

Abstract
We noted a marked increase in cyclooxygenase-2 (Cox2) and the activation of the endoplasmic reticulum (ER) stress pathway in newborn murine lung on exposure to hyperoxia and IFN-γ. We sought to evaluate Cox2-mediated ER stress pathway activation in hyperoxia-induced and IFN-γ–mediated injury in developing lungs. We applied in vivo genetic gain-of-function and genetic/chemical inhibition, as well as in vitro loss-of-function genetic strategies. Hyperoxia-induced and IFN-γ–mediated impaired alveolarization was rescued by Cox2 inhibition, using celecoxib. The use of small interfering RNA against the ER stress pathway mediator, the C/EBP homologous protein (CHOP; also known as growth arrest and DNA damage–inducible gene 153/GADD153), alleviated cell death in alveolar epithelial cells as well as in hyperoxia-induced and IFN-γ–mediated murine models of bronchopulmonary dysplasia (BPD). In addition, CHOP siRNA also restored alveolarization in the in vivo models. Furthermore, as evidence of clinical relevance, we show increased concentrations of Cox2 and ER stress pathway mediators in human lungs with BPD. Cox2, via CHOP, may significantly contribute to the final common pathway of hyperoxia-induced and IFN-γ–mediated injury in developing lungs and human BPD.
Keywords: newborn, oxygen, BPD, CHOP, cell death
Clinical Relevance
Exposure to hyperoxia and IFN-γ has been implicated in the pathogenesis of bronchopulmonary dysplasia (BPD). Cyclooxygenase-2 (Cox2) has been associated with human BPD in the developing lung. Using developmentally appropriate murine models of BPD, a novel signaling pathway of hyperoxia-induced IFN-γ and Cox2 caused endoplasmic reticulum stress–dependent cell death, resulting in impaired alveolarization in the lung. The clinical relevance of these findings is highlighted through our use of human lung tissue with BPD.
In the developing lung, injury attributable to hyperoxic exposure is an important component in the pathogenesis of bronchopulmonary dysplasia (BPD) (1, 2). The immature human lung during the saccular phase is most commonly exposed to such an exogenous insult, and is predisposed to BPD. The final result is a lung phenotype characterized by fewer and larger simplified alveoli (1–4). Hyperoxia-induced lung injury is characterized by an influx of inflammatory cells, along with endothelial and epithelial cell death (5, 6).
Cell death is said to be a key initiator of the process of alveolar simplification. The activation of key caspases (3, 8, 9) and components of the extrinsic/death receptor and intrinsic/mitochondrial cell death pathways underlies the molecular mechanisms of cell death (5–7). Another cell-death signaling pathway involves the endoplasmic reticulum (ER), which is the site for the folding and assembly of proteins destined for delivery to the extracellular space, plasma membrane, and the exocytic/endocytic compartments (8, 9). When cells are exposed to ER stress, malfolded or unfolded proteins accumulate in the ER lumen, giving rise to a synonymous term, the unfolded protein response (UPR) (8, 9). ER stress is sensed by three ER-resident transmembrane proteins, specifically, inositol-requiring enzyme–1 α (IRE-α), activating transcription factor–6α (ATF6α), and protein kinase regulated by RNA–like ER kinase (PERK), which are freed from binding immunoglobulin protein (BiP; also known as glucose-regulated protein–78, or GRP78) during ER stress (10). Whereas ER stress–induced cell death signaling can occur via multiple pathways, the pathway (i.e., via PERK) that induces transcription of the proapoptotic factor C/EBP homologous protein (CHOP; also known as growth arrest and DNA damage–inducible gene–153, or GADD153) can inhibit antiapoptotic B cell lymphoma (Bcl-2), leading to activation of the executioner caspase-3 and cell death (10).
Although the pathogenesis of hyperoxia-induced lung injury is not well understood, it is believed to be mediated by reactive oxygen species (5, 6, 11), with cytokines also playing an important role (12). IFN-γ is one such proinflammatory cytokine that is detected in lungs exposed to hyperoxia, before neutrophil infiltration in the lung (13, 14), thus qualifying as an early mediator of this process. IFN-γ induces cell death in lung epithelial cells and mediates lung injury (15–17). We have recently reported on the potential role of IFN-γ in murine and baboon models, as well as in human BPD (18). IFN-γ has been shown to induce cyclooxygenase-2 (Cox2) mRNA, protein, and activity in A549 and normal human bronchial epithelial cells (19–21). Cox2 has been detected in alveolar epithelia in the human fetal and preterm lung, and in the bronchial epithelia of infants with BPD (22).
We hypothesized that Cox2 mediates hyperoxia-induced and IFN-γ–dependent injury and impaired alveolarization in the developing lung via the ER stress–activated CHOP-dependent pathway.
We show an increased expression of Cox2 and CHOP in newborn (NB) wild-type (WT) murine lungs and in murine lung epithelial (MLE)-12 cells exposed to hyperoxia, as well as in our unique triple transgenic (TTG) IFN-γ–overexpressing, lung-targeted, externally regulatable NB murine model in room air, which has a lung phenotype of impaired alveolarization, resembling human BPD. The use of small interfering (si)RNA against CHOP alleviated cell death in MLE-12 cells, and in hyperoxia-induced and IFN-γ–mediated murine models of BPD. The effects on the pulmonary phenotype of the NB WT and IFN-γ TTG murine BPD models are Cox2-mediated, because we found evidence of rescue with the use of celecoxib, a selective Cox inhibitor. In addition, CHOP siRNA also restored alveolarization in the in vivo models. Finally, as an indication of clinical relevance, we show increased Cox2, BiP, and CHOP protein expression in human lungs with BPD.
Materials and Methods
Transgenic Mice
We used C57BL6/J mice in our experiments. All animal work was approved by the Institutional Animal Care and Use Committee at the Yale University School of Medicine. IFN-γ–overexpressing TTG CC10–rtTA/tetracycline-controlled transcriptional silencer (tTS)–IFN-γ mice were generated in our laboratory (18). The details of the genetic constructs, the methods of microinjection and genotypic evaluation, the inducibility, and the emphysematous and inflammatory phenotype of dual transgenic CC10–rtTA–IFN-γ mice have been previously described (16). We obtained the TTG IFN-γ–overexpressing mice by breeding the dual transgenic and the tTs mice (the tTs mice were kindly provided by Jack Elias, MD). For the Cox2 inhibition experiments, NB IFN-γ TTG and WT control mice received transmammary exposure to the drug via oral maternal dosing of 20 mg/kg/day of celecoxib (dissolved in water) for 7 days, concomitant with doxycycline (Dox) water administration.
Dox water administration, mRNA, Western blotting, histology, densitometry, morphometry, oxygen exposure, the murine model of BPD, the preparation and administration of CHOP siRNA, terminal deoxynucleotidyl transferase 2'-deoxyuridine 5'-triphosphate nick end labeling (TUNEL) staining, α–smooth muscle actin (α-SMA) staining, and the human lung samples of BPD are described in further detail in the online supplement.
Statistical Analysis
For the animal and human studies, values were expressed as means ± SEMs, or means ± SDs. As appropriate, groups were compared with the Student two-tailed unpaired t test or the Mann-Whitney test, using GraphPad Prism version 3.0 (GraphPad Software, Inc., San Diego, CA). In all analyses, P < 0.05 was considered statistically significant.
Results
Effect of Hyperoxia on the Expression of Cox2 and ER Stress Pathway Mediators in the NB WT Murine Lung
Given the critical role of hyperoxia in the pathogenesis of BPD, we initially decided to identify molecules regulated by hyperoxia, using a developmentally appropriate murine model. The lungs of NB mice (Postnatal Days 1–4) are at the saccular stage of lung development, similar to that of 23- to 28-week human preterm newborns with lungs at the late canalicular/early saccular stages of development. We exposed these NB mice to hyperoxia from birth to Postnatal Day 2, as described in Materials and Methods, and detected increased lung Cox2 mRNA expression, as shown in Figure 1A. Next, we selected early-phase (BiP) and late-phase (CHOP) mediators of the ER stress pathway. As noted in Figure 1B, a slight, but significant, increase was evident in BiP expression (Figure 1C), but without much change in CHOP expression.
Figure 1.

Effect of hyperoxia on the expression of Cox2 and the ER stress pathway mediators in the WT NB Postnatal Day 2 and 7 murine lung tissue. WT NB mice were exposed to hyperoxia for 48 hours after birth. (A) Cox2 mRNA expression is shown. (B) ER stress pathway mediators, BiP, and CHOP protein expression are shown. Each lane in the gel is representative of the condition described, with the corresponding β-actin as control sample. (C) Densitometry for BiP protein expression is shown. Each bar represents the mean ± SEM of a minimum of three animals. WT NB mice were exposed to hyperoxia from Postnatal Days 1–7 after birth. (D) Cox2 and CHOP mRNA expression is shown. (E) ER stress pathway mediators, BiP, and CHOP protein expression are shown. (F) XBP1 unspliced and spliced mRNA isoforms are shown. Each lane in the gel is representative of the condition described, with the corresponding β-actin as control sample. NB, newborn; WT, wild-type; Cox2, cyclooxygenase 2; ER, endoplasmic reticulum; BiP, binding immunoglobulin protein, also known as glucose-regulated protein–78 (GRP78); CHOP, C/EBP homologous protein, also known as growth arrest and DNA damage–inducible gene–153 (GADD153); IRE-α, inositol-requiring enzyme–1α; ATF6α, activating transcription factor–6α; PERK, protein kinase regulated by RNA-like ER kinase; XBP1, X-box binding protein–1; RA, room air; HYP, hyperoxia. *P < 0.05, t test.
After Postnatal Day 5, murine lungs are at the alveolar stage, similar to human newborn lungs at approximately 36 weeks of gestational age. Continued exposure of the NB mice to hyperoxia from Postnatal Days 1–7 led to an increased expression of Cox and CHOP mRNA (Figure 1D), of BiP, IRE-1α, ATF6α, PERK, and CHOP proteins (Figure 1E), and of the active spliced isoform of X-box binding protein–1 (XBP1) mRNA (Figure 1F).
Effect of Cox2 Inhibition on the Pulmonary Phenotype and the Expression of ER Stress Pathway Mediators in the NB WT Hyperoxia-Induced Murine Model of BPD
We (23) and others (24, 25) have reported on a murine model of BPD created by the exposure of NB WT mice to hyperoxia during the saccular stage of lung development (Postnatal Days 1–4), followed by recovery for 10 days. This model has clinical significance in human BPD, not only for its similarity in terms of pulmonary phenotype, but also in terms of long-term physiologic consequences (26, 27). In this murine model of BPD, we administered the selective Cox2 inhibitor celecoxib, and killed the mice on Postnatal Day 14. Alveolar size, as measured by chord length, demonstrated the rescue of altered alveolar architecture in the celecoxib-administered NB WT lungs in the hyperoxia-induced murine model of BPD (Figure 2A). In addition, we noted a decreased expression of ER stress pathway mediators on Cox2 inhibition in the NB WT murine model of BPD (Figure 2B).
Figure 2.

Effect of Cox2 inhibition on the pulmonary phenotype and ER stress pathway mediators in the hyperoxia-induced murine model of bronchopulmonary dysplasia (BPD). NB WT mice were exposed to room air or hyperoxia to induce the murine model of BPD, received the selective Cox2 inhibitor celecoxib, and were killed on Postnatal Day 14. (A) Alveolar size, as measured by chord length, demonstrated the rescue of altered alveolar architecture in the celecoxib-administered NB WT mice lungs in the hyperoxia-induced murine model of BPD. Each bar represents the mean ± SEM for a minimum of four animals. (B) The mRNA expression of Cox2, BiP, CHOP, and caspase-3 in lung tissue was noted on Postnatal Day 14 of the NB WT hyperoxia-induced murine model of BPD. Each lane in the gel is representative of the condition described, with the corresponding β-actin as control sample. For the definition of other abbreviations, see the legend for Figure 1 or the text. *P < 0.05 and **P ≤ 0.01, t test.
Effect of Hyperoxia on the Expression of ER Stress Pathway Mediators in Alveolar Epithelial Cells
For a better understanding of the cell-specific downstream effects of the activation of the ER stress pathway in alveolar epithelial cells (AECs), we exposed MLE-12 cells to hyperoxia for 24 hours. No change in BiP mRNA, but an increase in CHOP mRNA expression, was evident (Figure E1A in the online supplement). On Western blotting, the expression of BiP and CHOP were increased (Figure E1B).
Effect of CHOP Inhibition on Cell Death in AECs Exposed to Hyperoxia
The knockdown of CHOP in MLE-12 cells exposed to hyperoxia resulted in a decrease in cell death (Figure E2).
Effect of CHOP Inhibition on Cell Death and the Pulmonary Phenotype in the NB WT Hyperoxia-Induced Murine Model of BPD
In the NB WT murine model of BPD, the knockdown of CHOP resulted in a significant decrease in TUNEL-positive cells on Postnatal Day 14 (Figure 3A). Concomitantly, we noted a significant improvement in the pulmonary phenotype, as evidenced by decreased chord length (Figure 3B).
Figure 3.

Effect of CHOP inhibition on cell death and pulmonary phenotype in the NB WT hyperoxia-induced murine model of BPD. NB WT mice were exposed to room air or hyperoxia to induce the murine model of BPD, received the selective CHOP (or scrambled control) small interfering (si)RNA (as described in Materials and Methods), and were killed on Postnatal Day 14. (A) The quantification of TUNEL-positive cells was performed in the lungs (as described in Materials and Methods), and expressed as a percentage. (B) Alveolar size, as measured by chord length, demonstrated the rescue of altered alveolar architecture in the NB WT mice lungs receiving CHOP siRNA in the hyperoxia-induced murine model of BPD. Each bar represents the mean ± SEM for a minimum of four animals. BPD, bronchopulmonary dysplasia; RA, room air. For the definition of other abbreviations, see the legend of Figure 1 or the text. Each bar represents the mean ± SEM for a minimum of four animals. *P < 0.05, **P ≤ 0.01, and ***P ≤ 0.001, t test.
Taken together, these data suggest that hyperoxia increased Cox2 in the developing murine lung, and acted via the ER stress pathway to result in the BPD pulmonary phenotype. More specifically, our data suggest that Cox2 is found upstream of the ER stress pathway that is modulated via CHOP in hyperoxia-induced injury models of the developing murine lung. The inhibition of Cox2 and/or CHOP resulted in an improvement in the pulmonary phenotype of murine BPD. Hence, Cox2 is a critical mediator in the hyperoxia-induced pathway of injury to the developing murine lung.
To validate our premise further and establish the signaling pathway, we investigated an additional murine model of BPD. We have recently reported that increased IFN-γ in the developing lung leads to a pulmonary phenotype suggestive of human BPD (18).
Effect of the Lack of IFN-γ on the Pulmonary Phenotype, Cox2, and ER Stress Pathway Mediators in the NB Hyperoxia-Induced Murine Model of BPD
To validate the role of IFN-γ, we evaluated the impact of a genetic lack of IFN-γ on the murine model of hyperoxia-induced BPD. As noted in Figure E3, the absence of IFN-γ was significantly protective of the BPD pulmonary phenotype. These data reaffirm the importance of the role for IFN-γ in the murine model of BPD.
Effect of Increased IFN-γ in the NB Lung on Cox2 and ER Stress Pathway Mediators in the NB IFN-γ TTG Model of BPD
Next, we evaluated the mRNA expression of Cox2 and ER stress pathway mediators (Figure 4A) in our previously described NB IFN-γ TTG model of BPD (18). No difference in caspase-12 mRNA expression was evident (Figure 4A). On densitometry, the increase in CHOP mRNA was statistically significant in the NB IFN-γ TTG murine lungs (Figure 4B). Western blotting for protein yielded a marked increase in Cox2, BiP, and CHOP proteins in NB IFN-γ TTG murine lungs (Figure 4C). These data suggest that, as with hyperoxia-exposed NB WT lungs, Cox2 and ER stress pathway mediators are up-regulated in the NB IFN-γ TTG murine BPD model. In addition, the CHOP-dependent pathway, but not the caspase-12–dependent pathway, appears to be activated.
Figure 4.

Effect of increased IFN-γ in the NB lung on Cox2 and ER stress pathway mediators. (A) mRNA expression in the lung tissue of Cox2, BiP, CHOP, and caspase-12 was noted on Postnatal Day 7 of NB IFN-γ TTG (+) or WT (−) mice receiving regular water (−) or DOX (+) water. Each lane in the gel is representative of the condition described, with the corresponding β-actin as control sample. (B) The quantified mRNA expression of CHOP was noted on Postnatal Day 7 of NB IFN-γ TTG (+) or WT (−) mice receiving regular water (−) or DOX (+) water. Each bar represents the mean ± SEM for a minimum of four animals. (C) The protein expression of Cox2, BiP, and CHOP was detected by Western blotting. Each lane in the gel is representative of the condition described, with the corresponding β-tubulin as control sample. IFN-γ TTG, IFN-γ triple transgenic; DOX, doxycycline. For the definition of other abbreviations, see the legend for Figure 1 or the text. *P < 0.05 and **P ≤ 0.01, t test.
Effect of CHOP Inhibition on Cell Death and the Pulmonary Phenotype in the NB IFN-γ TTG Model of BPD
In the NB IFN-γ TTG model of BPD, the knockdown of CHOP resulted in a significant decrease in TUNEL-positive cells on Postnatal Day 7 (Figure 5A). Concomitantly, we noted a significant improvement in pulmonary phenotype, as evidenced by decreased chord length (Figure 5B).
Figure 5.

Effect of CHOP inhibition on cell death and the pulmonary phenotype in the IFN-γ–overexpressing murine model of BPD. NB IFN-γ TTG (+) or WT receiving DOX water in room air were administered the selective CHOP (or scrambled control) siRNA (as described in Materials and Methods), and killed on Postnatal Day 7. (A) The quantification of TUNEL-positive cells was performed in the lungs (as described in Materials and Methods), and expressed as a percentage. (B) Alveolar size, as measured by chord length, demonstrated the rescue of altered alveolar architecture in the CHOP siRNA–administered IFN-γ–overexpressing murine model of BPD. +ve, positive; IFN-γ TTG, IFN-γ triple transgenic; DOX, doxycycline; BPD, bronchopulmonary dysplasia. For the definition of other abbreviations, see the legend for Figure 1 or the text. Each bar represents the mean ± SEM for a minimum of four animals. *P < 0.05, **P ≤ 0.01, and ***P ≤ 0.001, t test.
Effect of Cox2 Inhibition on the Pulmonary Phenotype and the Expression of ER Stress Pathway Mediators in the IFN-γ–Overexpressing Murine Model of BPD
We administered the selective Cox2 inhibitor, celecoxib, as described in Materials and Methods, to the NB IFN-γ TTG mice. We were able to rescue the murine BPD pulmonary phenotype (Figure 6A). As previously described (18), we stained lung tissue with α-SMA for the relative proportion of myofibroblast-type cells, and found that the area of distribution for α-SMA staining was increased in NB IFN-γ TTG mice on Dox water (Figure 6B). This increased proportion may be attributable to the proliferation of existing myofibroblasts, or to the transition/transformation of other cells into myofibroblasts. We noted a partial normalization of the α-SMA staining distribution in NB IFN-γ TTG mice on Dox water and in those that had received celecoxib. This was quantified through the use of an SMA distortion index, as described in Materials and Methods, and as depicted in Figure 6B. To assess the impact of celecoxib on the ER stress pathway, we evaluated the expression of ER stress pathway mediators in NB IFN-γ TTG murine lungs. We noted a decreased mRNA expression of ER stress pathway mediators after Cox2 inhibition in the IFN-γ–overexpressing murine model of BPD (Figure 6C). As noted in Figure 6D, CHOP mRNA was restored (as compared with Figure 4B) to concentrations similar to those in control samples. Furthermore, we confirmed that Cox2 inhibition decreased the protein expression of ER stress pathway mediators in the IFN-γ–overexpressing murine model of BPD (Figure 6E). Hence, Cox2 activation would appear to occur upstream of CHOP activation in our NB IFN-γ TTG mice model of BPD.
Figure 6.

Effect of Cox2 inhibition on the pulmonary phenotype and ER stress pathway mediators in the IFN-γ–overexpressing murine model of BPD. (A) NB IFN-γ TTG (+) or WT (−) mice receiving regular (−) or DOX (+) water in room air were administered the selective Cox2 inhibitor celecoxib, and were killed on Postnatal Day 7. Alveolar size, as measured by chord length, demonstrated the rescue of altered alveolar architecture in the NB IFN-γ TTG (+) murine model of BPD. The data in the first four bars from the left were previously shown in Figures 5B and 5D, respectively, of Harijith and colleagues (18). (B) The quantification of α-smooth muscle actin (SMA) staining (by distortion index measurement) and the normalization of the stained area in the NB IFN-γ TTG murine lung tissue was undertaken via Cox2 inhibition. The data in the first four bars from the left were previously shown in Figures 5B and 5D, respectively, of Harijith and colleagues (18). (C) mRNA expression of Cox2, BiP, ATF4, CHOP, and caspase-3 was noted in the lung tissue on Postnatal Day 7 in the IFN-γ–overexpressing murine model of BPD. Each lane in the gel is representative of the condition described, with the corresponding β-actin as control sample. (D) Quantification of mRNA expression of CHOP in the lung tissue was noted on Postnatal Day 7 in the IFN-γ–overexpressing murine model of BPD. (E) Protein expression of BiP, phosphorylated and total eIFα, ATF3, CHOP, and total and cleaved caspase-3 in the lung tissue was noted on Postnatal Day 7 in the IFN-γ–overexpressing murine model of BPD. Each lane in the gel is representative of the condition described, with the corresponding β-actin as control sample. IFN-γ TTG, IFN-γ triple transgenic; DOX, doxycycline water; BPD, bronchopulmonary dysplasia. HYP, hyperoxia; eIF2α, eukaryotic initiation factor–2α; ATF4, activating transcription factor–4. For the definition of other abbreviations, see the legend for Figure 1 or the text. Each bar represents the mean ± SEM for a minimum of three animals. *P < 0.05, **P ≤ 0.01, and ***P ≤ 0.001, t test.
Association of Cox2, BiP, and CHOP in Human Neonatal Lungs with BPD
To ascertain the clinical relevance of our findings, we used immunohistochemistry to detect Cox2 in human neonatal lungs. As depicted in the representative microphotographs in Figure 7A, an increased brown staining of the epithelial and inflammatory cells in the BPD lung was evident, compared with control samples.
Figure 7.
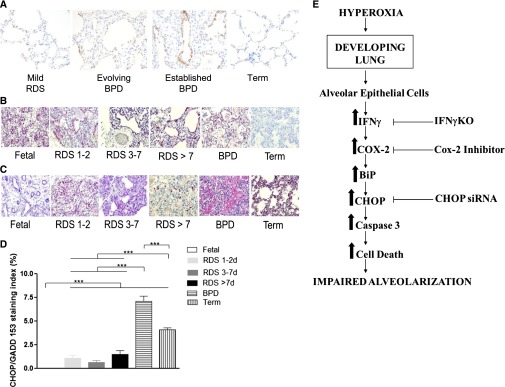
Expression of Cox2, BiP, and CHOP in human lungs with BPD. Immunohistochemical Cox2 staining was performed on human lung tissue from newborns (NB) with mild respiratory distress syndrome (Mild RDS), NB with evolving bronchopulmonary dysplasia (Evolving BPD), and established BPD. (A) NB lungs from a full-term infant (Term) are shown as control samples. Each microphotograph is representative of two lung samples from each of the four groups. Increased Cox2 (brown) staining of the epithelial and inflammatory cells was observed in the BPD lungs, compared with control samples. (B and C) Immunohistochemical BiP and CHOP staining of human lungs of newborn (NB) with early (RDS 1–2), evolving (RDS 3–7), and late (RDS > 7) phases of respiratory distress syndrome (RDS), and established bronchopulmonary dysplasia (BPD). NB lungs from fetal subjects (Fetal) and full-term infants (Term) are shown as control samples. Each microphotograph (×20 magnification) is representative of two lung samples from each of the six groups. BiP staining is shown in B, whereas CHOP staining is shown in C. (D) The quantification of CHOP staining is shown. Increased pink staining of the epithelial and inflammatory cells was observed in BPD lungs, compared with control samples. ***P ≤ 0.001, t test. (E) Proposed pathway for impaired alveolarization in the developing lung, the pathologic hallmark of BPD. Hyperoxic exposure to the developing lung (saccular/early alveolar phases) results in enhanced secretion of IFN-γ from alveolar epithelial cells. This increases Cox2, which activates the ER stress pathway. This results in an increase in BiP, with progression downstream via the CHOP-dependent ER stress pathway, leading to increased caspase-3 and enhanced cell death, in turn leading to impaired alveolarization. The loss of IFN-γ, selective Cox2 (by celecoxib), or CHOP (by siRNA) inhibition at different points in the pathway is able to rescue, in part, the pulmonary phenotype of BPD. BPD, bronchopulmonary dysplasia. For the definition of other abbreviations, see the legend of Figure 1 or the text.
We used immunohistochemistry to detect BiP and CHOP in human neonatal lungs. As depicted in the representative microphotographs in Figures 7B and 7C, respectively, increased pink staining of the epithelial and inflammatory cells in BPD lungs was evident, compared with control samples. We were able to quantify nuclear CHOP staining in neonatal lungs with BPD (Figure 7D).
Taken together, our data suggest that hyperoxia and ventilation-induced injury to the developing lung are accompanied by the activation of Cox2 and the ER stress pathway. The alveolar simplification phenotype noted in the NB WT and IFN-γ TTG murine lungs could be rescued by selective Cox2 inhibition, which occurs upstream from CHOP in the ER stress pathway. Hence, our data further suggest that impaired alveolarization attributable to increased IFN-γ in the developing NB lung is dependent, at least in part, on Cox2 and CHOP (Figure 7E). Furthermore, neonatal human lungs with BPD show an increased protein expression of Cox2, BiP, and CHOP.
Discussion
Injury to the developing lung in the saccular/early alveolar stages secondary to hyperoxia has been implicated in impaired alveolarization, the pathologic hallmark of “new” BPD (26). An important aspect of this pathological process involves cell death secondary to hyperoxia (6, 28, 29). Although a variety of mechanisms account for cell death in the lung (30), we decided to focus on the ER stress pathway, because limited information is available on this process in the context of BPD.
As already mentioned, ER stress is sensed by three ER-resident transmembrane proteins, IRE-1α, ATF6α, and PERK (10). Because all three are freed from BiP during ER stress, BiP binding is important in regulating the activation of each arm in the ER stress pathways (10). ER stress–induced cell death signaling can occur via multiple pathways (8, 10): (1) Activated IRE-1α can recruit c-Jun N-terminal inhibitory kinase (JIK) and tumor necrosis factor receptor–associated factor–2 (TRAF2) to activate apoptosis-signal regulating kinase–1 and c-Jun N-terminal kinase (JNK), leading to the activation of a mitochondria-dependent cell-death pathway (i.e., caspase-8, Bcl-2–associated X protein or Bax, the release of cytochrome c, and the activation of caspase-3 and caspase-9). IRE-1α also stimulates endonuclease activity, resulting in the formation of the active spliced isoform of XBP1 (31, 32). (2) The release of JIK from procaspase-12 allows for activation to caspase-12, which activates procaspase-9, which in turn activates procaspase-3, the executioner of cell death. (3) Finally, activated PERK phosphorylates the eukaryotic initiation factor–2α that enhances the translation of ATF4 mRNA, which is important for ATF3 induction, which in turn has been shown to induce CHOP (31, 33). CHOP can inhibit antiapoptotic Bcl-2, leading to the activation of the executioner caspase-3.
For the in vitro approach, we used alveolar epithelial MLE-12 cells. We were able to detect activation of the mediators (via the release of BiP and increased CHOP) of the ER stress pathway. Blocking CHOP decreased cell death. With our in vivo experiments, we confirmed in vitro observations by producing similar results with BiP and CHOP as well as IRE-1α, XBP1, ATF6α, and PERK in NB WT murine lungs exposed to hyperoxia. In addition, we noted a marked increase in Cox2. To investigate the role of Cox2 in the pulmonary phenotype of BPD, we studied NB WT and IFN-γ TTG murine models of BPD. We were able to rescue the phenotype as well as recover CHOP expression (and other ER stress pathway mediators) by administering the selective Cox2-inhibitor, celecoxib. In these two in vivo models of BPD, CHOP siRNA restored alveolarization. For translational significance, we were able to detect increased Cox2, BiP, and CHOP protein in human lungs with evolving and established BPD.
Recently, a number of papers have implicated ER stress–induced cell death in adult lungs of experimental models and humans with idiopathic pulmonary fibrosis (34–36). ER stress–induced cell death has also been reported in adult lungs, secondary to oxidative stress attributable to cigarette-smoke exposure and/or chronic obstructive pulmonary disease (37–40). Interestingly, most reports on adult lungs have implicated alveolar epithelial cells (34, 39–42) and the activation of CHOP (34, 38–40).
In terms of hyperoxic exposure, an earlier study evaluated MLE-12 cells for 24 hours, and noted little change in BiP mRNA and protein concentrations (43). We noted no change in BiP mRNA. However, BiP protein appeared to be increased in MLE-12 cells (Figures E1A and E1B). On the other hand, we noted a marked increase in CHOP protein (Figure E1B), as also reported in an earlier study with MLE-12 cells (43). We found that CHOP siRNA decreased cell death in MLE-12 cells (Figure E2).
Human lung adenocarcinoma cell lines (A549 and H1299), when exposed to hyperoxia for up to 72 hours, exhibited decreased BiP and did not activate the ER stress pathway (44). Using the human endothelial cell line EA.hy926, investigators have associated increased BiP expression with decreased hyperoxia-induced cell death (45). Konsavage and colleagues reported that the activation of mediators upstream in the ER stress pathway, upon exposure to hyperoxia for 24 hours, was evident in human small airway epithelial and pulmonary microvascular endothelial cells, but not in neonatal human lung fibroblasts (31).
Lozon and colleagues reported similar results in vivo, using adult mice exposed to hyperoxia for up to 72 hours. In that study, the induction of CHOP was considered independent of the ER stress pathway because BiP concentrations did not alter, but was considered dependent on RNA-dependent protein kinase (PKR), because PKR phosphorylation was increased for up to 72 hours (43). In addition, they reported that CHOP was protective in hyperoxia-induced lung injury among adult mice (43).
In a recent study by Konsavage and colleagues, the alteration in ER morphology was most prominent in Type II pneumocytes and interstitial fibroblasts in NB rat lungs exposed to hyperoxia for 24 hours (31). At 24 hours, the investigators did not note any statistically significant changes in BiP expression (31), but they did not report their results regarding BiP or CHOP expression at later time points. Interestingly, however, they did report increased ATF4 and ATF3 expression at 24 hours and 72 hours, respectively (31). Both these mediators are upstream from CHOP in the ER stress pathway (8, 31). Interestingly, PKR protein, but not PKR phosphorylation, was increased in NB rat lungs exposed to hyperoxia for 24 and 72 hours.
In sharp contrast to the adult murine data (43), our in vivo NB murine hyperoxic exposures on Postnatal Days 2 and 7 showed marked increases in both BiP and CHOP (Figure 1). Our results suggest that the increase in CHOP is related to the ER stress pathway, and highlight the developmental regulation of these processes. In addition, the data on PKR phosphorylation are in sharp contrast between adult murine lungs (43) and NB rat lungs (31). We previously reported on similar significant differences in the responses of adult versus NB mice upon exposure to hyperoxia (12, 46–48). CHOP can obviously be activated in NB lungs via both ER stress–independent and ER stress–dependent pathways. This could be reflective of the type, timing, and duration of injury. Given that only the developing lung develops BPD, the activation of CHOP reinforces the importance of evaluating developmentally appropriate systems independently, using specific experimental conditions and not extrapolating from adult in vivo data.
We noted increased Cox2 expression, concomitant with increased CHOP expression, upon exposure to hyperoxia on Postnatal Days 2 and 7 (Figure 1). Rogers and colleagues reported increased Cox2 protein in the lungs of NB mice exposed to hyperoxia on Postnatal Days 7 and 14, but not earlier (49). This difference in timing could be related to the different strain (C3H/HeN) of mice used (49). Because Cox2 has been suggested to play a potential role in infants with BPD (22), we investigated this role further in our recently described NB IFN-γ TTG murine model of BPD. We noted increased mRNA and protein expression of Cox2, concomitant with increased BiP and CHOP (Figure 4). Celecoxib, a selective Cox2 inhibitor, was able to rescue the pulmonary phenotype in both the NB IFN-γ TTG (Figure 6) and hyperoxia-induced (Figure 2) murine models of BPD. Because this rescue was accompanied by a normalization of CHOP mRNA (and other ER stress pathway mediators; Figures 2 and 6), we suggest that Cox2 occurs upstream from CHOP (Figure 7E). Our results are in contrast to those reported in the human lung cancer cell lines, A549 and H460, where Cox2 inhibition using niflumic acid increased CHOP protein expression (50). As already mentioned, such in vitro data would not be considered equivalent to NB developing lung in vivo data.
Our Cox2 staining data on human lungs are supported by a previous report implicating a potential role for Cox2 in the perinatal lung (22). To our knowledge, this is the first study to report on the characteristics of BiP and CHOP staining in human lungs with respiratory distress syndrome and BPD.
In conclusion, our experiments highlight the role of Cox2 in mediating hyperoxia-induced and IFN-γ–induced injury to the developing lung via the ER stress pathway mediator, CHOP. Moreover, they suggest a novel signaling connection between hyperoxia, IFN-γ, Cox2, and CHOP. The clinical relevance is supported by our human lung staining data. Additional investigations of the potential therapeutic utility of anti-Cox2–based and anti-CHOP–based approaches to BPD are warranted.
Footnotes
This work was supported by grant HL63039 (G.P.) from the National Heart, Lung, and Blood Institute of the National Institutes of Health, by the Sigrid Jusélius Foundation (S.A.), by grant 0755843T (V.B.) from the American Heart Association, by grant ATS-07-005 (V.B.) from the American Thoracic Society, and by grants HL74195 and HL85103 (V.B.) from the National Heart, Lung, and Blood Institute of the National Institutes of Health.
Author Contributions: R.C.-W., M.A.S., and V.B. contributed to the concept and design of this study. R.C.-W., M.A.S., A.H., B.B., G.P., C.J., S.A., R.J.H., and V.B. contributed to the acquisition of study data. R.C.-W., M.A.S., A.H., G.P., C.J., S.A., R.J.H., and V.B. contributed to the analysis and interpretation of study data. R.C.-W., M.A.S., A.H., B.B., G.P., C.J., S.A., R.J.H., and V.B. contributed to drafting and/or critically revising the manuscript for intellectual content. All authors approved the final version of the manuscript.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2012-0381OC on March 7, 2013
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Bhandari A, Bhandari V. Pathogenesis, pathology and pathophysiology of pulmonary sequelae of bronchopulmonary dysplasia in premature infants. Front Biosci. 2003;8:e370–e380. doi: 10.2741/1060. [DOI] [PubMed] [Google Scholar]
- 2.Bhandari A, Bhandari V. Bronchopulmonary dysplasia: an update. Indian J Pediatr. 2007;74:73–77. doi: 10.1007/s12098-007-0032-z. [DOI] [PubMed] [Google Scholar]
- 3.Bhandari V, Bizzarro MJ, Shetty A, Zhong X, Page GP, Zhang H, Ment LR, Gruen JR. Familial and genetic susceptibility to major neonatal morbidities in preterm twins. Pediatrics. 2006;117:1901–1906. doi: 10.1542/peds.2005-1414. [DOI] [PubMed] [Google Scholar]
- 4.Baraldi E, Filippone M. Chronic lung disease after premature birth. N Engl J Med. 2007;357:1946–1955. doi: 10.1056/NEJMra067279. [DOI] [PubMed] [Google Scholar]
- 5.Bhandari V. Molecular mechanisms of hyperoxia-induced acute lung injury. Front Biosci. 2008;13:6653–6661. doi: 10.2741/3179. [DOI] [PubMed] [Google Scholar]
- 6.Bhandari V. Hyperoxia-derived lung damage in preterm infants. Semin Fetal Neonatal Med. 2010;15:223–229. doi: 10.1016/j.siny.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pagano A, Barazzone-Argiroffo C. Alveolar cell death in hyperoxia-induced lung injury. Ann N Y Acad Sci. 2003;1010:405–416. doi: 10.1196/annals.1299.074. [DOI] [PubMed] [Google Scholar]
- 8.Zhang K, Kaufman RJ. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;66:S102–S109. doi: 10.1212/01.wnl.0000192306.98198.ec. [DOI] [PubMed] [Google Scholar]
- 9.Momoi T. Caspases involved in ER stress–mediated cell death. J Chem Neuroanat. 2004;28:101–105. doi: 10.1016/j.jchemneu.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 10.Rutkowski DT, Kaufman RJ. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem Sci. 2007;32:469–476. doi: 10.1016/j.tibs.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Zaher TE, Miller EJ, Morrow DM, Javdan M, Mantell LL. Hyperoxia-induced signal transduction pathways in pulmonary epithelial cells. Free Radic Biol Med. 2007;42:897–908. doi: 10.1016/j.freeradbiomed.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhandari V, Elias JA. Cytokines in tolerance to hyperoxia-induced injury in the developing and adult lung. Free Radic Biol Med. 2006;41:4–18. doi: 10.1016/j.freeradbiomed.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 13.Shea LM, Beehler C, Schwartz M, Shenkar R, Tuder R, Abraham E. Hyperoxia activates NF-kappaB and increases TNF-alpha and IFN-gamma gene expression in mouse pulmonary lymphocytes. J Immunol. 1996;157:3902–3908. [PubMed] [Google Scholar]
- 14.Yamada M, Kubo H, Kobayashi S, Ishizawa K, Sasaki H. Interferon-gamma: a key contributor to hyperoxia-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1042–L1047. doi: 10.1152/ajplung.00155.2004. [DOI] [PubMed] [Google Scholar]
- 15.Kim KB, Choi YH, Kim IK, Chung CW, Kim BJ, Park YM, Jung YK. Potentiation of Fas- and Trail-mediated apoptosis by IFN-gamma in A549 lung epithelial cells: enhancement of caspase-8 expression through IFN-response element. Cytokine. 2002;20:283–288. doi: 10.1006/cyto.2003.2008. [DOI] [PubMed] [Google Scholar]
- 16.Wang Z, Zheng T, Zhu Z, Homer RJ, Riese RJ, Chapman HA, Jr, Shapiro SD, Elias JA. Interferon gamma induction of pulmonary emphysema in the adult murine lung. J Exp Med. 2000;192:1587–1600. doi: 10.1084/jem.192.11.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wen LP, Madani K, Fahrni JA, Duncan SR, Rosen GD. Dexamethasone inhibits lung epithelial cell apoptosis induced by IFN-gamma and Fas. Am J Physiol. 1997;273:L921–L929. doi: 10.1152/ajplung.1997.273.5.L921. [DOI] [PubMed] [Google Scholar]
- 18.Harijith A, Choo-Wing R, Cataltepe S, Yasumatsu R, Aghai ZH, Janer J, Andersson S, Homer RJ, Bhandari V. A role for matrix metalloproteinase 9 in IFNgamma-mediated injury in developing lungs: relevance to bronchopulmonary dysplasia. Am J Respir Cell Mol Biol. 2011;44:621–630. doi: 10.1165/rcmb.2010-0058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitchell JA, Belvisi MG, Akarasereenont P, Robbins RA, Kwon OJ, Croxtall J, Barnes PJ, Vane JR. Induction of cyclo-oxygenase–2 by cytokines in human pulmonary epithelial cells: regulation by dexamethasone. Br J Pharmacol. 1994;113:1008–1014. doi: 10.1111/j.1476-5381.1994.tb17093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newton R, Kuitert LM, Slater DM, Adcock IM, Barnes PJ. Cytokine induction of cytosolic phospholipase A2 and cyclooxygenase-2 mRNA is suppressed by glucocorticoids in human epithelial cells. Life Sci. 1997;60:67–78. doi: 10.1016/s0024-3205(96)00590-5. [DOI] [PubMed] [Google Scholar]
- 21.Asano K, Nakamura H, Lilly CM, Klagsbrun M, Drazen JM. Interferon gamma induces prostaglandin G/H synthase–2 through an autocrine loop via the epidermal growth factor receptor in human bronchial epithelial cells. J Clin Invest. 1997;99:1057–1063. doi: 10.1172/JCI119233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lassus P, Wolff H, Andersson S. Cyclooxygenase-2 in human perinatal lung. Pediatr Res. 2000;47:602–605. doi: 10.1203/00006450-200005000-00008. [DOI] [PubMed] [Google Scholar]
- 23.Li Z, Choo-Wing R, Sun H, Sureshbabu A, Sakurai R, Rehan VK, Bhandari V. A potential role of the JNK pathway in hyperoxia-induced cell death, myofibroblast transdifferentiation and TGF-beta1–mediated injury in the developing murine lung. BMC Cell Biol. 2011;12:54. doi: 10.1186/1471-2121-12-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Reilly MA, Marr SH, Yee M, McGrath-Morrow SA, Lawrence BP. Neonatal hyperoxia enhances the inflammatory response in adult mice infected with influenza A virus. Am J Respir Crit Care Med. 2008;177:1103–1110. doi: 10.1164/rccm.200712-1839OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yee M, Chess PR, McGrath-Morrow SA, Wang Z, Gelein R, Zhou R, Dean DA, Notter RH, O’Reilly MA. Neonatal oxygen adversely affects lung function in adult mice without altering surfactant composition or activity. Am J Physiol Lung Cell Mol Physiol. 2009;297:L641–L649. doi: 10.1152/ajplung.00023.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhandari A, Bhandari V. The “new” bronchopulmonary dysplasia: a clinical review. Clin Pulm Med. 2011;18:137–143. [Google Scholar]
- 27.Yee M, White RJ, Awad HA, Bates WA, McGrath-Morrow SA, O’Reilly MA. Neonatal hyperoxia causes pulmonary vascular disease and shortens life span in aging mice. Am J Pathol. 2011;178:2601–2610. doi: 10.1016/j.ajpath.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alphonse RS, Vadivel A, Coltan L, Eaton F, Barr AJ, Dyck JR, Thebaud B. Activation of AKT protects alveoli from neonatal oxygen-induced lung injury. Am J Respir Cell Mol Biol. 2011;44:146–154. doi: 10.1165/rcmb.2009-0182OC. [DOI] [PubMed] [Google Scholar]
- 29.Das KC, Ravi D, Holland W. Increased apoptosis and expression of p21 and p53 in premature infant baboon model of bronchopulmonary dysplasia. Antioxid Redox Signal. 2004;6:109–116. doi: 10.1089/152308604771978417. [DOI] [PubMed] [Google Scholar]
- 30.Tang PS, Mura M, Seth R, Liu M. Acute lung injury and cell death: how many ways can cells die? Am J Physiol Lung Cell Mol Physiol. 2008;294:L632–L641. doi: 10.1152/ajplung.00262.2007. [DOI] [PubMed] [Google Scholar]
- 31.Konsavage WM, Zhang L, Wu Y, Shenberger JS. Hyperoxia-induced activation of the integrated stress response in the newborn rat lung. Am J Physiol Lung Cell Mol Physiol. 2012;302:L27–L35. doi: 10.1152/ajplung.00174.2011. [DOI] [PubMed] [Google Scholar]
- 32.Karar J, Dolt KS, Qadar Pasha MA. Endoplasmic reticulum stress response in murine kidney exposed to acute hypobaric hypoxia. FEBS Lett. 2008;582:2521–2526. doi: 10.1016/j.febslet.2008.06.019. [DOI] [PubMed] [Google Scholar]
- 33.Liu G, Su L, Hao X, Zhong N, Zhong D, Singhal S, Liu X. Salermide up-regulates death receptor 5 expression through the ATF4–ATF3–CHOP axis and leads to apoptosis in human cancer cells. J Cell Mol Med. 2012;16:1618–1628. doi: 10.1111/j.1582-4934.2011.01401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, Lang G, Fink L, Bohle RM, Seeger W, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178:838–846. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, Blackwell TR, Xu C, Markin C, Ware LB, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1119–L1126. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- 36.Baek HA, Kim DS, Park HS, Jang KY, Kang MJ, Lee DG, Moon WS, Chae HJ, Chung MJ. Involvement of endoplasmic reticulum stress in myofibroblastic differentiation of lung fibroblasts. Am J Respir Cell Mol Biol. 2012;46:731–739. doi: 10.1165/rcmb.2011-0121OC. [DOI] [PubMed] [Google Scholar]
- 37.Csordas A, Kreutmayer S, Ploner C, Braun PR, Karlas A, Backovic A, Wick G, Bernhard D. Cigarette smoke extract induces prolonged endoplasmic reticulum stress and autophagic cell death in human umbilical vein endothelial cells. Cardiovasc Res. 2011;92:141–148. doi: 10.1093/cvr/cvr165. [DOI] [PubMed] [Google Scholar]
- 38.Tagawa Y, Hiramatsu N, Kasai A, Hayakawa K, Okamura M, Yao J, Kitamura M. Induction of apoptosis by cigarette smoke via ROS-dependent endoplasmic reticulum stress and CCAAT/enhancer-binding protein–homologous protein (CHOP) Free Radic Biol Med. 2008;45:50–59. doi: 10.1016/j.freeradbiomed.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 39.Malhotra D, Thimmulappa R, Vij N, Navas-Acien A, Sussan T, Merali S, Zhang L, Kelsen SG, Myers A, Wise R, et al. Heightened endoplasmic reticulum stress in the lungs of patients with chronic obstructive pulmonary disease: the role of NRF2-regulated proteasomal activity. Am J Respir Crit Care Med. 2009;180:1196–1207. doi: 10.1164/rccm.200903-0324OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Geraghty P, Wallace A, D’Armiento JM. Induction of the unfolded protein response by cigarette smoke is primarily an activating transcription factor 4–c/EBP homologous protein mediated process. Int J Chron Obstruct Pulmon Dis. 2011;6:309–319. doi: 10.2147/COPD.S19599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanjore H, Cheng DS, Degryse AL, Zoz DF, Abdolrasulnia R, Lawson WE, Blackwell TS. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem. 2011;286:30972–30980. doi: 10.1074/jbc.M110.181164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weichert N, Kaltenborn E, Hector A, Woischnik M, Schams A, Holzinger A, Kern S, Griese M. Some ABCA3 mutations elevate ER stress and initiate apoptosis of lung epithelial cells. Respir Res. 2011;12:4. doi: 10.1186/1465-9921-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lozon TI, Eastman AJ, Matute-Bello G, Chen P, Hallstrand TS, Altemeier WA. PKR-dependent CHOP induction limits hyperoxia-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2011;300:L422–L429. doi: 10.1152/ajplung.00166.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gewandter JS, Staversky RJ, O’Reilly MA. Hyperoxia augments ER-stress–induced cell death independent of BIP loss. Free Radic Biol Med. 2009;47:1742–1752. doi: 10.1016/j.freeradbiomed.2009.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu D, Perez RE, Rezaiekhaligh MH, Bourdi M, Truog WE. Knockdown of ERP57 increases BIP/GRP78 induction and protects against hyperoxia and tunicamycin-induced apoptosis. Am J Physiol Lung Cell Mol Physiol. 2009;297:L44–L51. doi: 10.1152/ajplung.90626.2008. [DOI] [PubMed] [Google Scholar]
- 46.Choo-Wing R, Nedrelow JH, Homer RJ, Elias JA, Bhandari V. Developmental differences in the responses of IL-6 and IL-13 transgenic mice exposed to hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2007;293:L142–L150. doi: 10.1152/ajplung.00434.2006. [DOI] [PubMed] [Google Scholar]
- 47.Bhandari V. Developmental differences in the role of interleukins in hyperoxic lung injury in animal models. Front Biosci. 2002;7:1624–1633. doi: 10.2741/A866. [DOI] [PubMed] [Google Scholar]
- 48.Bhandari V, Choo-Wing R, Lee CG, Yusuf K, Nedrelow JH, Ambalavanan N, Malkus H, Homer RJ, Elias JA. Developmental regulation of NO-mediated VEGF-induced effects in the lung. Am J Respir Cell Mol Biol. 2008;39:420–430. doi: 10.1165/rcmb.2007-0024OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rogers LK, Tipple TE, Nelin LD, Welty SE. Differential responses in the lungs of newborn mouse pups exposed to 85% or >95% oxygen. Pediatr Res. 2009;65:33–38. doi: 10.1203/PDR.0b013e31818a1d0a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim BM, Maeng K, Lee KH, Hong SH. Combined treatment with the Cox-2 inhibitor niflumic acid and PPARgamma ligand ciglitazone induces ER stress/caspase-8–mediated apoptosis in human lung cancer cells. Cancer Lett. 2011;300:134–144. doi: 10.1016/j.canlet.2010.09.014. [DOI] [PubMed] [Google Scholar]