

Abstract
This report presents the synthesis and biological evaluation of a collection of 2-aminothiazoles as a novel class of compounds with the capability to reduce the production of PGE2 in HCA-7 human adenocarcinoma cells. A total of 36 analogs were synthesized and assayed for PGE2 reduction, and those with potent cellular activity were counter screened for inhibitory activity against COX-2 in a cell free assay. In general, analogs bearing a 4-phenoxyphenyl substituent in the R2 position were highly active in cells while maintaining negligible COX-2 inhibition. Specifically, compound 5l (R1=Me, R2=4-OPh-Ph, R3=CH(OH)Me) exhibited the most potent cellular PGE2 reducing activity of the entire series (EC50 = 90 nM) with an IC50 value for COX-2 inhibition of > 5 μM in vitro. Furthermore, the anti-tumor activity of analog 1a was analyzed in xenograft mouse models exhibiting promising anti-cancer activity.
Inflammatory processes are implicated in 25% of all cancers and chronic inflammation has been shown to promote the growth of malignant tissues. Indeed, prostaglandin E2 (PGE2) has been identified as a key mediator of pain and inflammation and is overexpressed in various cancers. Definitive evidence suggests that PGE2 is the dominant prostaglandin involved in the growth of tumors associated with colon, lung, breast, head, and neck cancers.1-4 Interestingly recent studies support a role for PGE2 in most if not all of the six hallmarks of cancer named by Hanahan and Weinberg; the mechanisms by which PGE2 induces these cellular capabilities can vary amongst cancer types.4-6
Traditionally, prostaglandin synthesis has been modulated primarily through the use of non-steroidal anti-inflammatory drugs (NSAIDs), including non-specific cyclooxygenase (COX) -1 and 2 inhibitors (Aspirin, Tylenol, and Ibuprofen) and specific COX-2 inhibitors (Bextra®, Celebrex®, and Vioxx®). Incidentally, patients taking NSAIDs for chronic inflammatory disorders such as rheumatoid arthritis are associated with a decreased incidence of colon cancer, and high doses of NSAIDs have been shown to cause colorectal tumor regression.1-4 Furthermore, exogenous treatment with PGE2 blocks NSAID-induced tumor regression.1 Therefore, it is believed that the anti-cancer effects of COX inhibitors are ultimately due to a reduction of PGE2 levels, although there is a downside of NSAID treatment in that the high doses and chronic usage required for anti-cancer activity lead to adverse target-based side effects including gastrointestinal intolerance, heart attacks, and blood clotting.1-4 Thus, these safety implications have motivated research efforts focused on the reduction of PGE2 levels through alternate mechanisms.7-11
Herein we report the biological evaluation of a series of 2-aminothiazole analogs as a novel class of small molecules with PGE2 reducing character in HCA-7 colon cancer cells. The general structure for analogs 1-5 is shown in Figure 1.
Figure 1.

General structure of aminothiazoles 1-5
2-Aminothiazole analogs depicted in Table 1 (1a-h, 2a-g, 3a-d, 4a-e) as well as structures 5c-i from Table 2 were assembled by employing a microwave assisted Hantzsch reaction (Scheme 1).12, 16 Compounds 5j and 5k-l were obtained by reduction of the ketone functionality of 5i and 5f, respectively (Scheme 1). Alternately, compounds 5a and 5b were synthesized in two-steps proceeding through an amidino-thiourea intermediate (6) (Scheme 2).13 Purities of all analogs were assessed by HPLC/MS analysis. All compounds showed purity level greater than 95%, as judged by UV absorbance at 254 nm and 214 nm as well as evaporative light scattering (ELS).
Table 1.
| Cmpd | R1 | R2 | R3 | R4 | PGE2 (%)c | COX-2 (%)d | COX-2 IC50μMe | PGE2 EC50 /μMf |
|---|---|---|---|---|---|---|---|---|
| 1a | 4-Cl-Ph | 4-OPh-Ph | H | H | 75.5 ± 7 | 32.1 ± 5 | > 10 | 0.28 ± 0.11 |
| 1b | 4-Cl-Ph | 3-OCH2O-4-Ph | H | H | 94.2 ± 3 | 68.8 ± 8 | 0.84 ± 0.20 | - |
| 1c | 4-Cl-Ph | 3-OMe-Ph | H | H | 78.3 ± 3 | 55.1 ± 3 | 1.39 ± 0.34 | - |
| 1d | 4-Cl-Ph | Ph | H | H | 51.0 ± 8 | - | - | - |
| 1e | 4-Cl-Ph | 1-naphthyl | H | H | 51.4 ± 8 | - | - | - |
| 1f | 4-Cl-Ph | 4-CN-Ph | H | H | 23.1 ± 10 | - | - | - |
| 1g | 4-Cl-Ph | 4-OH-Ph | H | Me | 85.8 ± 8 | 84.2 ± 13 | 1.08 ± 0.37 | - |
| 1h | 4-Cl-Ph | 4-OMe-Ph | Me | H | 89.3 ± 2 | 12.0 ± 1 | > 5 | 0.29 ± 0.10 |
| 2a | Ph | 4-OPh-Ph | H | H | 79.6 ± 10 | 31.0 ± 1 | > 5 | 0.33 ± 0.15 |
| 2b | 4-F-Ph | 4-OPh-Ph | H | H | 87.9 ± 1 | 43.4 ± 8 | > 5 | 0.12 ± 0.04 |
| 2c | 2-OMe-Ph | 4-OPh-Ph | H | H | 82.0 ± 3 | 38.9 ± 5 | > 5 | 0.18 ± 0.02 |
| 2d | 3-OMe-Ph | 4-OPh-Ph | H | H | 88.9 ± 1 | 40.3 ± 4 | > 5 | 0.29 ± 0.13 |
| 2e | 4-OMe-Ph | 4-OPh-Ph | H | H | 80.4 ± 2 | 40.8 ± 5 | > 5 | 0.24 ± 0.05 |
| 2f | 2-napthyl | 4-OPh-Ph | H | H | 53.5 ± 1 | - | - | - |
| 2g | 4-Cy-Ph | 4-OPh-Ph | H | H | 41.6 ± 16 | - | - | - |
| 3a | 4-Me-Ph | 4-OMe-Ph | H | H | 37.3 ± 6 | - | - | - |
| 3b | 3,4-diMe-Ph | 4-OMe-Ph | H | H | 73.9 ± 2 | 69.3 ± 5 | 1.20 ± 0.19 | - |
| 3c | 4-OMe-Ph | 4-OMe-Ph | H | H | 73.6 ± 3 | 66.8 ± 3 | 1.37 ± 0.08 | - |
| 3d | 4-OMe-Ph | 4-Me-Ph | H | H | 91.5 ± 1 | 73.1 ± 6 | 0.81 ± 0.16 | - |
| 4a | Ph | 3-OCH2O-4-Ph | H | H | 98.2 ± 1 | 114.2 ± 1 | 0.41 ± 0.11 | - |
| 4b | 2-Cl-Ph | 3-OCH2O-4-Ph | H | H | 85.9 ± 3 | 84.8 ± 3 | 0.91 ± 0.10 | - |
| 4c | 2-OMe-Ph | 3-OCH2O-4-Ph | H | H | 89.0 ± 4 | 91.3 ± 4 | 0.93 ± 0.02 | - |
| 4d | 3-OMe-Ph | 3-OCH2O-4-Ph | H | H | 94.4 ± 2 | 86.0 ± 8 | 0.92 ± 0.38 | - |
| 4e | 4-OMe-Ph | 3-OCH2O-4-Ph | H | H | 96.2 ± 1 | 94.4 ± 2 | 0.91 ± 0.06 | - |
Celecoxib was used as a positive control (PGE2, 5.70 ± 3.28% and EC50 = 1.15 ± 0.06 nM; COX-2, 85.6 ± 4.4% inhibition and IC50 = 0.03 ± 0.01)12.
- not determined.
% of inhibition of PGE2 levels in HCA-7 cells at 1μM concentration ± SD (n = 3).
% of inhibition of COX-2 levels in vitro at 5 μM concentration ± SD (n = 3).
in vitro IC50 for COX-2 inhibition ± SD (n = 3).
EC50 for PGE2 level reduction in HCA-7 cells ± SD (n = 3).
Table 2.
| Cmpd | R1 | R2 | R3 | PGE2 (%)d | COX-2 (%)e | COX-2 IC50 /μMf | PGE2 EC50/μMg |
|---|---|---|---|---|---|---|---|
| 5a | Ph | 3-OCH2O-4-Ph | C(O)Ph | NR | - | - | - |
| 5b | Ph | 4-CN-Ph | C(O)Ph | NR | - | - | - |
| 5c | Ph | 4-OMe-Ph | C(O)Ph | NR | - | - | - |
| 5d | Ph | 4-OPh-Ph | C(O)Ph | 42.0 ± 6 | - | - | - |
| 5e | Me | Ph | C(O)Me | 29.9 ± 1 | - | - | - |
| 5f | Me | 4-OPh-Ph | C(O)Me | 87.9 ± 1 | 28.9 ± 15 | > 5 | 0.39 ± 0.07 |
| 5g | Me | 3-OCH2O-4-Ph | C(O)Me | 25.3 ± 3 | - | - | - |
| 5h | Me | 4-CN-Ph | C(O)Me | 22.4 ± 4 | - | - | - |
| 5i | Me | 4-OMe-Ph | C(O)Me | 58.5 ± 4 | - | - | - |
| 5j | Me | 4-OMe-Ph | Et | 94.4 ± 1 | 49.3 ± 4 | > 5 | 0.32 ± 0.14 |
| 5k | Me | 4-OPh-Ph | Et | 87.1 ± 3 | 41.3 ± 5 | > 5 | 0.37 ± 0.07 |
| 5l | Me | 4-OPh-Ph | CH(OH)Me | 80.4 ± 3 | -16.9 ± 10 h | > 5 | 0.09 ± 0.04 |
Celecoxib was used as a positive control (PGE2, 5.70 ± 3.28% and EC50 = 1.15 ± 0.06 nM; COX-2, 85.6 ± 4.4% inhibition and IC50 = 0.03 ± 0.01)12.
NR, no observed reduction.
- not determined.
% of inhibition of PGE2 levels in HCA-7 cells at 1 μM concentration ± SD (n = 3).
% of inhibition of COX-2 levels in vitro at 5 μM concentration ± SD (n = 3).
in vitro IC50 for COX-2 inhibition ± SD (n = 3).
EC50 for PGE2 level reduction in HCA-7 cells ± SD (n = 3).
to be considered as no inhibition of activity rather than induction of activity.
Scheme 1.

Synthesis of aminothiazoles 1a-h, 2a-g, 3a-d, 4a-e, 5c-l
Scheme 2.
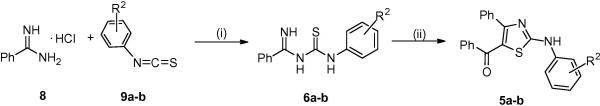
Synthesis of aminothiazoles 5a-b
Initially, all compounds (Figure 2-3) were screened for their ability to reduce PGE2 production in HCA-7 colon cancer cells at 1 μM concentration; activities are summarized in Tables 1-2 as percentage reduction of PGE2 levels.17 As a means for tuning out COX-2 activity, compounds that exhibited reduction of PGE2 levels higher than 70% were tested for COX-2 inhibition at 5 μM in an in vitro cell free assay,18 with Celecoxib incorporated as a positive control in both PGE2 and COX-2 assays.14 IC50 values for COX-2 inhibition were determined only for compounds that exhibited inhibitory activity against COX-2 greater than 50%. Only compounds that exhibited more than 70% reduction of PGE2 levels, but did not show more than 50% COX-2 inhibition were tested further for EC50 determination.
Figure 2.

Structure of 2-aminothiazoles analogs 1-4.
Figure 3.

Structure of 2-aminothiazoles analogs 5.
Aminothiazole analogs 1a-h consist of a para-chlorophenyl group at the C-4 position (R1) of the thiazole ring and varying substituents at other positions. Presence of 4-phenoxyphenyl (1a), 3,4-methylenedioxyphenyl (1b), or 3-methoxyphenyl (1c) at the R2 position leads to good activity (reduction in PGE2 levels = 76-94%). Compounds 1d-e were characterized by lower activity (reduction in PGE2 levels = 51%) when compared to compounds 1a-c. Substitution with a polar cyano group (1f) also resulted in significant loss of activity (reduction in PGE2 levels = 23%) possibly due to a decreased ability to permeate the cellular membrane. Surprisingly, analog 1g bearing a methyl group on the amino functionality (R4), resulted in good cellular activity despite the polar 4-hydroxyphenyl substituent at R2 (reduction in PGE2 levels = 86%). Similarly, substitution of a methyl group at the C-5 position (R3) of the aminothiazole ring, as in compound 1h, is also well tolerated (reduction in PGE2 levels 89%). Compounds from this group exhibited relatively strong inhibition of COX-2 in vitro (IC50 = 0.84-1.39 μM), with exceptions represented by 1a and 1h (IC50 > 5 μM). These two compounds were then tested for EC50 determination and show similar EC50 values for cellular PGE2 reduction (0.28 μM and 0.29 μM, respectively).
Compounds 1a and 2a-g consist of a para-phenoxyphenyl group on the amino functionality (R2). These compounds show a moderate to high reduction of PGE2 levels (42-89%). In this series, compounds characterized by a para-fluorophenyl (2b) or a methoxyphenyl (2c-e) substituent at C-4 of the 2-aminothiazole core (R1) exhibited the highest level of PGE2 reduction (80-89%). Movement of the methoxy group amongst ortho, meta, and para positions on the phenyl ring at R1 did not significantly influence the observed biological activity (2c-e). However, the activity of compound 2a (reduction of PGE2 levels 80%) suggested that the presence of a substituent on the phenyl ring of R1 was not essential. Presence of bulky lipophilic substituents (naphthyl, 2f; 4-cyclohexylphenyl, 2g) at the R1 position results in lower activity (reduction in PGE2 levels = 54% and 42%, respectively) when compared to compound 2a. None of these compounds 2a-g showed significant COX-2 inhibitory activity (IC50 values > 5 μM). Notably, analogs 2b and 2c exhibited the lowest EC50 values for PGE2 reduction of this group (0.12 and 0.18 μM, respectively). Other analogs showed EC50 values similar to analog 1a (0.24-0.33 μM). In general, analogs bearing a para-phenoxyphenyl substituent at R2 (1a and 2a-e) showed negligible inhibitory activity on COX-2 while still strongly reducing PGE2 levels in cells.
Replacement of the previous para-phenoxyphenyl group at R2 (2a-g) with a para-methoxphenyl group (3a-c) resulted in an analogous activity trend. Compounds 2e and 3c bearing a para-methoxyphenyl at R1 both show similar reduction of PGE2 levels (80% and 74%, respectively). Additionally, compounds 3a and 3b bearing 4-methylphenyl and 3,4-dimethylphenyl substituents, respectively, showed cellular activities comparable to analogs 2a, 2f, and 2g also containing hydrophobic moieties at C-4 (reduction in PGE2 levels = 37-74%, 3a-b and 42-80%, 2a, f-g). Interestingly, when the 4-methoxyphenyl group on R2 of analog 3c was exchanged with a 4-methylphenyl to give analog 3d, an increase in cellular PGE2 reducing activity was observed (reduction in PGE2 levels = 74% and 92%, respectively). Although transitioning from a para-phenoxyphenyl (2a-g) to a para-methoxyphenyl (3a-c) at R2 does not significantly affect PGE2 reducing activity, the COX-2 inhibition of 3b-c (IC50 = 1.20 and 1.37 μM, respectively) is considerably stronger than for 2a-e (IC50 values > 5 μM).
Introduction of a 3,4-methylenedioxyphenyl substituent on the amine nitrogen (R2) resulted in a dramatic reduction of cellular PGE2 levels (4a-e, 98-86%). Compound 4a, characterized by an un-substituted phenyl ring at the C-4 position of the 2-aminothiazole (R1) showed the highest activity (reduction in PGE2 levels = 98%). Unfortunately, the high levels of cellular PGE2 reducing activity observed with this group of compounds is always associated with relatively strong inhibition of COX-2 activity (4a-e; IC50 = 0.41- 0.93 μM). Thus, the strong cellular activity of 4a-e as well as analogs 3b-c may arise, at least in part, from inhibitory effects on COX-2. However the overall cellular PGE2 reducing activity of these analogs cannot be fully explained by COX-2 inhibition and rather is likely the result of inhibitory activity on more than one target. Furthermore, although tuning out COX-2 inhibition has proven to be clinically important in terms of safety, PGE2 reducing therapeutics functioning primarily through a mechanism not involving COX, but maintaining weak COX-2 inhibition, may still be therapeutically useful if PGE2 biosynthesis can be blocked while COX activity remains at least at a basal level.1-4
Compounds 5a-i containing a keto moiety (acetyl or benzyl group) at the R3 position (Table 2) were moderately active to inactive (0-59%; except 5f, reduction of PGE2 levels = 88%). Overall, analogs characterized by the presence of a methyl at C-4 (R1) and an acetyl group at C-5 (R3) of the aminothiazole (5e-i) exhibited higher PGE2 reducing character than analogs 5a-d. Of interest, when the acetyl group of 5i was reduced to the corresponding ethyl chain (R3, 5j) a marked reduction of PGE2 levels was observed (94%). In contrast, reduction of the ketone functionality of 5f to the corresponding ethyl group (5k) did not lead to a significant change in activity (reduction in PGE2 levels = 88% and 87%, respectively). Interestingly, compound 5l with -CH(OH)Me at R3 was the most potent compound identified in this report (EC50 = 0.09 μM). Compound 5l was slightly more active than its parent analog 5f (EC50 = 0.39 μM) and its fully reduced derivative 5k (EC50 = 0.37 μM). Active compounds 5f and 5j-l were analyzed for COX-2 activity, showing little to no inhibition at 5 μM.
In conclusion, we have synthesized and tested a series of 2-aminothiazole congeners with the ability to reduce the production of PGE2 in HCA-7 cells. A total of 36 aminothiazoles were evaluated, and active compounds with limited COX-2 inhibition were identified. Compound 1a was also evaluated for its effect on tumor growth in mouse xenograft models under three different cell lines, and was confirmed to have promising anti-cancer activity. Tumor vs. control values (T/C) were 61% and 40% for HCA-7 human colonic adenocarcinoma and SW837 rectum adenocarcinoma cell lines, respectively, under a dosing schedule of 200 mg/kg over 5 days (P<0.05). Additionally, a T/C value of 38% was observed in A549 human lung adenocarcinoma cells after treatment with 100 mg/kg over a period of 10 days (P<0.05).15 Work is on-going to identify alternate modes of action that may feasibly lead to reduced PGE2 levels. Future synthetic efforts will be focused on further development of aminothiazoles with diverse substitution patterns that will continue to reveal important structure activity relationship features and aid in the identification of more potent compounds with anticancer activity in vivo.
Acknowledgements
The authors would like to thank financial support provided by an NIH training grant research fellowship (BCP Fellowship, Grant # 5T32GM008804-09), the University of Arizona College of Pharmacy for start-up funds and SR01CA138702. Finally, the authors graciously acknowledge Kristen Keck for selected compound purification, Alex Laetsch for compound management and Nicole Schechter for group coordination.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Iyer JP, Srivastava PK, Dev R, Dastidar SG, Ray A. Expert Opin. Ther. Tar. 2009;13:849. doi: 10.1517/14728220903018932. [DOI] [PubMed] [Google Scholar]
- 2.Friesen RW, Mancini JA. J. Med. Chem. 2008;51:4059. doi: 10.1021/jm800197b. [DOI] [PubMed] [Google Scholar]
- 3.Tawab MA, Zettl H, Zsilavecz MS. Curr. Med. Chem. 2009;16:2042. doi: 10.2174/092986709788682209. [DOI] [PubMed] [Google Scholar]
- 4.Wang D, DuBois R. Nat. Rev. Cancer. 2010;10:181. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a Hanahan D, Weinberg RA. Cell. 2000;100:57. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]; b Hanahan D, Weinberg RA. Cell. 2011;144:646. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Greenhough A, Smartt HJM, Moore AE, Roberts HR, Williams AC, Paraskeva C, Kaidi A. Carcinogenesis. 2009;30:377. doi: 10.1093/carcin/bgp014. [DOI] [PubMed] [Google Scholar]
- 7.Riendeau D, Aspiotis R, Ethier D, Gareau Y, Grimm EL, Guay J, Guiral S, Juteau H, Mancini JA, Methot N, Rubin J, Friesen W. Bioorg. Med. Chem. Lett. 2005;15:3352. doi: 10.1016/j.bmcl.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 8.Cote B, Boulet L, Brideau C, Claveau D, Ethier D, Frenette R, Gagnon M, Giroux A, Guay J, Guiral S, Mancini J, Martins E, Masse F, Methot N, Riendeau D, Rubin J, Xu D, Yu H, Ducharme Y, Friesen RW. Bioorg. Med. Chem. Lett. 2007;17:6816. doi: 10.1016/j.bmcl.2007.10.033. [DOI] [PubMed] [Google Scholar]
- 9.Giroux A, Boulet L, Brideau C, Chau A, Claveau D, Cote B, Ethier D, Frenette R, Gagnon M, Guay J, Gurial S, Mancini JA, Martins E, Masse F, Methot N, Riendeau D, Rubin J, Xu D, Yu H, Ducharme Y, Friesen RW. Bioorg. Med. Chem. Lett. 2009;19:5837. doi: 10.1016/j.bmcl.2009.08.085. [DOI] [PubMed] [Google Scholar]
- 10.Moon JT, Jeon JY, Park HA, Noh Y-S, Lee K-T, Kim J, Choo DJ, Lee JY. Bioorg. Med. Chem. 2010;20:734. doi: 10.1016/j.bmcl.2009.11.067. [DOI] [PubMed] [Google Scholar]
- 11.Hamza A, Zhao X, Tong M, Tai HH, Zhan CG. Bioorg. Med. Chem. 2011;19:6077. doi: 10.1016/j.bmc.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kabalka GW, Mereddy AR. Tetrahedron Lett. 2006;47:5171. [Google Scholar]
- 13.Masquelin T, Obrecht DA. Tetrahedron. 2001;57:153. [Google Scholar]
- 14.Isakson PC, Zhang YY, Veenhuizen AW, Seibert K, Perkins WE, Koboldt CM, Gregory SA, Cogburn JN, Burton EG, Anderson GD, Yu SS, Rogier DJ, Rogers RS, Miyashiro JM, Malecha JW, Lee JF, Graneto MJ, Docter S, Collins PW, Carter JS, Bertenshaw SR, Talley JJ, Penning TDJ. Med. Chem. 1997;40:1347. doi: 10.1021/jm960803q. [DOI] [PubMed] [Google Scholar]
- 15.Chang HH, Song Z, Wisner L, Tripp T, Gokhale V, Meuillet EJ. Invest. New Drugs. doi: 10.1007/s10637-011-9748-8. doi: 10.1007/s10637-011-9748-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.General procedure for the preparation of 2e: 2-bromo-1-(4-methoxyphenyl)ethanone 7 (0.437 mmol, 100 mg) and N-(4-phenoxyphenyl)thiourea 8 (0.437 mmol, 107 mg) were suspended in ethanol (2 mL) and subjected to microwave irradiation at 110 °C for 10 min. The resulting solution was diluted with CH2Cl2 (10 mL) and washed with a saturated aqueous solution of Na2CO3 (20 mL). The organic layer was dried (MgSO4) and evaporated under reduced pressure. The resulting crude residue was purified by silica gel column chromatography (EtOAc-hexanes, gradient) using a ISCO™ system to give aminothiazole 2e as a light orange solid (0.246 mmol, 92 mg, 56%). mp: 125.5-127 °C; 1H NMR (300 MHz, DMSO-d6) δ 10.25 (br s, 1H), 7.85 (d, J = 8.7 Hz, 2H), 7.77 (d, J = 8.9 Hz, 2H), 7.37 (t, J = 7.8 Hz, 2H), 7.14 (s, 1H), 7.13 – 7.02 (m, 3H), 7.02 – 6.94 (m, 4H), 3.79 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 163.9, 159.7, 158.6, 150.9, 150.8, 138.4, 130.8, 128.3, 127.9, 123.5, 120.9, 119.2, 118.4, 114.8, 101.4, 56.0. LC-MS [MH]+ 375.1.
- 17.PGE2 production assay: Cells were seeded in 6-well plates and incubated overnight in DMEM/ 10% FBS. They were serum starved for the next 18 h. Cells were then treated with 10 ng/ml IL-1β and increasing concentration of compounds (dissolved in DMSO) in 1 mL serum-free medium. After 72 h of incubation, the supernatants were collected for PGE2 level detection using the PGE2 EIA kit (R&D Systems).
- 18.COX-2 cell-free assay: COX-2 activity was measured by a COX Fluorescent inhibitor screen assay kit following the manufacturer's instructions (Cayman Chemical, http://www.caymanchem.com).