

Abstract
Procalcitonin (PCT) is expressed in nonthryoidal tissues of humans during severe infections. Serum PCT levels are measured to diagnose and guide therapy, and there is some evidence that PCT may also contribute to the pathogenesis of sepsis. We tested whether disruption of the gene encoding PCT in mice affected the course of sepsis. Mice with exons 2–5 of the gene encoding calcitonin/calcitonin gene–related polypeptide α (Calca) knocked out and congenic C57BL/6J control mice were challenged with aerosolized Streptococcus pneumoniae or Pseudomonas aeruginosa, or injected intraperitoneally with S. pneumoniae. There were no significant differences in the survival of knockout and control mice in the two pneumonia models, and no significant differences in weight loss, splenic bacterial counts, or blood leukocyte levels in the peritoneal sepsis model. To verify disruption of the Calca gene in knockout mice, the absence of calcitonin in the serum of knockout mice and its presence and inducibility in control mice were confirmed. To evaluate PCT expression in nonthyroidal tissues of control mice, transcripts were measured in multiple organs. PCT transcripts were not significantly expressed in liver or spleen of control mice challenged with aerosolized P. aeruginosa or intraperitoneal endotoxin, and were expressed in lung only at low levels, even though serum IL-6 rose 3,548-fold. We conclude that mice are not an ideal loss-of-function model to test the role of PCT in the pathogenesis of sepsis because of low nonendocrine PCT expression during infection and inflammation. Nonetheless, our studies demonstrate that nonendocrine PCT expression is not necessary for adverse outcomes from sepsis.
Keywords: procalcitonin, calcitonin, sepsis, pneumonia
The CALCA gene encodes the polypeptides procalcitonin (PCT) and PCT gene–related peptide α (proCGRPα), which are differentially expressed by alternative splicing (1, 2). In normal healthy humans, PCT is selectively produced by C cells of the thyroid and undergoes proteolytic cleavage intracellularly before being released as the mature calcitonin (CT) polypeptide. In pathologic inflammatory conditions, such as sepsis, pneumonia, burn, trauma, and pancreatitis, PCT is widely produced by nonendocrine parenchymal cells, but does not undergo proteolytic cleavage before being released. Circulating PCT is metabolized only slowly, and can therefore be used as a biomarker of inflammatory conditions. Measurement of PCT levels has been used to help guide the diagnosis of pneumonia and sepsis, prognosticate outcome, and guide antibiotic usage (3–5).
Aside from its role as a biomarker, there is evidence that PCT may participate in the pathophysiology of sepsis. In animal models, both gain-of-function and loss-of-function studies support a causal role of PCT in worsening outcomes. In a hamster model of sepsis, the administration of human PCT worsened mortality (6), whereas, in hamster, rat, and pig models of sepsis, the immunoneutralization of PCT reduced the severity of hypotension and rate of mortality (6–9). These findings are surprising, because patients with thyroidal and other tumors may have very elevated PCT levels without apparent adverse effects, although it is possible that elevated PCT levels exert an adverse effect only in septic conditions or when they rise rapidly. However, the gain-of-function experimental approach was subject to artifact in that endogenous PCT may not naturally achieve the level or rate of rise reached by exogenous administration, and the exogenous PCT polypeptide was from a different species, and so could trigger an immune recognition response (6). The immunoneutralization loss-of-function approach was subject to artifact in that PCT–antibody complexes might have immunomodulatory roles, and the antibodies were from different species, and so could themselves trigger an immune recognition response (6–9). To test further the role of PCT in the outcome of sepsis with a genetic loss-of-function approach that is free from these possible artifacts, we used gene-targeted mice that lack expression of PCT and proCGRPα.
Materials and Methods
Mice
Mice with exons 2–5 of the Calca gene knocked out (KO mice) were generated as previously described (2), and crossed for 10 generations to a C57BL/6J background. Control wild-type (WT) C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and bred within our colony. All mice were handled in accordance with the Institutional Animal Care and Use Committee of the M. D. Anderson Cancer Center. Mice were examined twice daily by veterinarians blinded to their genotypes, and, if distressed, were killed with an intraperitoneal injection (5 ml/kg) of a mixture of ketamine (37.5 mg/ml), xylazine (1.9 mg/ml), and acepromazine (0.37 mg/ml).
Infectious Challenges
For the gram-positive pneumonia model, Streptococcus pneumoniae serotype 4 isolated from the blood of a patient with pneumonia and passaged serially through mice to select for virulence toward mice was stored as frozen stock (1 × 109 CFU/ml), as previously described (10). Thawed stock was grown in Todd-Hewett broth in logarithmic phase, then washed and resuspended in PBS. For the pneumonia model, 10 ml of the suspension (5–6 × 1010 CFU/ml) was placed in an Aerotech nebulizer (Biodex Medical Systems, Shirley, NY) driven by 10 L/min of 5% CO2 in air to promote deep ventilation, connected by polyethylene tubing to a 10-L polyethylene exposure chamber with an efflux tube containing a low-resistance microbial filter vented to a biosafety hood, all as previously described (10). Mice of both genotypes were placed in the exposure chamber together so the bacterial challenge was similar for all mice, and aerosolization was continued for 1 hour. For the gram-negative pneumonia model, Pseudomonas aeruginosa strain PA103 was grown in tryptic soy broth as described previously (11), then washed and resuspended in PBS to 3–4 × 106 CFU/ml, and aerosolized as above. For the infectious peritonitis model, 50–100 CFU of S. pneumoniae were injected intraperitoneally in 0.1 ml PBS, and weight loss after 24 hours was measured as an indicator of sepsis severity. Mice were then killed, blood was obtained by cardiac puncture, and leukocyte counts were measured using a Coulter counter (Beckman Coulter, Danvers, MA). The spleen was homogenized in PBS, and bacterial titers were determined by plating serial dilutions on blood agar (Remel, Lenexa, KS), as previously described (10).
Measurement of Serum CT and PCT Immunoreactivity
Combined serum levels of CT and PCT were measured by two different techniques that do not distinguish between them. The first technique was a two-site immunoradiometric assay using a kit (no. 50-5000; Immutopics, San Clemente, CA) with two different antibodies to rat CT—a mouse monoclonal antibody immobilized on plastic beads to capture CT and PCT polypeptides, and radiolabeled affinity-purified polyclonal goat antibodies for detection. As rat and mouse CT are highly homologous in their coding sequence, this assay detects both. Because the relative abundance of CT and PCT in the samples is not known, the results are reported as pooled CT/PCT molar concentrations rather than the more conventional mass concentrations. The second technique was a standard radioimmunoassay using rabbit polyclonal antibodies to human CT with cross-reactivity to mouse CT (12). Because the relative sensitivity for mouse CT is not known, the results are reported as arbitrary units. To stimulate CT release independent of PCT release, mice were injected intraperitoneally with human parathyroid hormone (0.5 μg/g body weight) to elevate serum calcium levels (2). To stimulate both CT and PCT release, mice were exposed to aerosolized S. pneumoniae or P. aeruginosa as described previously here. We attempted to measure mouse PCT separately from CT using the Brahms PCT Sensitive Kryptor Assay (Thermo Fisher Scientific, Waltham, MA) designed for measurement of PCT in human subjects, and a Mouse PCT ELISA (M7878; TSZ Scientific, Biotang, Inc., Waltham, MA), both according to the manufacturers’ recommendations.
Measurement of PCT Transcripts and Serum Cytokine Levels
Mice were challenged with aerosolized P. aeruginosa as described previously here, and killed after 7 hours when the first mouse died suddenly and the others showed signs of distress. Thyroid, lung, liver, and spleen were harvested, frozen in liquid nitrogen, and then placed in a −80°C freezer until purification of total RNA using an RNeasy Mini Kit (Qiagen, Valencia, CA). Mice were also challenged in a noninfectious inflammation model by intraperitoneal injection of 100 μg of Escherichia coli endotoxin (Sigma-Aldrich, St. Louis, MO) in 0.1 ml PBS. After 4 or 24 hours, they were killed, organs were harvested for measurement of PCT transcripts, and blood was obtained by cardiac puncture for measurement of cytokines by two multiplex methods: Luminex xMAP (EMD Millipore, Billerica, MA), which uses custom-made fluorescent magnetic beads in a microtiter plate format, and a Mouse Pro-Inflammatory Cytokine 7-Plex Array (Meso Scale Discovery, Gaithersburg, MD), which uses electrochemiluminescence of binding events on patterned arrays. Results with both methods were similar, and only results using Luminex xMAP are illustrated. Quantitative RT-PCR (qRT-PCR) of PCT transcripts was performed using biological cDNA replicates and technical qRT-PCR triplicates. Reverse transcription was performed with qScript cDNA SuperMix (Quanta Biosciences, Gaithersburg, MD). The qRT-PCR reactions were performed in a 20-μl reaction mix that included 8 ng of cDNA, FAM-labeled PCT exon 3–4 assay primers (assay ID: Mm00801464_m1; Life Technologies, Carlsbad, CA), VIC-labeled glyceraldehyde 3-phosphate dehydrogenase endogenous control assay primers (catalog no. 4352339E; Life Technologies), PerfeCta qRT-PCR FastMix (Quanta Biosciences), and uracil-N-glycolase (Quanta Biosciences). Following uracil-N-glycolase incubation and AmpliTaq (Life Technologies) activation, 40 cycles of 95°C for 3 seconds and 60°C for 30 seconds were performed using a OneStep real-time PCR system (Life Technologies). Relative quantitation values were calculated using the ΔΔCT method subtracting glyceraldehyde 3-phosphate dehydrogenase cycle threshold values from PCT values.
Statistical Analyses
Proportions of mice surviving pathogen challenges were compared using Fisher’s exact test. A Kaplan-Meier analysis was not performed, because almost all the mice died within a single observation period. Summary statistics for changes in mouse weight, bacterial and leukocyte counts, CT and PCT polypeptide levels, cytokine levels, and PCT transcripts were compared using Student’s t test with Bonferroni’s adjustment for multiple comparisons. Calculations were performed using SPSS Statistics (IBM Software, Armonk, NY), and P values less than 0.05 were considered significant. Error bars illustrate the SEM in all figures. All data shown are representative of at least two independent experiments, and were not combined for analysis because of differences in the rates of death in WT mice among experiments that could not be controlled.
Results
Pneumonia Models
Mice of both genotypes were challenged with aerosolized S. pneumoniae or P. aeruginosa using concentrations titrated to cause the deaths of 20–80% of WT mice so that either increased or decreased survival could be detected in KO relative to WT mice. Most mice challenged with S. pneumoniae died by the third day, and most mice challenged with P. aeruginosa died by the second day (data not shown), but small numbers died as late as the fourth day, so overall survival was followed for 7 days. In the gram-positive (S. pneumoniae) challenge (Figure 1A), the 7-day survival rate of WT mice was 3/13 (23%) versus 3/14 (21.4%) for KO mice (P = 1.0). In the gram-negative (P. aeruginosa) challenge (Figure 1B), the 7-day survival rate of WT mice was 2/10 (20%) versus 2/11 (18.2%) for KO mice (P = 1.0). There were no significant differences in the survival of WT and KO mice in any of the gram-positive or gram-negative pneumonic challenges.
Figure 1.
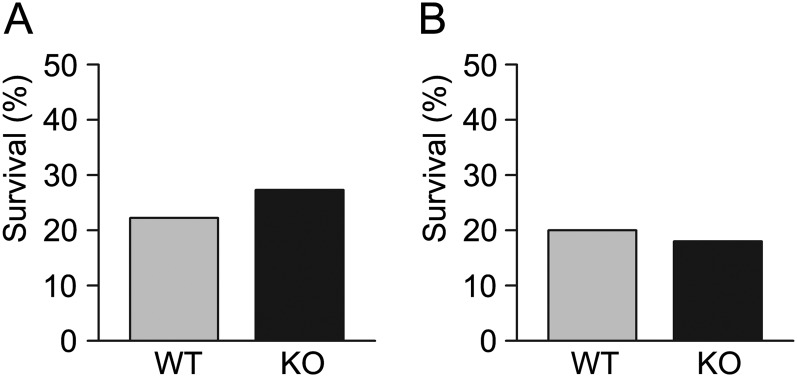
Responses of wild-type (WT) and Calca knockout (KO) mice to pneumonic challenges. (A) WT (n = 13) and KO (n = 14) mice were exposed to aerosolized Streptococcus pneumoniae (6.0 × 1010 CFU/ml for 60 min), and survival was measured after 7 days. (B) WT (n = 10) and KO (n = 11) mice were exposed to aerosolized Pseudomonas aeruginosa (3.7 × 106 CFU/ml for 60 min), and survival was measured after 7 days.
Peritonitis Model
After intraperitoneal challenge with the same virulent strain of S. pneumoniae, mice die between 24 and 48 hours, and weight loss and splenic bacterial counts measured at 24 hours strongly predict mortality (10). Baseline weights (30.4 g versus 32.1 g) and leukocyte counts (8.0 × 106/μl versus 7.8 × 106/μl) were similar in WT and KO mice, and there were no significant differences between WT and KO mice 24 hours after challenge in weight loss (4.8% versus 4.6%; P = 0.24; Figure 2A), splenic bacterial counts (1.9 × 107 versus 1.6 × 107; P = 0.31; Figure 2B), or leukocyte counts (5.8 × 106/μl versus 6.2 × 106/μl; P = 0.68; Figure 2C). Lymphocyte counts did fall significantly in challenged WT mice compared with unchallenged WT mice (1.3 × 106/μl versus 4.6 × 106/μl; P = 0.01), but there were no significant differences between challenged WT and KO mice or between challenged and unchallenged KO mice (Figure 2C), and no significant differences in other leukocyte subsets.
Figure 2.

Responses of WT and Calca KO mice to septic peritoneal challenge. WT and KO mice (five per group) were injected intraperitoneally with 100 μl of an S. pneumonia suspension (50–100 CFU), then killed and evaluated after 24 hours. (A) Weight change 24 hours after challenge measured as the percent decrease from baseline weight. (B) Splenic bacterial counts measured in CFU 24 hours after challenge. (C) Leukocyte counts in blood obtained by cardiac puncture 24 hours after challenge. *P = 0.01 compared with unchallenged WT mice.
Serum CT/PCT Immunoreactivity
The Calca gene KO mice used in these experiments have been shown previously to lack detectable CT or CGRPα in the thyroid gland by RT-PCR, immunohistochemistry, and two-site immunoradiometric assay (2). Because of the lack of differences in outcomes of infectious challenges in the current study, we sought to confirm the absence of CT/PCT in the circulation of KO mice and its presence in WT mice. We found no detectable CT/PCT immunoreactivity in the serum of KO animals at baseline by two-site immunoradiometric assay, whereas it was detectable in the serum of WT mice (Figure 3A). At 24 hours after S. pneumoniae infection by aerosol challenge, serum CT/PCT immunoreactivity rose 2.1-fold (P < 0.001) in WT mice, but remained undetectable in KO mice (Figure 3A). Samples were also assayed using a standard radioimmunoassay for CT in a separate experiment (12). Serum CT/PCT immunoreactivity was detectable at baseline in WT mice and rose 4.4-fold (P < 0.001) after stimulation with parathyroid hormone and 2.6-fold (P < 0.001) 24 hours after S. pneumoniae infection, but was undetectable in KO mice in any of these conditions (Figure 3B).
Figure 3.

Serum procalcitonin (PCT) and calcitonin (CT) immunoreactivity in WT and Calca KO mice. (A) Combined levels of serum PCT and CT were measured in WT and KO mice (five per group) using a two-site immunoradiometric assay for rat CT, both under baseline (Bsln) conditions and 24 hours after challenge with 5.0 × 1010 CFU/ml of aerosolized S. pneumoniae (S.p.) that kills 20–80% of infected mice after 3 days. *P < 0.001 compared with unchallenged WT mice. (B) Combined levels of serum PCT and CT were measured in WT and Calca KO mice (five per group) using a standard radioimmunoassay for human CT under baseline control conditions, 24 hours after challenge with 5.0 × 1010 CFU/ml of aerosolized S. pneumoniae, or 2 hours after intraperitoneal challenge with human parathyroid hormone (0.5 μg/g body weight) to elevate serum calcium levels. *P < 0.001 compared with unchallenged WT mice.
PCT Expression
We next sought to determine whether mice express PCT in nonthyroidal tissue during infection or inflammation. We were unable to detect PCT in the serum of WT mice 24 hours after aerosol challenge with S. pneumonia or P. aeruginosa using either a clinical assay for PCT in human subjects or an assay for mouse PCT (data not shown). However the sensitivity of these assays for mouse PCT is not known.
To evaluate further the expression of PCT in WT mice, we used a qRT-PCR assay targeting the exon 3/4 splicing junction. At baseline, PCT transcripts were four orders of magnitude more abundant in thyroid than in lung (Figure 4A), which contains neuroendocrine cells that express CGRP and low levels of PCT (5, 13, 14). Transcripts were not detectable in liver, spleen (Figure 4A), or other tissues (data not shown). PCT transcripts did not rise significantly in any tissue 7 hours after aerosol challenge with P. aeruginosa, and remained undetectable in liver and spleen (data not shown).
Figure 4.

Expression of tissue PCT transcripts and serum cytokines in mice challenged with intraperitoneal endotoxin. (A) WT mice (five per time point) were injected intraperitoneally with 100 μg of Escherichia coli endotoxin, killed after 4 or 24 hours, and organs were harvested for the measurement of PCT transcripts by quantitative RT-PCR. The levels of PCT transcripts were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) transcripts in the same organs, and are expressed relative to PCT transcripts in thyroid at baseline. *P < 0.001 compared with PCT transcripts in thyroid of unchallenged mice; #P < 0.001 for lung transcripts and P = 0.004 for thyroid transcripts compared with unchallenged mice. (B) Blood from the same mice as in (A) was obtained by cardiac puncture for measurement of serum cytokines by multiplexed assay. *P < 0.001 compared with unchallenged mice.
To be certain that mice were being tested during a temporal window in which they were both sufficiently healthy to mount an inflammatory response and sufficiently stimulated to induce a response, WT mice were challenged intraperitoneally with endotoxin and their cytokine responses were measured. None of the mice died from the challenge or displayed behavioral changes, such as huddling together or ruffling their fur that might suggest endotoxic shock. Levels of TNF-α, IL-10, IL-6, and keratinocyte chemoattractant (CXCL1) were very low in unchallenged WT mice (Figure 4B). At 4 hours after challenge, levels of all four cytokines rose dramatically (TNF-α rose 22-fold, from 4.1 to 91.8 ng/ml [P < 0.001]; IL-10 rose 147-fold, from 3.1 to 448.9 ng/ml [P < 0.001]; IL-6 rose 3,548-fold, from 8.2 to 29,069.8 ng/ml [P < 0.001]; CXCL1 rose 240-fold, from 102.0 to 24,516.0 ng/ml [P < 0.001]), confirming the sufficiency of the stimulus. However, PCT transcripts remained unchanged in livers and spleens from the same mice (Figure 4A). There was a 3.8-fold (P = 0.004) rise in PCT transcripts in thyroid at 24 hours (Figure 4A), similar to the rise in serum CT after infectious challenge (Figures 3A and 3B), and an 11.4-fold (P < 0.001) rise in the low level of PCT transcripts in lungs at 4 hours, which returned to baseline at 24 hours (Figure 4A). Transcripts in thyroid tissue were more than 1,000-fold greater than in nonthyroidal tissues at all time points (Figure 4A), in contrast to differences of less than 100-fold in septic hamsters that have a measureable rise in serum PCT (14).
Discussion
Sepsis is a common and serious disorder, with more than 700,000 cases per year in North America and a mortality of 30–50% (15). Therefore, a therapy that could alter mortality from sepsis would have great medical importance. Prior preclinical studies in hamster, pig, and rat models of sepsis in which PCT levels were manipulated by administration of exogenous polypeptide or immunoneutralization of endogenous polypeptide suggested that PCT plays an important role in sepsis outcomes as follows. In a hamster model of sepsis induced by surgical implantation into the peritoneal cavity of toxigenic E. coli embedded in agar, the administration of human PCT increased mortality from 56 to 93% (6). Conversely, the prophylactic administration of goat antiserum against PCT reduced mortality from 62 to 6% in the same model, and the therapeutic administration of antiserum 1 hour after microbial challenge reduced mortality from 82 to 54% (6). In a pig model of sepsis induced by surgical implantation into the peritoneal cavity of a mixture of autologous cecal contents and toxigenic E. coli suspended in water, prophylactic intravenous administration of pooled Igs from rabbits immunized against a porcine PCT peptide improved physiologic and metabolic parameters and reduced mortality from 100 to 14% (9). Therapeutic administration of Igs 3 hours after challenge in the same model reduced mortality from 100 to 20% (7). In a rat model of sepsis induced by intraperitoneal injection of bacterial LPS, prophylactic intraperitoneal injection of rabbit antibodies or a mouse monoclonal antibody against human PCT reduced mortality from 100% to 15% and 30%, respectively, and therapeutic injection 2 hours after LPS reduced mortality to 45% (8). Taken together, these results, in a variety of animal models, challenged with a variety of infectious and inflammatory insults, suggest that PCT or its metabolic products play an important role in the pathogenesis of mortality from sepsis. However, all of these experiments are subject to the possibility of artifacts, including immune reactions to foreign PCT and foreign antibodies, the formation of immune complexes, and nonphysiologic kinetics of changes in PCT levels.
To assess the role of PCT in sepsis free from these possible artifacts, we used a mouse loss-of-function genetic model. There were no significant differences between WT and Calca KO mice in the course of serious infection in any of three separate models of bacterial challenge (Figures 1 and 2). However, our failure to detect PCT in the serum of WT mice or of PCT transcripts in nonthyroidal tissue, aside from lung, in a variety of infectious and inflammatory challenges limits the utility of the mouse as an animal model to test whether PCT contributes to the pathophysiology of sepsis. In particular, the nonlethal challenge with endotoxin establishes the inability of mice to express PCT in nonthyroidal tissue (Figure 4). This experiment was performed because we considered the possibility that mice in our infectious challenges transitioned from a localized infection to septic death too rapidly to mount a nonthyroidal PCT response. Therefore, we wanted to be certain that mice were being tested during a temporal window in which they were both sufficiently stimulated to induce a strong response and sufficiently healthy to mount the response. The sufficiency of the inflammatory stimulus is indicated by the marked rise in cytokines 4 hours after endotoxin injection (Figure 4B), and the health of the animals is indicated by the fact that none of the mice displayed distressed behavior or died spontaneously before they were killed at 4 or 24 hours after endotoxin injection (Figure 4B). The fact that PCT transcripts did not rise in livers or spleens from these mice (Figure 4A), yet were readily detectable in thyroids from the same animals where they rose with inflammatory (Figure 4A) and infectious (Figure 3) challenges, strongly supports the inability of mice to express PCT in nonthyroidal tissue.
Despite this limitation of the mouse as an experimental model to evaluate the role of PCT in the pathogenesis of sepsis, the fact that KO mice in our infectious challenges experienced acute weight loss (Figure 2A) and mortality (Figure 1) suggests that circulating PCT is not necessary for severe physiologic compromise or death in mammals. However, whether elevated circulating PCT causes worse outcomes could not be evaluated. It would be feasible to introduce exogenous mouse PCT during infectious challenges to test the latter possibility, but a negative result might not be informative, because, if mice do not release PCT into the circulation as our results suggest, they may also lack receptors that could mediate PCT activity. Of interest, rats also appear to have weak or absent nonthyroidal PCT expression, because the area under the curve for serum PCT rose less than twofold in response to LPS or muramyl dipeptide challenge in a prior study, whereas the areas under the curve for several cytokines had fold increases in the hundreds to thousands (16).
Alternative interpretations of our results include the possibility that simultaneous ablation of the expression of PCT and other Calca gene products induced opposing effects that canceled each other out. For example, injection of CGRPα protected mice against lethal endotoxemia in one model system (17). However, the fact that CGRPα has proinflammatory effects in other model systems (18) suggests that pretreatment of mice with CGRPα may have conditioned them to tolerate endotoxin, rather than acting directly to mitigate sepsis. Similarly, the carboxyterminus peptides encoded by the CT transcript (CCP-I) may also be capable of signaling (19). Another possibility is that lifelong absence of the polypeptides encoded by the Calca gene in our KO mice results in compensatory effects that undermine the straightforward interpretation of our results. Finally, the mouse genome encodes a closely related gene, Calcb, which expresses proCGRPβ, but does not contain an open reading frame for exon 4 that is homologous to PCT, so is unlikely to confound our results (20).
In view of the importance of advancing therapies for sepsis, further preclinical investigations of the role of PCT seem warranted. These might include the use of a neutralizing monoclonal antibody genetically modified to minimize differences from the experimental host outside the region of antigen recognition. This approach would have the advantage of modeling a plausible therapeutic strategy in humans. Furthermore, multiple PCT epitopes could be targeted to support the possibility of a class effect of this therapeutic strategy, and a single neutralizing monoclonal antibody could be engineered to be compatible with multiple experimental animal species. Additional genetic approaches might include disruption of the entire Calca gene or selective regions encoding individual peptides in an animal model that experiences a rise in circulating PCT during sepsis, such as the pig.
In summary, deletion of the gene encoding PCT did not influence the course of sepsis in three separate mouse models of infection. However, the significance of these findings is limited by the fact that PCT does not appear to be substantially expressed in nonthyroidal tissues in mice.
Acknowledgments
Acknowledgments
The authors thank Luis Vence of the Immune Monitoring Core Laboratory for performing the cytokine assays, and Mark Munsell of the Department of Biostatistics for assistance with data analysis, both from the M. D. Anderson Cancer Center.
Footnotes
This work was supported by institutional funds and M. D. Anderson Cancer Center support grant CA16672 (M.J.T., B.F.D.), and by the Department of Veterans Affairs (L.J.D.).
Originally Published in Press as DOI: 10.1165/rcmb.2012-0489OC on March 22, 2013
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Christ-Crain M, Muller B. Calcitonin peptides—the mediators in sepsis or just another fairy tale? Crit Care Med. 2008;36:1684–1687. doi: 10.1097/CCM.0b013e3181726819. [DOI] [PubMed] [Google Scholar]
- 2.Hoff AO, Catala-Lehnen P, Thomas PM, Priemel M, Rueger JM, Nasonkin I, Bradley A, Hughes MR, Ordonez N, Cote GJ, et al. Increased bone mass is an unexpected phenotype associated with deletion of the calcitonin gene. J Clin Invest. 2002;110:1849–1857. doi: 10.1172/JCI200214218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schuetz P, Albrich W, Mueller B. Procalcitonin for diagnosis of infection and guide to antibiotic decisions: past, present and future. BMC Med. 2011;9:107. doi: 10.1186/1741-7015-9-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jensen JU, Hein L, Lundgren B, Bestle MH, Mohr TT, Andersen MH, Thornberg KJ, Loken J, Steensen M, Fox Z, et al. Procalcitonin-guided interventions against infections to increase early appropriate antibiotics and improve survival in the intensive care unit: a randomized trial. Crit Care Med. 2011;39:2048–2058. doi: 10.1097/CCM.0b013e31821e8791. [DOI] [PubMed] [Google Scholar]
- 5.Riedel S. Procalcitonin and the role of biomarkers in the diagnosis and management of sepsis. Diagn Microbiol Infect Dis. 2012;73:221–227. doi: 10.1016/j.diagmicrobio.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 6.Nylen ES, Whang KT, Snider RH, Jr, Steinwald PM, White JC, Becker KL. Mortality is increased by procalcitonin and decreased by an antiserum reactive to procalcitonin in experimental sepsis. Crit Care Med. 1998;26:1001–1006. doi: 10.1097/00003246-199806000-00015. [DOI] [PubMed] [Google Scholar]
- 7.Martinez JM, Wagner KE, Snider RH, Nylen ES, Muller B, Sarani B, Becker KL, White JC. Late immunoneutralization of procalcitonin arrests the progression of lethal porcine sepsis. Surg Infect (Larchmt) 2001;2:193–202. doi: 10.1089/109629601317202678. [DOI] [PubMed] [Google Scholar]
- 8.Tavares E, Minano FJ. Immunoneutralization of the aminoprocalcitonin peptide of procalcitonin protects rats from lethal endotoxaemia: neuroendocrine and systemic studies. Clin Sci (Lond) 2010;119:519–534. doi: 10.1042/CS20100007. [DOI] [PubMed] [Google Scholar]
- 9.Wagner KE, Martinez JM, Vath SD, Snider RH, Nylen ES, Becker KL, Muller B, White JC. Early immunoneutralization of calcitonin precursors attenuates the adverse physiologic response to sepsis in pigs. Crit Care Med. 2002;30:2313–2321. doi: 10.1097/00003246-200210000-00021. [DOI] [PubMed] [Google Scholar]
- 10.Clement CG, Evans SE, Evans CM, Hawke D, Kobayashi R, Reynolds PR, Moghaddam SJ, Scott BL, Melicoff E, Adachi R, et al. Stimulation of lung innate immunity protects against lethal pneumococcal pneumonia in mice. Am J Respir Crit Care Med. 2008;177:1322–1330. doi: 10.1164/rccm.200607-1038OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans SE, Scott BL, Clement CG, Larson DT, Kontoyiannis D, Lewis RE, LaSala PR, Pawlik J, Peterson JW, Chopra AK, et al. Stimulated innate resistance of lung epithelium protects mice broadly against bacteria and fungi. Am J Respir Cell Mol Biol. 2010;42:40–50. doi: 10.1165/rcmb.2008-0260OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deftos LJ. Immunoassay for human calcitonin. I. Method. Metabolism. 1971;20:1122–1128. doi: 10.1016/0026-0495(71)90037-0. [DOI] [PubMed] [Google Scholar]
- 13.Russwurm S, Stonans I, Stonane E, Wiederhold M, Luber A, Zipfel PF, Deigner HP, Reinhart K. Procalcitonin and CGRP-1 mRNA expression in various human tissues. Shock. 2001;16:109–112. doi: 10.1097/00024382-200116020-00004. [DOI] [PubMed] [Google Scholar]
- 14.Muller B, White JC, Nylen ES, Snider RH, Becker KL, Habener JF. Ubiquitous expression of the calcitonin-I gene in multiple tissues in response to sepsis. J Clin Endocrinol Metab. 2001;86:396–404. doi: 10.1210/jcem.86.1.7089. [DOI] [PubMed] [Google Scholar]
- 15.Ward PA, Bosmann M. A historical perspective on sepsis. Am J Pathol. 2012;181:2–7. doi: 10.1016/j.ajpath.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tavares E, Maldonado R, Ojeda ML, Minano FJ. Circulating inflammatory mediators during start of fever in differential diagnosis of gram-negative and gram-positive infections in leukopenic rats. Clin Diagn Lab Immunol. 2005;12:1085–1093. doi: 10.1128/CDLI.12.9.1085-1093.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gomes RN, Castro-Faria-Neto HC, Bozza PT, Soares MB, Shoemaker CB, David JR, Bozza MT. Calcitonin gene–related peptide inhibits local acute inflammation and protects mice against lethal endotoxemia. Shock. 2005;24:590–594. doi: 10.1097/01.shk.0000183395.29014.7c. [DOI] [PubMed] [Google Scholar]
- 18.Springer J, Geppetti P, Fischer A, Groneberg DA. Calcitonin gene–related peptide as inflammatory mediator. Pulm Pharmacol Ther. 2003;16:121–130. doi: 10.1016/S1094-5539(03)00049-X. [DOI] [PubMed] [Google Scholar]
- 19.Becker KL, Nylen ES, White JC, Muller B, Snider RH., Jr Clinical review 167: procalcitonin and the calcitonin gene family of peptides in inflammation, infection, and sepsis: a journey from calcitonin back to its precursors. J Clin Endocrinol Metab. 2004;89:1512–1525. doi: 10.1210/jc.2002-021444. [DOI] [PubMed] [Google Scholar]
- 20.Thomas PM, Nasonkin I, Zhang H, Gagel RF, Cote GJ. Structure of the mouse calcitonin/calcitonin gene–related peptide alpha and beta genes. DNA Seq. 2001;12:131–135. doi: 10.3109/10425170109047567. [DOI] [PubMed] [Google Scholar]