiviral vector that specifically targets dendritic cells through a modified sindbis virus glycoprotein, SVGmu 14. These vectors are unique in that they show high specificity to dendritic cells and generate stronger immune responses than nonspecific, VSVg-pseudotyped vectors.
Here, we report a method for producing large quantities of these DC-targeted lentiviral vectors. We demonstrate that these DC-LVs can infect DCs and generate strong CD8+ T cell immune responses. All procedures involving animals were performed humanely, under the approval of the USC Intitutional Animal Care and Use Committee. In order to perform in vivo and clinical experiments, it is critical to create a cell line that can produce virus at a high titer. Performing the transduction and transfection steps exactly as described will maximize the chances of generating such a clone.
Protocol
DC-LV stable producer cells are constructed based on the GPR packaging cell line 11 that contains the necessary lentiviral components gagpol, rev and the tet-off system. First, retroviral transduction is used to generate a GPRS packaging cell line that encodes a tet-dependent SVGmu glycoprotein. Then, concatemer array transfection is used to transfect the GPRS cell line with a lentiviral vector transgene such as GFP. This stable producer cell line, designated as LV-MGFP, can be tested in vitro and in vivo for its ability to produce a DC-LV vaccine against GFP.
1. Generating a Tet-dependent SVGmu Cell Line
Plasmid pRX-SVGmu is a construct in which DC-specific glycoprotein SVGmu is cloned downstream of the tTA-advanced promoter of retroviral plasmid pRetroX-Tet-off 1 (Figure 1C). Culture 293T cells in D10 (Dulbecco's modified Eagle's medium with 10% fetal bovine serum, 2 mM L-glutamine). Culture GPR packaging cell line in D10 with doxycycline (1 ng/ml) and puromycin (2 μg/ml). All cells are cultured in a humidified 37 °C incubator with 5% CO2.
16-18 hr before transient transfection, plate 2 x 106 293T cells in 4 ml of D10 in a 6-cm tissue culture dish such that the cells approach 90% confluency at the time of transfection.
Transfection of retroviral plasmids: mix 100 μl 1.25 M CaCl2 solution, sterile Milli-Q water and the following plasmids: 5 μg pRX-SVGmu, 2.5 μg pGag-Pol, 2.5 μg pVSV-G. The final volume is 500 μl. To a 5 ml polystyrene round-bottom tube, add 500 μl 2X HBS (50 mM HEPES, 10 mM KCl, 12 mM Dextrose, 280 mM NaCl, 1.5 mM Na2HPO4*7H2O, pH 7.05). Add the plasmid mixture dropwise into the 2X HBS buffer while bubbling the buffer vigorously with a glass Pasteur pipette. After adding the mixture, continue bubbling for another 30 sec. Then, add the whole mixture onto the 293T cells in the 6-cm culture dish, and incubate at 37 °C.
4 hr after transfection, carefully replace the medium in the culture dish with 4 ml pre-heated D10.
Spin infection: 48 hr after transfection, harvest SVGmu-encoding retroviral particles in the supernatant by passing the supernatant through a 0.45-μm filter. Plate the GPR packaging cells in a 24-well dish at 2 x 104 cells/well. Add filtered supernatant to GPR packaging cells in the dish (2 ml/well) and centrifuge cells for 90 min at 1,050 x g and 25 °C. Change medium into fresh D10 with doxycycline (1 ng/ml) after spin-infection (2 ml/well).
72 hr post-transfection, expand the culture of transduced packaging cells in D10 with doxycycline (1 ng/ml) and puromycin (2 ng/ml).
To confirm expression of SVGmu, culture the cells without doxycycline for 48 hr. Measure surface expression of SVGmu with flow cytometry using anti-Sindbis serum. These cells are designated as GPRS packaging cell line.
2. Construct DC-LV Producer Cells by Concatemer Array Transfection
Lentiviral transfer plasmid TL20-GFP is a self-inactivating lentiviral transfer vector plasmid based on pCL20c-MSCV-GFP with a Dox-regulatable viral RNA genome expression system and replacement of the cytomegalovirus (CMV) enhancer with 7 tet operators(Figure 1C) 11,12,15. Plasmid PGK-ble is a bleomycin resistant (ble) cassette driven by a weak PGK promoter 2.
Digest 20 μg plasmid TL20-GFP with the restriction enzyme SfiI at 50 °C. Digest 20 μg plasmid PGK-ble with PflMI at 37 °C.
Purify the DNA fragments (the PGK-ble cassette is 1011 bp and the vector TL20-GFP is 6861 bp) by agarose gel electrophoresis.
Ligate the TL20-GFP vector and PGK-ble cassette at a molar ratio of 25:1 (NEB T4 DNA Ligase). Incubate overnight at room temperature.
After overnight ligation, purify DNA from the ligation mixture (Qiagen DNeasy Blood and Tissue Kit). Ensure the amount of purified DNA is around 5 μg.
16-18 hr before transfection, plate GPRS packaging cells in a 6-cm tissue culture dish such that the confluency will be about 90% at the time of transfection.
Use the calcium phosphate transfection method described in step 1.2 to transfect 5 μg of the purified concatemeric DNA into the GPRS packaging cells in the 6-cm tissue culture dish. 4 hr after transfection, carefully remove the medium and replace with 4 ml pre-heated D10 with doxycycline (1 ng/ml).
48 hr after transfection, change the medium into D10 with doxycycline (1 ng/ml) and puromycin (2 μg/ml). Select for transfected clones by adding zeocin (50 μg/ml) in the culture medium. Culture the cells for about 2 weeks until cell colonies can be seen at the bottom of the dishes.
Label the cell colonies at the bottom of the dishes. Take 24-well tissue culture dishes, number the wells and add to each well 2 ml D10 containing zeocin (50 μg/ml), doxycycline (1 ng/ml), and puromycin (2 μg/ml). Aspirate the medium of the transduced cells, then add one drop of trypsin onto each of the colonies for less than one minute. Then, add one or more drops of D10 on the same colonies. Pick up colonies one by one with a pipette and transfer them into separate wells of the 24-well tissue culture plate.
Culture and expand all cell clones in D10 containing zeocin (50 μg/ml), doxycycline (1 ng/ml) and puromycin (2 μg/ml) for the evaluation of viral producing ability.
3. Evaluate Viral Production of Each Cell Clone
Trypsinize the producer cells and plate around 4x106 cells in 6-cm tissue culture dish in D10 without doxycycline such that the confluency exceeds 90%. Replace the medium with fresh pre-heated D10 daily and collect the medium for the titer assay 72 hr after removing dox.
Harvest the viral supernatant by filtering the medium with a 0.45-μm filter.
Plate target cells expressing the DC-SIGN receptor (e.g. 293T.hDC-SIGN or mouse bone marrow derived dendritic cells 16) and 293T as a negative control in 96-well culture dishes, 1 x 104 cells / well. Add 2-fold serial dilutions of the viral supernatant to the wells (100 μl/well). Usually, 6-8 dilutions are sufficient to reach the range in which transduced GFP-positive cells are in a linear relationship to the amount of virus added.
Centrifuge cells and replace the medium as described in step 1.4. BMDCs should be cultured in RPMI medium with 10% FBS and GM-CSF (1:20 J558L conditioned medium).
Measure the GFP expression in transduced cells by flow cytometry 4-6 days post-transduction. The lentiviral vector titer is calculated in the vector dilution range when the percentage of GFP-positive cells and vector amount are in a linear relationship. Choose the cell clone with the highest viral production for subsequent applications.
4. Produce and Concentrate Lentiviral Vectors
Culture the producer cell line (LV-MGFP) in 15-cm tissue culture dishes.
Trypsinize the producer cells and plate cells in 15-cm tissue culture dishes at greater than 90% confluency in fresh D10 without doxycycline. Change medium with fresh, pre-heated D10 daily.
At the time of peak viral production (as determined by the user), harvest the viral supernatant and filter it using a 0.45-μm filter. Load the filtered supernatant into thick wall 32.5 ml ultracentrifuge tubes, seal with parafilm and centrifuge at 50,000 x g, 4 °C for 90 min. Thoroughly resuspend the pellet in 50 μl or an appropriate volume of PBS or HBSS depending on the application.
5. Immunize Mice In vivo and Analyze Antigen-specific Immune Response
Produce high-titer virus as described in section 4.
Inject BALB/c mice (6-8 week old, female) via a footpad route with the vector suspension (25 μl per footpad).
2 weeks after immunization, isolate spleen and harvest splenocytes. Culture splenocytes with GFP epitope peptides and analyze the presence of GFP-specific CD8+ T cells using intracellular cytokine staining, as previously described 17 (Figure 3B).
Representative Results
The stable cell line described in this method can produce large quantities of lentiviral vectors that are specifically targeted to dendritic cells. As shown in Figure 1B, isolation of individual clones yielded stable cell lines of varying quality 12. Among 26 clones tested, 8 produced lentiviral particles at a titer of greater than 106 transduction units per ml (TU/ml), which is a typical benchmark for SVG-pseudotyped lentiviral vectors produced by transient transfection. At the same time, several clones produced less than 104 TU/ml; it is important to isolate multiple clones in order to obtain a strong producer.
One clone reliably produced lentiviral vectors at a titer of greater than 107 TU/ml. Further testing on this clone demonstrated that this level of viral production was stable for multiple days, unaffected by the presence/absence of serum in the culture medium, and achievable in cells cultured for more than 3 months (Figure 2) 12.
To confirm that these viruses were not only effective in vitro, they were also used to immunize mice. Analysis of the GFP-specific immune response by intracellular cytokine staining found that stably-produced DC-LV could stimulate the expansion of GFP-specific CD8+ T cells to nearly 4% of all CD8+ Splenocytes (Figure 3) 12.
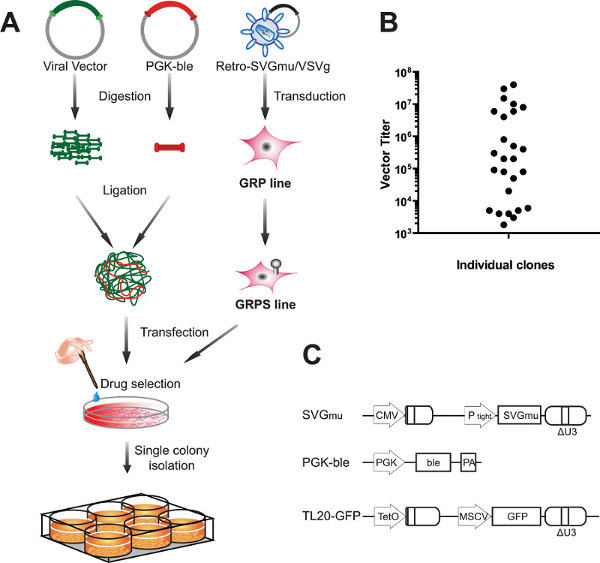 Figure 1. Generation of tetracycline-regulated lentiviral vector producer cells.(A) Schematic outline of the procedure for making DC-targeted lentiviral vector producer cells, including transduction of GRP lines with a retroviral vector containing the SVGmu envelope gene and transfection of the gene of interest (viral vector) by concatemeric transfection. (B) Single clones selected by zeocin treatment were expanded over 1-2 weeks, and the titer of the viral supernatant was measured 3 days after removal of doxycycline. As shown, viral titers varied widely by clone. (C) Plasmid maps. The viral vector plasmid, TL20-GFP, encodes green fluorescent protein under the control of a murine stem cell virus promoter and the vector genome under the control of a tetracycline-repressible promoter. PGK-ble encodes the zeocin resistance gene (Sh ble) under the constitutive phosphoglycerate kinase 1 promoter (PGK). Retro-SVGmu contains the SVGmu glycoprotein under the Ptight tet responsive element. Click here to view larger figure.
Figure 1. Generation of tetracycline-regulated lentiviral vector producer cells.(A) Schematic outline of the procedure for making DC-targeted lentiviral vector producer cells, including transduction of GRP lines with a retroviral vector containing the SVGmu envelope gene and transfection of the gene of interest (viral vector) by concatemeric transfection. (B) Single clones selected by zeocin treatment were expanded over 1-2 weeks, and the titer of the viral supernatant was measured 3 days after removal of doxycycline. As shown, viral titers varied widely by clone. (C) Plasmid maps. The viral vector plasmid, TL20-GFP, encodes green fluorescent protein under the control of a murine stem cell virus promoter and the vector genome under the control of a tetracycline-repressible promoter. PGK-ble encodes the zeocin resistance gene (Sh ble) under the constitutive phosphoglycerate kinase 1 promoter (PGK). Retro-SVGmu contains the SVGmu glycoprotein under the Ptight tet responsive element. Click here to view larger figure.
 Figure 2. Characteristics of viral vector production.(A) After removal of dox, the viral vector titer of culture supernatants was measured each day for 7 days. Virus production peaked between 48 - 72 hr post induction. Cells cultured in serum-free media produced nearly as much virus as those cultured with 10% FBS. (B) Producer cells were cultured over 3 months and aliquots were tested periodically for virus production. No significant loss of virus production was observed.
Figure 2. Characteristics of viral vector production.(A) After removal of dox, the viral vector titer of culture supernatants was measured each day for 7 days. Virus production peaked between 48 - 72 hr post induction. Cells cultured in serum-free media produced nearly as much virus as those cultured with 10% FBS. (B) Producer cells were cultured over 3 months and aliquots were tested periodically for virus production. No significant loss of virus production was observed.
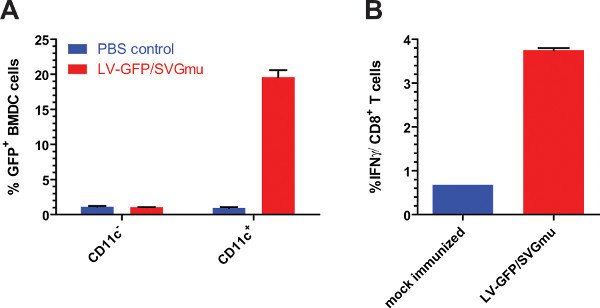 Figure 3. Lentiviral vectors produced by stable DC-targeted cell lines can selectively transduce DCs in vitro and generate an immune response in vivo.(A) Mouse bone marrow-derived dendritic cells were infected with LV-GFP and GFP expression was measured with flow cytometry. GFP expression was specific to CD11c+ cells, which include dendritic cells. (B) Mice were immunized with 2 x 107 TUs of purified and concentrated LV-GFP. Two weeks later, nearly 4% of all CD8+ T cells were activated and expressed IFNγ in response to the presentation of the GFP dominant peptide.
Figure 3. Lentiviral vectors produced by stable DC-targeted cell lines can selectively transduce DCs in vitro and generate an immune response in vivo.(A) Mouse bone marrow-derived dendritic cells were infected with LV-GFP and GFP expression was measured with flow cytometry. GFP expression was specific to CD11c+ cells, which include dendritic cells. (B) Mice were immunized with 2 x 107 TUs of purified and concentrated LV-GFP. Two weeks later, nearly 4% of all CD8+ T cells were activated and expressed IFNγ in response to the presentation of the GFP dominant peptide.
Discussion
Here, we have outlined a method for producing large quantities of lentiviral vectors using 293T cells stably transduced with all lentiviral components under the tet off regulatory system. To date, most protocols to produce lentiviral vectors rely on standard calcium phosphate transient transfection (see 18, for example). This approach has been successful on a clinical scale, but it may suffer from some limitations not likely to be present in scaling up production from stable cell lines. For example, co-transfection with large quantities of LV plasmids could theoretically increase the chance for the development of replication competent lentivirus 19. Furthermore, transient transfections are more likely to produce variable results on a large scale than utilizing a stable producer line due to the large quantities of DNA inputs required 12. Under the right conditions, such producer lines have been used to generate other γ-retroviral vectors on large scales 20,21. However, studies utilizing lentiviral producer lines are limited, and thus the inclusion of robust assays 22 to detect replication competent lentivirus will be critical in developing any cell line for clinical use.
Other groups have published descriptions for lentivirus-producing stable cell lines 6-11,23. However, a common limitation of these reports is that many of them rely on transducing the producer cells with lentiviral vectors encoding the gene of interest, which may reduce their safety for clinical use. Here, we utilize the concatemeric array transfection method 11 to transduce cells that can produce self-inactivating lentivirus. Furthermore, our use of the tet off system improves the method's suitability to clinical use. This regulatory system prevents toxicity from SVGmu during cell culture maintenance and allows for the collection of lentiviral vectors under antibiotic-free conditions.
During the procedure, it is important that laboratory workers use appropriate precautions when handling the lentiviral vectors. Lentiviral vectors are derived from HIV-1, a human pathogen, and all procedures should be performed under the guidance of an institution biosafety committee. According to the NIH, BL2 containment is generally appropriate for working with advanced lentiviral systems such as the one described here, although enhanced BL2 containment may be indicated if experiments are scaled up for clinical practice 24.
There are several critical steps in the procedure that require special care. First, cell culture must be performed with great attention to detail. We have found that virus titers decline noticeably if producer cells are not passaged every 24 hr. Similarly, some lots of FBS can lead to significant declines in virus production, so it is important to test multiple lots of FBS before scaling up an experiment. The selection of a clone that can produce high virus titers is essential for downstream applications. In agreement with previous work 25, we have found that clones that appear to divide more rapidly tend to produce more virus. Once a high-titer clone has been found, these cells should be aliquoted and frozen in FBS + 10% DMSO as soon as possible. Then, using freshly thawed aliquots helps to generate the highest titer possible. By following this rule of thumb, we have had the most success producing virus through transient transfections, and we expect that similar cell line maintenance will benefit stable virus-producing cells.
This method's major limitation is that it requires a significant amount of up front effort to generate a stable producer line. Thus, it is not likely to be appropriate in situations where multiple types of lentiviral vectors will be tested. Transient transfections may be more convenient for these types of applications. We generally foresee using this method to generate large quantities of viral vector after the vector has already been tested in a small format. Another limitation of our specific cell line is that vectors produced by the GPRS cells will only be specific for cells expressing DC-SIGN because the envelope protein, SVGmu, binds to this receptor. To target other cellular receptors, the procedure can be modified by introducing other glycoprotein plasmids into the original GPR cell line. Similarly, other genes of interest can be transfected into the producer cell lines so that lentiviral vectors can deliver more relevant proteins than GFP.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The authors would like to acknowledge Michael Chou, Bingbing Dai, and Liang Xiao for contributing data for this manuscript. We also acknowledge Dr. John Gray for the generous gifts of reagents used in this study. P.B. is supported by a postdoctoral fellowship from the National Cancer Center. This research was supported by grants from the National Institute of Health (R01AI68978, P01CA132681 and RCA170820A), a grant from the Bill and Melinda Gates Foundation, a translational acceleration grant from the Joint Center for Translational Medicine and a grant from the California HIV/AIDS Research Program.
References
- Naldini L, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- Kootstra NA, Verma IM. Gene therapy with viral vectors. Annu. Rev. Pharmacol. Toxicol. 2003;43:413–439. doi: 10.1146/annurev.pharmtox.43.100901.140257. [DOI] [PubMed] [Google Scholar]
- Montini E, et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 2006;24:687–696. doi: 10.1038/nbt1216. [DOI] [PubMed] [Google Scholar]
- Montini E, et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Invest. 1172;119:964–975. doi: 10.1172/JCI37630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Tai A, Wang P. Immunization delivered by lentiviral vectors for cancer and infectious diseases. Immunological Reviews. 2011;239:45–61. doi: 10.1111/j.1600-065X.2010.00967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broussau S, et al. Inducible packaging cells for large-scale production of lentiviral vectors in serum-free suspension culture. Molecular Therapy: the journal of the American Society of Gene Therapy. 2008;16:500–507. doi: 10.1038/sj.mt.6300383. [DOI] [PubMed] [Google Scholar]
- Cockrell AS, Ma H, Fu K, McCown TJ, Kafri T. A trans-lentiviral packaging cell line for high-titer conditional self-inactivating HIV-1 vectors. Molecular Therapy: the journal of the American Society of Gene Therapy. 2006;14:276–284. doi: 10.1016/j.ymthe.2005.12.015. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, et al. Continuous high-titer HIV-1 vector production. Nature Biotechnology. 2003;21:569–572. doi: 10.1038/nbt815. [DOI] [PubMed] [Google Scholar]
- Kafri T, van Praag H, Ouyang L, Gage FH, Verma IM. A packaging cell line for lentivirus vectors. Journal of Virology. 1999;73:576–584. doi: 10.1128/jvi.73.1.576-584.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strang BL, et al. Human immunodeficiency virus type 1 vectors with alphavirus envelope glycoproteins produced from stable packaging cells. Journal of Virology. 2005;79:1765–1771. doi: 10.1128/JVI.79.3.1765-1771.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Throm RE, et al. Efficient construction of producer cell lines for a SIN lentiviral vector for SCID-X1 gene therapy by concatemeric array transfection. Blood. 2009;113:5104–5110. doi: 10.1182/blood-2008-11-191049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CL, Chou M, Dai B, Xiao L, Wang P. Construction of stable producer cells to make high-titer lentiviral vectors for dendritic cell-based vaccination. Biotechnol. Bioeng. 2012;109:1551–1560. doi: 10.1002/bit.24413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- Yang L, et al. Engineered lentivector targeting of dendritic cells for in vivo immunization. Nature Biotechnology. 2008;26:326–334. doi: 10.1038/nbt1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawa H, et al. Efficient gene transfer into rhesus repopulating hematopoietic stem cells using a simian immunodeficiency virus-based lentiviral vector system. Blood. 2004;103:4062–4069. doi: 10.1182/blood-2004-01-0045. [DOI] [PubMed] [Google Scholar]
- Yang L, Baltimore D. Long-term in vivo provision of antigen-specific T cell immunity by programming hematopoietic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:4518–4523. doi: 10.1073/pnas.0500600102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai B, et al. HIV-1 Gag-specific immunity induced by a lentivector-based vaccine directed to dendritic cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20382–20387. doi: 10.1073/pnas.0911742106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Husic N, Lin Y, Snider BJ. Production of lentiviral vectors for transducing cells from the central nervous system. J. Vis. Exp. 2012. p. e4031. [DOI] [PMC free article] [PubMed]
- Kochan G, Escors D, Stephenson H, Breckpot K. Clinical Grade Lentiviral Vectors. Lentiviral Vectors and Gene Therapy. 2012. pp. 69–85.
- Loew R, et al. A new PG13-based packaging cell line for stable production of clinical-grade self-inactivating gamma-retroviral vectors using targeted integration. Gene Ther. 2010;17:272–280. doi: 10.1038/gt.2009.134. [DOI] [PubMed] [Google Scholar]
- Gaspar HB, et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet. 2004;364:2181–2187. doi: 10.1016/S0140-6736(04)17590-9. [DOI] [PubMed] [Google Scholar]
- Cornetta K, et al. Replication-competent lentivirus analysis of clinical grade vector products. Molecular Therapy: the journal of the American Society of Gene Therapy. 2011;19:557–566. doi: 10.1038/mt.2010.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart HJ, et al. A stable producer cell line for the manufacture of a lentiviral vector for gene therapy of Parkinson's disease. Human Gene Therapy. 2011;22:357–369. doi: 10.1089/hum.2010.142. [DOI] [PubMed] [Google Scholar]
- Biosafety Considerations for Research with Lentiviral Vectors [Internet] National Institute of Health Recombinant DNA Advisory Committee; 2006. Available from: http://oba.od.nih.gov/oba/rac/Guidance/LentiVirus_Containment/pdf/Lenti_Containment_Guidance.pdf. [Google Scholar]
- Coroadinha AS, et al. Retrovirus producer cell line metabolism: implications on viral productivity. Appl. Microbiol. Biotechnol. 2006;72:1125–1135. doi: 10.1007/s00253-006-0401-y. [DOI] [PubMed] [Google Scholar]