

Abstract
Mechanical ventilation with high tidal volumes has been associated with pulmonary alveolar flooding. Understanding the mechanisms underlying cyclic stretch–induced increases in alveolar epithelial permeability may be important in designing preventive measures for acute lung injury. In this work, we assessed whether cyclic stretch leads to the generation of reactive oxygen species in type I–like alveolar epithelial cells, which increase monolayer permeability via activation of NF-κB and extracellular signal–regulated kinase (ERK). We cyclically stretched type I–like rat primary alveolar epithelial cells at magnitudes of 12, 25, and 37% change in surface area (ΔSA) for 10 to 120 minutes. High levels of reactive oxygen species and of superoxide and NO specifically were detected in cells stretched at 37% ΔSA for 10 to 120 minutes. Exogenous superoxide and NO stimulation increased epithelial permeability in unstretched cells, which was preventable by the NF-κB inhibitor MG132. The cyclic stretch–induced increase in permeability was decreased by the superoxide scavenger tiron and by MG132. Furthermore, tiron had a dramatic protective effect on in vivo lung permeability under mechanical ventilation conditions. Cyclic stretch increased the activation of the NF-κB signaling pathway, which was significantly decreased with the ERK inhibitor U0126. Altogether, our in vitro and in vivo data demonstrate the sensitivity of permeability to stretch- and ventilation-induced superoxide production, suggesting that using antioxidants may be helpful in the prevention and treatment of ventilator-induced lung injury.
Keywords: acute lung injury, MAPK, mechanical deformation, p65, ventilator-induced lung injury
Clinical Relevance
Mechanical ventilation is associated with increases in the pulmonary epithelial barrier permeability, yet the underlying mechanisms are unknown. Oxidative injury has been associated independently with stretch and increased permeability in other tissues, but these mechanisms have not been linked in type I–like alveolar epithelial cells. Our data demonstrate for the first time that cyclic stretch of primary type I–like rat alveolar epithelial cells generates reactive oxygen species that lead to increases in the cell monolayer permeability via NF-κB activation and ERK phosphorylation. Our in vitro and in vivo data demonstrate the sensitivity of permeability to stretch- and ventilation-induced superoxide production, suggesting that using antioxidants may be helpful in the prevention and treatment of ventilator-induced lung injury.
Mechanical ventilation–induced lung injury is associated with a high mortality rate in patients with adult respiratory distress syndrome and is characterized by acute respiratory failure, alveolar cell dysfunction, and a profound increase in the epithelial barrier permeability (1–3). The cellular events associated with increased alveolar epithelial permeability include increased activity of Na+–K+ ATPase pumps (3), MAPK activation (4), cytoskeleton remodeling (5), and substantial changes in the structure and content of tight junction proteins (6). Recently, we have published mRNA data from primary type I–like rat alveolar epithelial cells (RAECs) that were exposed to physiological cyclic stretch (7). These data associated a high number of genes affected by stretch with regulation of cellular metabolic processes. Because oxygen is essential for all cellular metabolic reactions, we hypothesize that cyclic stretch induces oxidative stress by generating reactive oxygen species (ROS), which lead to signaling cascades that increase epithelial permeability.
Stretching cells cyclically has been previously associated with the generation of superoxide and the release of nitric oxide (NO) to the cell culture medium (8–11); however, this has never been demonstrated in type I–like RAECs, which are responsible for maintaining the epithelial barrier properties. Exposing lung cells and tissues to ROS without stretch has been widely shown to increase permeability (12–15), and in intestinal and kidney epithelial monolayers, NO dilated the tight junctions (16, 17). However, the mechanistic pathways that link stretch, ROS generation, and lung barrier dysfunction have yet to be elucidated.
Previous studies, including our own, have demonstrated the activation of NF-κB in lung epithelial cells by cyclic stretch (18–20), and associated stretch-induced NF-κB activation with ROS in other tissues (9). However, the vast majority of studies that link oxidative stress and NF-κB activation in the lung (21–24) have not used alveolar type I cells. In addition, we have recently reported increased phosphorylation of ERK in response to cyclic stretch (4), and the ERK pathway has been reported to be involved in NF-κB activation by oxidants (25).
The goal of this study was to determine if cyclic stretch–induced oxidative stress could alter the barrier properties of the alveolar epithelium via NF-κB pathway and ERK activation. After identifying the specific ROS generated by cyclic stretch, we tested their effect on permeability in unstretched cells. Then, we showed that the cyclic stretch–induced increase in permeability via ROS generation can be attenuated by ROS scavengers and by NF-κB and ERK inhibitors. To confirm the causal interactions between ROS, NF-κB, and ERK, we tested the response of NF-κB to the same inhibitors.
Materials and Methods
Isolation and Culture of Type I–Like RAECs
Primary rat type II alveolar epithelial cells were isolated from male Sprague-Dawley rats and cultured as previously described (26, 27). All protocols were approved by our institutional ethics committee. The cells were cultured for 4 to 5 days in modified Eagle’s medium with 10% FBS, after which they had adopted alveolar type I–like phenotype (28) and had grown to a confluent monolayer.
Cyclic Stretch of RAEC Monolayers
The cells were stretched biaxially at a frequency of 0.25 Hz and across a range of physiologically relevant magnitudes, including at 12, 25, and 37% change in surface area (ΔSA), roughly corresponding to 64, 86, and 100% total lung capacity, respectively, as previously described (4, 27).
Detection of General ROS, Superoxide, and NO
A broad spectrum of ROS were detected in living cells using the fluorogenic indicator CellRox (29). Mitochondrial superoxide was detected in living cells using the fluorogenic dye MitoSOX (30, 31). These probes were purchased from Invitrogen (Carlsbad, CA) and used according to the manufacturer’s instructions. To prevent uptake of these tracers due to transient membrane permeabilization or mixing during stretch, all the tracers were added to the cell culture after stretch. Images were captured on a Nikon TE-300 inverted epifluorescence microscope with Metamorph imaging software (Universal Imaging, West Chester, PA) and a microscope-mounted Hamamatsu camera and controller (Hamamatsu, Shizuoka, Japan). Identical image acquisition times and uniform settings for intensity were used for all images of each type acquired. Average pixel intensity was computed using Matlab (version 7.11.0 R2010b; The MathWorks, Natick, MA).
Measuring NO concentration in medium samples that were collected immediately before and after stretch was done using the Griess reaction assay, which detects the nitrite ion NO2− (11, 16). This assay kit was purchased from Cayman Chemical Co. (Ann Arbor, MI) and used according to the manufacturer’s instructions. To eliminate color interference of the NO scavenger carboxy-PTIO (cPTIO) (Cayman Chemical Co.), the NO concentration data in each culture after stretch were normalized to that of the same culture immediately before stretch.
Permeability Studies
Permeability in type I–like RAEC monolayers was estimated using a fluorescent Bodipy-tagged Ouabain (radius ∼15–20 Å; Invitrogen, Carlsbad, CA), which selectively binds to the Na+–K+ ATPase pumps on the basolateral surface of the plasma membrane when the tight junctions are breached (32–34). Bodipy fluorescence was captured as detailed above and quantified as previously described (35).
In vivo permeability studies were conducted with male Sprague-Dawley rats weighing 240 to 260 g. Treated animals were injected intraperitoneally with 1 ml of tiron at a concentration of 400 mg/ml solution suspended in sterile normal saline daily over a course of 5 days in a protocol approved by the University of Pennsylvania Institutional Animal Care and Use Committee. Untreated animals were injected with saline. On Day 6, the animals were sedated with isoflurane, and tracheostomy was performed. Animal ventilation was performed using a Harvard Apparatus small-animal ventilator at 25 ml/kg Vt (36) and zero positive end-expiratory pressure at a frequency of 35 breaths/min for 2 hours, all under isoflurane. Spontaneously breathing controls received isoflurane but not mechanical ventilation. At the initiation of mechanical ventilation or spontaneous respiration, all rats were injected with 0.5 ml of a 25 mg/ml solution of FITC-conjugated albumin (A9771; Sigma-Aldrich, St. Louis, MO) in the lateral tail vein. At the end of 2 hours, bronchoalveolar lavage (BAL) was performed with 5 ml of PBS, and reference whole blood was collected via direct cardiac puncture and allowed to clot. The animals were killed by exsanguination. Albumin fluorescence in BAL and serum was determined using a spectrophotometer with absorption/emission wavelengths of 480/520 nm. Aleveolar epithelial permeability was defined as the ratio of BAL to serum fluorescence, and each ventilated group was normalized by the average value from the spontaneously breathing group of animals.
Quantification of Cytoplasmic and Nuclear p65 Components
Two different methods were used to quantify the cytoplasmic and nuclear content of the p65 subunit of NF-κB. The first method was based on an image processing algorithm implemented in Matlab and applied on immunofluorescent images (captured as detailed above), double-labeled for p65 and DAPI. The DAPI channel was used as a mask to generate two p65 images: cytoplasmic image and nuclear image. The sum of pixel values in each image was then computed and divided by the number of nonzero pixels in the image. In the second method, cell lysates were separated into cytoplasmic and nuclear fractions as previously described (37), and p65 was quantified by Western blotting. The cytoplasmic and nuclear p65 band intensities were normalized to the band intensities of the cytoplasmic protein marker MEK1/2 and the nuclear protein marker Jun, respectively.
Statistical Analysis
All experiments were performed at least three times, each time with cells from a different animal. At least three culture wells were considered for each experimental group, and at least three measurements were done per well. The data presented are expressed as mean ± SEM of fold change above unstretched cells treated with vehicle control.
One-way ANOVA with repeated measures followed by a Tukey post hoc test was applied to determine statistical significance between three or more data sets (JMP, version 9.0.0). Statistical significance of differences between two groups was determined using a two-tailed, paired t test. Data were deemed statistically significant at P ≤ 0.05.
Results
ROS Generation in RAECs due to Cyclic Stretch
High levels of ROS were detected due to cyclic stretch in type I–like RAECs using the cell-permeant fluorogenic probe for detecting cellular oxidative stress (i.e., CellRox). Representative images of CellRox stain in stretched and unstretched cells are presented in Figures 1a through 1f, and computed average pixel intensity of the images is shown in Figure 1g. The average pixel intensity of CellRox stain was increased after 10 and 60 minutes of cyclic stretch at 37% ΔSA in comparison to unstretched cells (Figures 1c and 1e versus 1a; Figure 1g). Incubation with the superoxide scavenger tiron (10 mM; 120 min) dramatically decreased the ROS in cultures stretched at 37% ΔSA for 10 and 60 minutes in comparison to vehicle control (distilled deionized water)-treated cells (Figures 1d and 1f versus 1c and 1e, respectively; Figure 1g), demonstrating that superoxide is a major constituent of the ROS generated. These data were in accordance with data from images of cells stained with another fluorogenic indicator, carboxy H2DCFDA, showing increased ROS stain in cells stretched for 60 minutes at 37% ΔSA in comparison to unstretched cells and decreased stain in tiron-treated stretched cultures (data not shown).
Figure 1.
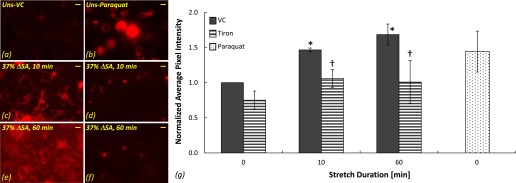
Reactive oxygen species (ROS) stained with CellRox in type I–like rat alveolar epithelial cells (RAECs). Representative images of ROS in unstretched cells treated with vehicle control (VC) (a) and the superoxide donor paraquat (b) and in cells that were stretched at 37% change in surface area (ΔSA) for 10 minutes (c and d) and 60 minutes (e and f) treated with VC (c and e) and the superoxide scavenger tiron (d and f). Bar = 10 μm. (g) Average pixel intensity computed in CellRox images of stretched and unstretched cells expressed as mean ± SEM of fold change above unstretched cells treated with VC. *P < 0.05 versus unstretched VC. †P < 0.05 versus stretched VC.
To identify the source of superoxide production within the type I–like cells, we used MitoSOX, which selectively targets the mitochondria in living cells and fluoresces upon oxidation. The average pixel intensity in the MitoSOX stain images was increased significantly after 60 and 120 minutes of stretch at 37% ΔSA in comparison to unstretched cells (Figures 2d and 2f versus 2a; Figure 2g) and after 120 minutes of stretch at 12% ΔSA (Figures 2e and 2g). Used as a positive control, paraquat significantly increased superoxide in unstretched cells compared with vehicle control–treated cells (Figure 2b versus 2a; Figure 2g).
Figure 2.
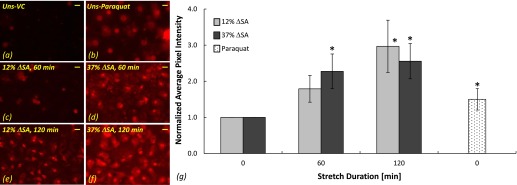
Superoxide stained with Mitosox in type I–like RAECs. Representative images of superoxide in unstretched cells treated with VC (a) and the superoxide donor paraquat (b) and in cells that were stretched for 60 minutes at 12% ΔSA (c) and 37% ΔSA (d) and for 120 minutes at 12% ΔSA (e) and 37% ΔSA (f). Bar = 10 μm. (g) Average pixel intensity computed in Mitosox images of stretched and unstretched cells expressed as mean ± SEM of fold change above unstretched cells treated with VC. *P < 0.05 versus unstretched VC.
The release of NO from the cells due to stretch was detected using the Griess reaction assay applied on culture medium that was collected immediately before and after stretch and normalized as described in the Materials and Methods section. Significantly elevated levels of NO were found in the culture medium of type I–like RAECs stretched for 60 minutes at 37% ΔSA in comparison to unstretched cells (Figure 3). Significantly higher levels of NO were also detected in the culture medium of cells that were stretched at magnitudes of 37% ΔSA for 10 minutes (1.46 ± 0.22) and of 25% ΔSA for 10 and 60 minutes (1.52 ± 0.24 and 1.68 ± 0.29, respectively) in comparison to unstretched cells (1.00 ± 0.00 by definition), whereas stretch at 12% ΔSA for 10 or 60 minutes did not yield a significant increase in the NO level above unstretched cells (data not shown). The NO scavenger cPTIO (1 mM; 90 min) decreased the levels of NO in the medium of stretched cultures to that of unstretched cells treated with vehicle control (PBS; Figure 3). As a positive control, exposing unstretched cells to the NO donor SNAP (10 μM; 60 min) yielded a significant and even more dramatic increase in the NO level in the medium (Figure 3).
Figure 3.

Nitric oxide release to the culture medium of type I–like RAECs that were stretched for 60 minutes at 37% ΔSA in comparison to unstretched cells. Stretched and unstretched cells were treated with VC or with the nitric oxide scavenger cPTIO. Unstretched cells were additionally treated with the nitric oxide donor SNAP. Data are expressed as mean ± SEM of fold change above unstretched cells treated with VC. *P < 0.05 versus unstretched VC. †P < 0.05 versus stretched VC.
ROS-Induced Increase in RAEC Monolayer Permeability via NF-κB and ERK Activation
The type I–like RAEC monolayer permeability was estimated using the Bodipy Ouabain assay as described previously and analyzed in detail below (32). In unstretched cells, the NO donor SNAP (2 mM; 60 min) and the superoxide donor paraquat (10 μM; 24 h) significantly increased Bodipy Ouabain binding in comparison to vehicle control (DMSO)-treated cells (Figure 4). Combining each of these treatments with MG132 (1 μM; 120 min), an inhibitor of p65 activation, significantly decreased the binding compared with the SNAP- or paraquat-treated cultures, suggesting that NF-κB contributes to ROS-induced increases in permeability. Combining SNAP with the ERK phosphorylation blocker U0126 (20 μM; 60 min) significantly reduced Bodipy Ouabain binding in comparison to cells treated only with SNAP, but U0126 did not help preserve the monolayer integrity in the paraquat-treated cultures, suggesting that ERK contributes more to NO-induced increases in permeability than to permeability increases after exposure to superoxide. Combining the MG132 and U0126 treatments significantly reduced the binding in SNAP- and paraquat-treated cultures. This reduction was not significantly larger than the reduction in the cultures treated with SNAP or paraquat combined with MG132 alone, suggesting that there was no synergistic effect of MG132 and U0126 on the monolayer permeability.
Figure 4.

Permeability measured using Bodipy Ouabain in unstretched type I–like RAECs. The cells were treated with VC or with the nitric oxide donor SNAP or with the superoxide donor paraquat. Simultaneously with each donor, we treated the cells with the NF-κB activation inhibitor MG132, extracellular signal–regulated kinase pathway blocker U0126, or a combination of both. Data are expressed as mean ± SEM of fold change above unstretched cells treated with VC. *P < 0.05 versus unstretched VC. †P < 0.05 versus unstretched SNAP. ††P < 0.05 versus unstretched paraquat.
The binding of the Bodipy Ouabain tracer significantly increased in type I–like RAECs that were stretched at 37% ΔSA for 60 minutes (Figure 5). Treating the cells with MG132 (1 μM; 120 min) significantly reduced the binding compared with vehicle control (DMSO)-treated cultures, suggesting that NF-κB mediates the stretch-induced barrier disruption in these cells. Tiron (10 mM; 120 min) also significantly decreased Bodipy Ouabain binding in the stretched cells almost to the level of unstretched cells; however, the NO scavenger cPTIO (1 mM; 90 min) did not reduce stretch-induced binding, suggesting that superoxide contributes more substantially than NO to the stretch-induced epithelial barrier dysfunction in type I–like RAECs. Testing the effect of each of the above-mentioned inhibitors on unstretched cells did not yield significant differences in comparison to unstretched cells treated with vehicle control, suggesting that these treatments do not affect the integrity of the tight junctions between the cells.
Figure 5.

Permeability measured using Bodipy Ouabain in type I–like RAECs that were stretched for 60 minutes at 37% ΔSA in comparison to unstretched (Uns) cells. The cells were treated with VC or with the NF-κB activation inhibitor MG132; the nitric oxide and superoxide scavengers cPTIO and Tiron, respectively; or with the extracellular signal–regulated kinase pathway blocker U0126. Data are expressed as mean ± SEM of fold change above unstretched cells treated with VC. *P < 0.05 versus unstretched VC; †P < 0.05 versus stretched VC. Inset: Permeability measured using Bodipy Ouabain in unstretched and stretched (37% ΔSA for 60 min) type I–like RAECs with and without treatment with 2 mM 2-deoxy-d-glucose and 10 μM antimycin A to reduce the expression of tight junction proteins and to increase cell–cell spacing (6). Data expressed as mean ± SEM of fold change above unstretched cells treated with VC. *P < 0.05 versus unstretched VC; #P < 0.05 versus unstretched AMA. NS = no significant difference between the marked groups.
Finally, we tested permeability at injurious tidal volume (Vt) levels by mechanically ventilating rats at 25 ml/kg (36) for 2 hours with or without pretreatment with tiron. Whereas transport of FITC-conjugated albumin into the airspace was significantly and dramatically increased in animals under mechanical ventilation compared with spontaneously breathing rats (Figure 6), pretreatment with tiron significantly attenuated permeability to a level not different from the permeability level in the spontaneously breathing rats.
Figure 6.

Permeability to FITC-conjugated albumin in rats treated with saline or 400 mg/ml tiron daily over a course of 5 days. Data from rats mechanically ventilated for 2 hours at 25 ml/kg Vt and zero positive end-expiratory pressure at a frequency of 35 breaths/min under isoflurane are compared with spontaneously breathing rats. Data are expressed as mean ± SEM of BAL/serum fluorescence ratio fold change above spontaneously breathing rats. *P < 0.05 versus spontaneously breathing rats; †P < 0.05 versus saline-treated mechanically ventilated rats.
ERK-Dependent Activation of NF-κB due to Cyclic Stretch
Immunofluorescent stains of p65 after cyclic stretch for 60 minutes at 37% ΔSA revealed translocation of the p65 subunit to the nucleus (Figures 7b and 7c). Using two different methods (i.e., image processing of p65 immunofluorescent stain images and Western blotting of cell lysates that were separated into cytoplasmic and nuclear fractions), we quantified the cytoplasmic and nuclear content of p65 as described in the Materials and Methods section. Data obtained from the two methods are presented in the bar graphs in Figure 7, expressed as fold change above unstretched cells treated with vehicle control (DMSO). The two methods were in good agreement (P > 0.05 between the corresponding unstretched and stretched data in Figure 7a versus 7g) and showed that stretch increased nuclear p65 significantly in comparison to unstretched cells (Figures 7a and 7g) but had no effect on the cytoplasmic component. Whereas stretching the cells for 60 minutes at 12% ΔSA did not increase the nuclear p65 levels significantly compared with unstretched cells (1.03 ± 0.13 above unstretched cells), stretching for 60 minutes at 25% ΔSA significantly increased the nuclear p65 levels (1.33 ± 0.07 above unstretched cells), comparable to the nuclear p65 levels observed at 37% ΔSA (1.29 ± 0.08 above unstretched cells) (Figure 7a).
Figure 7.
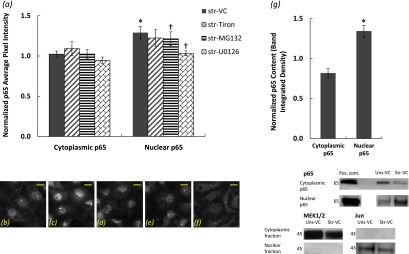
(a) Quantification of cytoplasmic and nuclear p65 subunit of NF-κB content in type I–like RAECs that were stretched at 37% ΔSA for 60 minutes and treated with VC (n = 19), the superoxide scavenger tiron (n = 7), the NF-κB activation inhibitor MG132 (n = 14), and the ERK inhibitor U0126 (n = 3) based on an image-processing algorithm applied on the p65-DAPI images. (b–f) Representative images of the p65 subunit of NF-κB in type I–like RAECs that remained unstretched and treated with VC (b). (c–f) Cells that were stretched for 60 minutes at 37% ΔSA and treated with VC (c), tiron (d), MG132 (e), and U0126 (f). Bar = 10 μm. (g) Quantification of cytoplasmic and nuclear p65 subunit of NF-κB content in type I–like RAECs that were stretched at 37% ΔSA for 60 minutes based on Western blotting (see representative bands below the graph). Cytoplasmic p65 was normalized to the cytoplasmic marker protein MEK1/2, and nuclear p65 was normalized to the nuclear marker protein Jun. Data are expressed as mean ± SEM of fold change above unstretched cells treated with VC. *P < 0.05 versus unstretched VC; †P < 0.05 versus stretched VC.
Treating the cells with MG132 (1 μM; 120 min) significantly attenuated the nuclear p65 levels compared with vehicle control–stretched cells (Figure 7a). The superoxide scavenger tiron (10 mM; 120 min) did not significantly reduce the p65 in the cytoplasm or the nucleus of the stretched cells, suggesting that superoxide does not play an important role in p65 regulation due to stretch. Conversely, the ERK inhibitor U0126 (20 μM; 60 min) significantly reduced the nuclear p65 compared with vehicle control–treated stretched cells, suggesting that ERK is upstream of the p65. Here too, treating unstretched cells with the specific inhibitors did not result in significant differences in comparison to unstretched cells treated with vehicle control.
Detailed Analysis of Permeability Measurement Using the Bodipy Ouabain Assay
To assess the portion of binding of the Bodipy Ouabain compound to the Na+–K+ ATPase pumps on the basolateral surface of the plasma membrane that is attributed to breaching of the tight junctions between the type I–like RAECs and not to stretch-induced trafficking of the pumps to the basolateral surface of the membrane (3, 33), we performed the following studies. We treated unstretched and stretched cells (37% ΔSA; 60 min) for 1 hour with 2 mM 2-deoxy-D-glucose and 10 μM antimycin A (AMA) to reduce the expression of tight junctional proteins and to increase cell–cell spacing (6). Permeability to Bodipy Ouabain was measured in both groups and compared with vehicle control stretched and unstretched cells. The results from these studies show that the amount of Bodipy Ouabain binding in unstretched cells treated with AMA is significantly larger than in unstretched cells treated with vehicle control, suggesting that the AMA treatment opens the route for Bodipy Ouabain passage between the cells (Figure 5, inset).
Second, let the amount of Bodipy Ouabain binding due to stretch-induced trafficking of the Na+–K+ ATPase pumps to the basolateral surface of the plasma membrane be Tstr, the amount of Bodipy Ouabain binding due to stretch-induced opening of gaps between the cells be Ostr, and the amount of Bodipy Ouabain binding due to AMA-induced opening of gaps between the cells be OAMA. Then, the amount of Bodipy Ouabain binding due to stretch-induced trafficking equals the difference between the binding in stretched cells treated with AMA (Tstr + OAMA) and the binding in unstretched cells treated with AMA (OAMA). We assume that the pretreatment with AMA before stretch opens the gaps between the cells almost completely, and the remaining stretch-induced Bodipy Ouabain binding can be attributed to trafficking of the pumps rather than further opening of paracellular routes. If we subtract this difference from the binding in stretched cells treated with vehicle control (Tstr + Ostr), we can find the Bodipy Ouabain binding that is attributed exclusively to stretch-induced opening of gaps between the cells (Ostr).
Substituting the data from these studies reveals a value greater than zero for Ostr, suggesting that Bodipy Ouabain binding is attributed to stretch-induced opening of gaps between the cells and not only to trafficking of the pumps to the basolateral surface of the plasma membrane. In fact, this calculated Ostr is 60% higher than the difference in Bodipy Ouabain binding between stretched and unstretched cells treated with AMA, which reflects the binding attributed to trafficking alone (4.59 versus 2.76, respectively). We have previously performed other systematic tests to ensure that the Bodipy Ouabain binding is not due to endocytosis, cell death, denuding of cells from the membrane, or to nonspecific Bodipy binding (32).
Other researchers have shown that ouabain has a deteriorating effect on the integrity of tight junctions in other types of cells (38). However, the Bodipy Ouabain signal in unstretched monolayers treated with vehicle control was very low (∼1–3% of the pixels in the image). Moreover, because we normalize all the Bodipy Ouabain data to signals from unstretched monolayers treated with vehicle control, any minor damage to the tight junctions that might be attributed to the exposure to ouabain is eliminated. Differences between the studies, such as incubation time, type of cells, and substrate used, may be responsible for the different outcomes to ouabain exposure. Altogether these studies ensure that the major component of the Bodipy Ouabain binding can be attributed to paracellular permeability in alveolar epithelial cell monolayers.
Discussion
To identify the mechanisms responsible for cyclic stretch–induced increases in alveolar epithelial permeability and opportunities for intervention, we proposed that oxidative stress mediates the increase in permeability of type I–like RAECs exposed to mechanical deformation. We demonstrate that cyclic stretch generates general ROS, superoxide, and NO in RAECs, which can be inhibited by superoxide and NO scavengers. Using superoxide and NO donors in unstretched cells, we show that each ROS increases permeability in unstretched cells, which can be eliminated by the NF-κB inhibitor MG132. In addition, we show that the NO-induced increase in permeability can also be eliminated by blocking ERK phosphorylation with U0126. Next, we show that MG132 and tiron attenuate cyclic stretch–induced increases in the monolayer permeability and that tiron pretreatment has a dramatic protective effect on in vivo lung permeability under mechanical ventilation conditions. Finally, we demonstrate stretch-induced activation of the NF-κB signaling pathway, which can be attenuated significantly with MG132 and U0126.
RAECs Generate ROS Rapidly during Cyclic Stretch
Significant increases in ROS were measured in type I–like RAECs after only 10 minutes of cyclic stretch at 37% ΔSA, and this elevated ROS state was sustained at 60 minutes of stretch (Figure 1), when we also measured significantly increased monolayer permeability (Figure 5). The levels of ROS after 10 and 60 minutes of stretch were not significantly different, suggesting that the system reached ROS saturation by 10 minutes of stretch or that antioxidant pathways managed to eliminate further increase of the ROS level after that time and a redox steady state was achieved. Consistently, the level of NO released by the type I–like cells to the culture medium after stretch at 25 or 37% ΔSA for 10 minutes was significantly higher than in unstretched cultures, and this elevated NO level was also present after 60 minutes of stretch (Figure 3).
The generation of ROS in the type I–like RAECs was also differentially affected by the stretch magnitude. The level of NO in stretched cells after 10 or 60 minutes was elevated only after exposure to magnitudes of 25 and 37% ΔSA and not in response to stretch 12% ΔSA, suggesting that the generation of ROS in type I–like RAECs in response to cyclic stretch is magnitude sensitive. Furthermore, both low and high stretch magnitudes (12 and 37% ΔSA, respectively) yielded significant superoxide generation after 120 minutes of stretch (Figure 2), consistent with data obtained by other labs in type II RAECs and in the A549 cell line after 120 minutes of stretch at 15 to 20% ΔSA (8). Like NO, stretching type I–like RAECs for 60 minutes yielded increases in superoxide at 37% ΔSA but not at 12%, again demonstrating a dose response to stretch (Figure 2). Tiron, the superoxide scavenger, was very efficient in preventing the increase in general ROS level in type I–like cells exposed to cyclic stretch at 37% ΔSA for short and long durations (i.e., 10 and 60 min; Figure 1), suggesting that superoxide is a major contributor to the ROS generated by stretch in these cells.
Superoxide Is the Major ROS Causing Increases in RAEC Monolayer Permeability
In unstretched cells, the NO donor SNAP and the superoxide donor paraquat significantly increased permeability (Figure 4). However, in stretched cells (37% ΔSA; 60 min) treated with the NO scavenger cPTIO and the superoxide scavenger tiron, only tiron was able to significantly reduce the stretch-induced increase in permeability to unstretched levels (Figure 5). Tiron did not significantly affect cell viability in unstretched or stretched preparations, assessed by counting ethidium homodimer-1–stained cells in 78 monolayer images from 26 wells with monolayers of comparable density obtained from three different animals (P > 0.05). This suggests that the superoxide-induced increase in permeability is not via cell death. In fact, we have previously shown that although there might be a modest contribution of cell death to stretch-induced increases in the epithelial barrier permeability, cell death mechanism contribution is lesser than increases due to tight junction dysfunction (32). Because we found that both scavengers were very efficient in preventing the stretch-induced generation of general ROS and NO (Figures 1 and 3), we conclude that superoxide contributes considerably more to stretch-induced permeability increases than NO. We postulate that stretch may not generate enough NO to cause a significant change in permeability because 10 μM of SNAP was enough to elevate the NO level 20-fold in the culture medium of unstretched cells (Figure 3), but 2 mM SNAP was required to cause a change in permeability in unstretched cells (Figure 4) that was in the same order of magnitude as the stretch-induced permeability change (Figure 5).
Finally, our in vivo permeability data showed a complete preservation of the pulmonary epithelial barrier function in tiron-treated animals that were subjected to injurious Vt values compared with untreated animals that presented extremely high permeability levels (Figure 6). These results are in very good agreement with other studies that showed protective effects of different antioxidants, such as superoxide dismutase (39) or Heme oxygenase-1 (40), in ventilator-induced lung injury. Future studies should examine changes in lung function and histological evidence of injury in ventilated animals treated with tiron or in spontaneously breathing animals treated with superoxide donors. This supports the conclusion that superoxide is an important contributor to ventilation-induced increases in permeability. Altogether, our in vitro and in vivo data demonstrated the sensitivity of permeability to ventilation/stretch-induced superoxide production, suggesting that using antioxidants may be helpful in the prevention and treatment of ventilator-induced lung injury.
Role of NF-κB and ERK in Cyclic Stretch-ROS Induced Increases in Permeability
Because the p65 subunit of NF-κB translocated from the cytoplasm to the nucleus after cyclic stretch at 37% ΔSA for 60 minutes (Figure 7) and because inhibition of NF-κB with MG132 significantly attenuated the stretch-induced increase in permeability (Figure 5), we conclude that NF-κB mediates the increase in permeability due to cyclic stretch. Furthermore, in unstretched cells exposed to the NO donor SNAP or the superoxide donor paraquat, MG132 prevented the permeability increases we obtained in ROS-challenged preparations (Figure 4), suggesting that NF-κB is activated by oxidative stress. Because tiron significantly attenuated the stretch-induced increase in permeability (Figure 5) but did not decrease the nuclear NF-κB levels significantly, we propose that superoxide increases permeability directly and indirectly via the NF-κB pathway. In other words, our data suggest that superoxide increases permeability via NF-κB activation but not exclusively via this pathway. In fact, other researchers have shown in melanoma cell lines that exposure to tiron at the same concentration used here (10 mM) but for 24 hours (instead of 120 min) resulted in increased NF-κB–DNA binding (41). However, when we treated unstretched cell monolayers with tiron, we found that the cytoplasmic p65 component (0.86 ± 0.08) and the nuclear p65 component (0.95 ± 0.07) were not significantly different from the values found in unstretched cells treated with vehicle control.
Although the data presented here were collected in normal cells, high levels of NO have been associated with septic conditions (42) that often present in patients with adult respiratory distress syndrome (43, 44). In 2006 we showed that, in type I–like RAECs isolated from septic rats, NF-κB is translocated even in unstretched cells (19). Future studies should therefore examine whether oxidative stress mediates NF-κB activation in cells from septic animals and whether NF-κB activation-induced increase in permeability in these cells can be prevented by MG132, tiron, or other ROS scavengers. Moreover, the exact pathway through which activation of NF-κB, induced by stretch or sepsis, leads to increased permeability is yet to be explored. In fact, publications that link NF-κB activation and permeability or changes in tight junction proteins in lung cells are scarce (45, 46).
We demonstrated that ERK inhibitor U0126 significantly decreased the nuclear p65 levels in stretched type I–like cells compared with vehicle control–treated stretched cells (Figure 7), suggesting that ERK is upstream of NF-κB activation. Similar interrelations between NF-κB and ERK were reported in studies with A549 cells that measured other cellular responses, such as release of inflammatory mediators due to exposure to neutrophil elastase (47) or regulation of toll-like receptor 2 and surfactant protein-A due to LPS stimulation (48). Although permeability to our approximately 15 to 20 Å tracer was not significantly different in U0126-treated cells that were stretched at 37% ΔSA for 60 minutes than in vehicle control–stretched cells (Figure 5), U0126 did help preserve the barrier integrity in unstretched cells treated with the NO donor SNAP, but it did not eliminate the increase in permeability in unstretched cells treated with the superoxide donor paraquat (Figure 4). We conclude that NO, but not superoxide, affects ERK activation.
However, we have previously published that U0126 did reduce permeability significantly in cells that were stretched at 37% ΔSA for 60 minutes in comparison to cells treated with vehicle control when permeability was measured using an approximately 5 Å–radius carboxyfluorescein (4). In that study, permeability in the U0126-treated cultures was still significantly higher than in unstretched cells, and U0126 did not prevent the stretch-induced reduction in the content of the tight junction protein occludin. Taken together with our current study, inhibition of ERK confers only partial protection of the RAEC monolayer, specifically to small molecules.
Study Limitations
The cells used in this study were isolated primary type II RAECs that were cultured for 4 to 5 days on fibronectin-coated substrates to ensure their adoption of alveolar type I–like phenotype (28); freshly isolated type I cells were not used in this study. Although the Day 4 to 5 type I–like cells have been shown to express the type I AEC markers RTI40 and aquaporin 5 (49) and to grow to form confluent monolayers with tight junctions between the cells (6) and were shown to preserve the epithelial barrier function under low magnitudes of stretch (32), they also exhibit somewhat different gene expression than freshly isolated type I cells (50). The contribution of these genes to the cell response to stretch should be explored to ensure that the responses of the in vitro cultured type I–like cells are similar to those of the in vivo type I cells.
In the present work, we did not directly measure the distribution of the Na+–K+ ATPase pumps, to which the Bodipy Ouabain compound binds, in response to the different treatments used. However, we have tested the effect of each treatment (and their combinations if used) on the Bodipy Ouabain signal in unstretched cells, and in all cases the signal was not significantly different from that in unstretched cells treated with the appropriate vehicle control, suggesting that these treatments do not affect the integrity of the tight junctions between the cells. Moreover, we show that stretch affects cell–cell junctions and increases the binding of the Bodipy Ouabain to the pumps (see Results and Figure 5, inset). Because MG132 and tiron, and also with U0126 when a different permeability tracer was used (4), reduce the stretch-induced Bodipy Ouabain signal, we conclude that these inhibitors do not activate the Na+–K+ ATPase pumps in the cells, which would result in their altered distribution. The same conclusion is drawn for the treatments with the ROS donors SNAP and paraquat because the inhibition treatments with U0126 and MG132, which were shown to have no effect on the pump distribution, attenuated the Bodipy Ouabain signal in the SNAP- and paraquat-treated cells (Figure 4), suggesting that these donors do not alter the pump distribution but affect cell–cell junctions. In addition, although we have shown that the Bodipy Ouabain assay provides a very good estimate of paracellular permeability in type I–like RAEC monolayers, it does not exclusively measure permeability, which may limit the interpretation of the results (see Detailed Analysis of Permeability Measurement using the Bodipy Ouabain Assay in the Results section).
Finally, although the gain and loss aspects of our experimental design using pharmacological agents confirm our hypothesized associations between cyclic stretch, ROS generation, NF-κB, and permeability in RAEC monolayers, we cannot exclude the possibility of broader off-target effects of these agents on other cellular processes.
Conclusions
In the present work, we demonstrate for the first time that cyclic stretch of type I–like RAECs generates ROS, which lead to increases in the cell monolayer permeability. Moreover, in animals treated with tiron over the course of 5 days, permeability was significantly attenuated, suggesting that tiron protects the pulmonary epithelial barrier under mechanical ventilation conditions. The ROS-induced increase in permeability was shown to be mediated by NF-κB activation and is associated with ERK phosphorylation. Using antioxidants such as tiron or NF-κB pathway–specific inhibitors may therefore be an effective strategy for preventing or treating ventilator-induced lung injury.
Acknowledgments
Acknowledgments
The authors thank Dianne Weeks (University of Pennsylvania) for technical assistance with the Griess assays.
Footnotes
This study was supported by National Institutes of Health/National Heart, Lung and Blood Institute grant R01-HL057204.
Author Contributions: Conception and design: N.D. and S.S.M. Performance of experiments: N.D., B.C.D., G.G.L., P.C., and N.Y. Analysis, interpretation, and drafting the manuscript: N.D. and S.S.M.
Originally Published in Press as DOI: 10.1165/rcmb.2012-0252OC on March 22, 2013
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Parker JC, Hernandez LA, Peevy KJ. Mechanisms of ventilator-induced lung injury. Crit Care Med. 1993;21:131–143. doi: 10.1097/00003246-199301000-00024. [DOI] [PubMed] [Google Scholar]
- 2.Slutsky AS, Ranieri VM. Mechanical ventilation: lessons from the ARDSNET trial. Respir Res. 2000;1:73–77. doi: 10.1186/rr15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waters CM, Ridge KM, Sunio G, Venetsanou K, Sznajder JI. Mechanical stretching of alveolar epithelial cells increases Na(+)-K(+)-ATPase activity. J Appl Physiol. 1999;87:715–721. doi: 10.1152/jappl.1999.87.2.715. [DOI] [PubMed] [Google Scholar]
- 4.Cohen TS, Gray Lawrence G, Khasgiwala A, Margulies SS. MAPK activation modulates permeability of isolated rat alveolar epithelial cell monolayers following cyclic stretch. PLoS ONE. 2010;5:e10385. doi: 10.1371/journal.pone.0010385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DiPaolo BC, Lenormand G, Fredberg JJ, Margulies SS. Stretch magnitude and frequency-dependent actin cytoskeleton remodeling in alveolar epithelia. Am J Physiol Cell Physiol. 2010;299:C345–C353. doi: 10.1152/ajpcell.00379.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cavanaugh KJ, Oswari J, Margulies SS. Role of stretch on tight junction structure in alveolar epithelial cells. Am J Respir Cell Mol Biol. 2001;25:584–591. doi: 10.1165/ajrcmb.25.5.4486. [DOI] [PubMed] [Google Scholar]
- 7.Yerrapureddy A, Tobias J, Margulies SS. Cyclic stretch magnitude and duration affect rat alveolar epithelial gene expression. Cell Physiol Biochem. 2010;25:113–122. doi: 10.1159/000272056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chapman KE, Sinclair SE, Zhuang D, Hassid A, Desai LP, Waters CM. Cyclic mechanical strain increases reactive oxygen species production in pulmonary epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2005;289:L834–L841. doi: 10.1152/ajplung.00069.2005. [DOI] [PubMed] [Google Scholar]
- 9.Ali MH, Pearlstein DP, Mathieu CE, Schumacker PT. Mitochondrial requirement for endothelial responses to cyclic strain: implications for mechanotransduction. Am J Physiol Lung Cell Mol Physiol. 2004;287:L486–L496. doi: 10.1152/ajplung.00389.2003. [DOI] [PubMed] [Google Scholar]
- 10.Kuebler WM, Uhlig U, Goldmann T, Schael G, Kerem A, Exner K, Martin C, Vollmer E, Uhlig S. Stretch activates nitric oxide production in pulmonary vascular endothelial cells in situ. Am J Respir Crit Care Med. 2003;168:1391–1398. doi: 10.1164/rccm.200304-562OC. [DOI] [PubMed] [Google Scholar]
- 11.Mohammed KA, Nasreen N, Tepper RS, Antony VB. Cyclic stretch induces PlGF expression in bronchial airway epithelial cells via nitric oxide release. Am J Physiol Lung Cell Mol Physiol. 2007;292:L559–L566. doi: 10.1152/ajplung.00075.2006. [DOI] [PubMed] [Google Scholar]
- 12.Shasby DM, Vanbenthuysen KM, Tate RM, Shasby SS, McMurtry I, Repine JE. Granulocytes mediate acute edematous lung injury in rabbits and in isolated rabbit lungs perfused with phorbol myristate acetate: role of oxygen radicals. Am Rev Respir Dis. 1982;125:443–447. doi: 10.1164/arrd.1982.125.4.443. [DOI] [PubMed] [Google Scholar]
- 13.Tasaka S, Amaya F, Hashimoto S, Ishizaka A. Roles of oxidants and redox signaling in the pathogenesis of acute respiratory distress syndrome. Antioxid Redox Signal. 2008;10:739–753. doi: 10.1089/ars.2007.1940. [DOI] [PubMed] [Google Scholar]
- 14.Chapman KE, Waters CM, Miller WM. Continuous exposure of airway epithelial cells to hydrogen peroxide: protection by KGF. J Cell Physiol. 2002;192:71–80. doi: 10.1002/jcp.10115. [DOI] [PubMed] [Google Scholar]
- 15.Sun Y, Minshall RD, Hu G. Role of claudins in oxidant-induced alveolar epithelial barrier dysfunction. Methods Mol Biol. 2011;762:291–301. doi: 10.1007/978-1-61779-185-7_21. [DOI] [PubMed] [Google Scholar]
- 16.Salzman AL, Menconi MJ, Unno N, Ezzell RM, Casey DM, Gonzalez PK, Fink MP. Nitric oxide dilates tight junctions and depletes ATP in cultured Caco-2BBe intestinal epithelial monolayers. Am J Physiol. 1995;268:G361–G373. doi: 10.1152/ajpgi.1995.268.2.G361. [DOI] [PubMed] [Google Scholar]
- 17.Cuzzocrea S, Mazzon E, De Sarro A, Caputi AP. Role of free radicals and poly(ADP-ribose) synthetase in intestinal tight junction permeability. Mol Med. 2000;6:766–778. [PMC free article] [PubMed] [Google Scholar]
- 18.Ning Q, Wang X. Role of Rel A and IkappaB of nuclear factor kappaB in the release of interleukin-8 by cyclic mechanical strain in human alveolar type II epithelial cells A549. Respirology. 2007;12:792–798. doi: 10.1111/j.1440-1843.2007.01166.x. [DOI] [PubMed] [Google Scholar]
- 19.Levine GK, Deutschman CS, Helfaer MA, Margulies SS. Sepsis-induced lung injury in rats increases alveolar epithelial vulnerability to stretch. Crit Care Med. 2006;34:1746–1751. doi: 10.1097/01.CCM.0000218813.77367.E2. [DOI] [PubMed] [Google Scholar]
- 20.Inoh H, Ishiguro N, Sawazaki S, Amma H, Miyazu M, Iwata H, Sokabe M, Naruse K. Uni-axial cyclic stretch induces the activation of transcription factor nuclear factor kappaB in human fibroblast cells. FASEB J. 2002;16:405–407. doi: 10.1096/fj.01-0354fje. [DOI] [PubMed] [Google Scholar]
- 21.Ahmed MN, Codipilly C, Hogg N, Auten RL. The protective effect of overexpression of extracellular superoxide dismutase on nitric oxide bioavailability in the lung after exposure to hyperoxia stress. Exp Lung Res. 2011;37:10–17. doi: 10.3109/01902148.2010.497893. [DOI] [PubMed] [Google Scholar]
- 22.Dagher Z, Garcon G, Billet S, Verdin A, Ledoux F, Courcot D, Aboukais A, Shirali P. Role of nuclear factor-kappa B activation in the adverse effects induced by air pollution particulate matter (PM2.5) in human epithelial lung cells (l132) in culture. J Appl Toxicol. 2007;27:284–290. doi: 10.1002/jat.1211. [DOI] [PubMed] [Google Scholar]
- 23.Pardo A, Barrios R, Maldonado V, Melendez J, Perez J, Ruiz V, Segura-Valdez L, Sznajder JI, Selman M. Gelatinases A and B are up-regulated in rat lungs by subacute hyperoxia: pathogenetic implications. Am J Pathol. 1998;153:833–844. doi: 10.1016/S0002-9440(10)65625-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simeonova PP, Toriumi W, Kommineni C, Erkan M, Munson AE, Rom WN, Luster MI. Molecular regulation of IL-6 activation by asbestos in lung epithelial cells: role of reactive oxygen species. J Immunol. 1997;159:3921–3928. [PubMed] [Google Scholar]
- 25.Janssen-Heininger YM, Macara I, Mossman BT. Cooperativity between oxidants and tumor necrosis factor in the activation of nuclear factor (NF)-kappaB: requirement of ras/mitogen-activated protein kinases in the activation of NF-kappaB by oxidants. Am J Respir Cell Mol Biol. 1999;20:942–952. doi: 10.1165/ajrcmb.20.5.3452. [DOI] [PubMed] [Google Scholar]
- 26.Tschumperlin DJ, Oswari J, Margulies SS. Deformation-induced injury of alveolar epithelial cells: effect of frequency, duration, and amplitude. Am J Respir Crit Care Med. 2000;162:357–362. doi: 10.1164/ajrccm.162.2.9807003. [DOI] [PubMed] [Google Scholar]
- 27.Tschumperlin DJ, Margulies SS. Equibiaxial deformation-induced injury of alveolar epithelial cells in vitro. Am J Physiol. 1998;275:L1173–L1183. doi: 10.1152/ajplung.1998.275.6.L1173. [DOI] [PubMed] [Google Scholar]
- 28.Oswari J, Matthay MA, Margulies SS. Keratinocyte growth factor reduces alveolar epithelial susceptibility to in vitro mechanical deformation. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1068–L1077. doi: 10.1152/ajplung.2001.281.5.L1068. [DOI] [PubMed] [Google Scholar]
- 29.Hansen-Hagge TE, Baumeister E, Bauer T, Schmiedeke D, Renne T, Wanner C, Galle J. Transmission of oxLDL-derived lipid peroxide radicals into membranes of vascular cells is the main inducer of oxLDL-mediated oxidative stress. Atherosclerosis. 2008;197:602–611. doi: 10.1016/j.atherosclerosis.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 30.Han Z, Varadharaj S, Giedt RJ, Zweier JL, Szeto HH, Alevriadou BR. Mitochondria-derived reactive oxygen species mediate heme oxygenase-1 expression in sheared endothelial cells. J Pharmacol Exp Ther. 2009;329:94–101. doi: 10.1124/jpet.108.145557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee SJ, Ryter SW, Xu J, Nakahira K, Kim HP, Choi AM, Kim YS. Carbon monoxide activates autophagy via mitochondrial reactive oxygen species formation. Am J Respir Cell Mol Biol. 2011;45:867–873. doi: 10.1165/rcmb.2010-0352OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cavanaugh KJ, Margulies SS. Measurement of stretch-induced loss of alveolar epithelial barrier integrity with a novel in vitro method. Am J Physiol Cell Physiol. 2002;283:C1801–C1808. doi: 10.1152/ajpcell.00341.2002. [DOI] [PubMed] [Google Scholar]
- 33.Fisher JL, Margulies SS. Na(+)-K(+)-ATPase activity in alveolar epithelial cells increases with cyclic stretch. Am J Physiol Lung Cell Mol Physiol. 2002;283:L737–L746. doi: 10.1152/ajplung.00030.2001. [DOI] [PubMed] [Google Scholar]
- 34.Khimenko PL, Barnard JW, Moore TM, Wilson PS, Ballard ST, Taylor AE. Vascular permeability and epithelial transport effects on lung edema formation in ischemia and reperfusion. J Appl Physiol. 1994;77:1116–1121. doi: 10.1152/jappl.1994.77.3.1116. [DOI] [PubMed] [Google Scholar]
- 35.Dipaolo BC, Margulies SS. Rho kinase signaling pathways during stretch in primary alveolar epithelia. Am J Physiol Lung Cell Mol Physiol. 2012;302:L992–L1002. doi: 10.1152/ajplung.00175.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caironi P, Langer T, Carlesso E, Protti A, Gattinoni L. Time to generate ventilator-induced lung injury among mammals with healthy lungs: a unifying hypothesis. Intensive Care Med. 2011;37:1913–1920. doi: 10.1007/s00134-011-2388-9. [DOI] [PubMed] [Google Scholar]
- 37.de Oliveira-Marques V, Cyrne L, Marinho HS, Antunes F. A quantitative study of NF-kappaB activation by H2O2: relevance in inflammation and synergy with TNF-alpha. J Immunol. 2007;178:3893–3902. doi: 10.4049/jimmunol.178.6.3893. [DOI] [PubMed] [Google Scholar]
- 38.Larre I, Lazaro A, Contreras RG, Balda MS, Matter K, Flores-Maldonado C, Ponce A, Flores-Benitez D, Rincon-Heredia R, Padilla-Benavides T, et al. Ouabain modulates epithelial cell tight junction. Proc Natl Acad Sci USA. 2010;107:11387–11392. doi: 10.1073/pnas.1000500107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fu P, Murley JS, Grdina DJ, Birukova AA, Birukov KG. Induction of cellular antioxidant defense by amifostine improves ventilator-induced lung injury. Crit Care Med. 2011;39:2711–2721. doi: 10.1097/CCM.0b013e3182284a5f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.An L, Liu CT, Qin XB, Liu QH, Liu Y, Yu SY. Protective effects of hemin in an experimental model of ventilator-induced lung injury. Eur J Pharmacol. 2011;661:102–108. doi: 10.1016/j.ejphar.2011.04.032. [DOI] [PubMed] [Google Scholar]
- 41.Yang J, Su Y, Richmond A. Antioxidants tiron and N-acetyl-L-cysteine differentially mediate apoptosis in melanoma cells via a reactive oxygen species-independent NF-kappaB pathway. Free Radic Biol Med. 2007;42:1369–1380. doi: 10.1016/j.freeradbiomed.2007.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chuang CY, Chen TL, Cherng YG, Tai YT, Chen TG, Chen RM. Lipopolysaccharide induces apoptotic insults to human alveolar epithelial A549 cells through reactive oxygen species-mediated activation of an intrinsic mitochondrion-dependent pathway. Arch Toxicol. 2011;85:209–218. doi: 10.1007/s00204-010-0585-x. [DOI] [PubMed] [Google Scholar]
- 43.Fein AM, Calalang-Colucci MG. Acute lung injury and acute respiratory distress syndrome in sepsis and septic shock. Crit Care Clin. 2000;16:289–317. doi: 10.1016/s0749-0704(05)70111-1. [DOI] [PubMed] [Google Scholar]
- 44.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 45.Illek B, Fu Z, Schwarzer C, Banzon T, Jalickee S, Miller SS, Machen TE. Flagellin-stimulated Cl- secretion and innate immune responses in airway epithelia: role for p38. Am J Physiol Lung Cell Mol Physiol. 2008;295:L531–L542. doi: 10.1152/ajplung.90292.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han X, Fink MP, Uchiyama T, Yang R, Delude RL. Increased iNOS activity is essential for hepatic epithelial tight junction dysfunction in endotoxemic mice. Am J Physiol Gastrointest Liver Physiol. 2004;286:G126–G136. doi: 10.1152/ajpgi.00231.2003. [DOI] [PubMed] [Google Scholar]
- 47.Chen HC, Lin HC, Liu CY, Wang CH, Hwang T, Huang TT, Lin CH, Kuo HP. Neutrophil elastase induces IL-8 synthesis by lung epithelial cells via the mitogen-activated protein kinase pathway. J Biomed Sci. 2004;11:49–58. doi: 10.1007/BF02256548. [DOI] [PubMed] [Google Scholar]
- 48.Wu TT, Chen TL, Loon WS, Tai YT, Cherng YG, Chen RM. Lipopolysaccharide stimulates syntheses of toll-like receptor 2 and surfactant protein-a in human alveolar epithelial A549 cells through upregulating phosphorylation of MEK1 and ERK1/2 and sequential activation of NF-kappaB. Cytokine. 2011;55:40–47. doi: 10.1016/j.cyto.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 49.Gonzalez RF, Allen L, Dobbs LG. Rat alveolar type I cells proliferate, express OCT-4, and exhibit phenotypic plasticity in vitro. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1045–L1055. doi: 10.1152/ajplung.90389.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gonzalez R, Yang YH, Griffin C, Allen L, Tigue Z, Dobbs L. Freshly isolated rat alveolar type I cells, type II cells, and cultured type II cells have distinct molecular phenotypes. Am J Physiol Lung Cell Mol Physiol. 2005;288:L179–L189. doi: 10.1152/ajplung.00272.2004. [DOI] [PubMed] [Google Scholar]