

Abstract
The actin cytoskeleton is constantly assembling and disassembling. Cells harness the energy of these turnover dynamics to drive cell motility and organize cytoplasm. Although much is known about how cells control actin polymerization, we do not understand how actin filaments depolymerize inside cells. I briefly describe how the combination of imaging actin filament dynamics in cells and using in vitro biochemistry progressively altered our views of actin depolymerization. I describe why I do not think that the prevailing model of actin filament turnover—cofilin-mediated actin filament severing—can account for actin filament disassembly detected in cells. Finally, I speculate that cells might be able to tune the mechanism of actin depolymerization to meet physiological demands and selectively control the stabilities of different actin arrays.
INTRODUCTION
Actin filaments in cells are highly dynamic, rapidly assembling in some regions of the cell while disassembling in others. Fast actin depolymerization allows cells to rapidly reconfigure their cytoskeleton in response to both internal and external cues. At the biochemical level, fast actin depolymerization is necessary to replenish the pool of polymerizable actin monomer that is used to rapidly assemble actin filaments and perform work (Pollard and Borisy, 2003). Therefore actin depolymerization is critical for actin-dependent processes, but the mechanism of actin depolymerization operating in cells is not known.
TREADMILLING, COFILIN, AND SEVERING
Actin filaments in pure solution turn over slowly at steady state through an ATP-dependent process know as treadmilling (Wegner, 1982). To treadmill, actin uses a small portion of the energy from ATP hydrolysis on actin to destabilize the filament to a slight extent, resulting in the slow release of ADP-actin subunits from the pointed ends of filaments. Classic photobleaching experiments performed at the leading edge of migrating cells were consistent with a model in which actin filaments assembled at the tip of the lamellipodium and disassembled at the base—hence treadmilling (Wang, 1985). Protrusion of the leading edge could thus be neatly explained by the intrinsic properties of F-actin. The model is very appealing, and treadmilling is a popular model for describing actin filament turnover in cells. There are, however, two caveats.
The first is that actin filaments in cells are too densely packed to directly visualize the mode of disassembly at the single-filament level by light microscopy. The extent to which this problem limited interpretation of mechanism became very apparent with the application of fluorescence speckle microscopy to actin arrays (Watanabe and Mitchison, 2002; Ponti et al., 2003). Speckling revealed that actin assembly and disassembly reactions are not restricted to the tip and base of the lamellipodium. Instead, both reactions are distributed throughout the structure from the tip to the base. Some of the filaments are polymerizing, whereas others right next to them are disassembling. Therefore we can no longer unequivocally state the mechanism of depolymerization at the single-filament level. Any and all modes of actin filament disassembly depicted in Figure 1 could be operating in cells.
FIGURE 1:
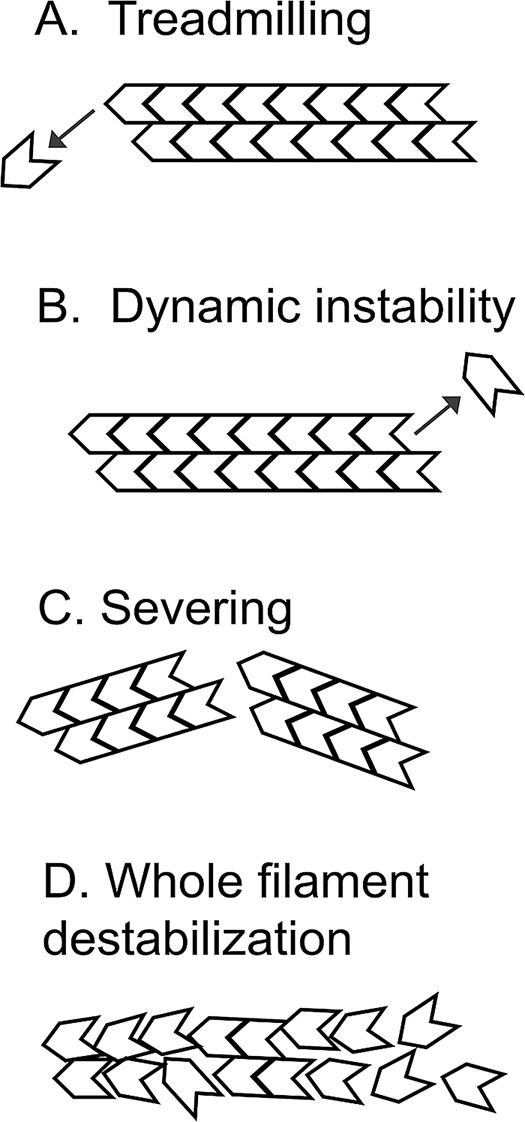
Alternative modes of actin disassembly. (A) Treadmilling filaments lose actin subunits from the pointed ends of filaments. Pure actin treadmills at steady state, which is a popular model for describing actin turnover in cells. (B) Dynamic instability would occur if ATP hydrolysis were to convert the barbed end from one that grows to one that shrinks. Microtubules and prokaryotic ParM filaments undergo dynamic instability, but dynamic instability has not been seen with actin. (C) Severing cuts a filament to produce two daughter filaments without loss of polymer mass. Cofilin severs actin filaments in vitro. (D) Whole-filament destabilization proposes a highly cooperative process in which long stretches of polymer abruptly convert to monomer. Whole-filament destabilization might describe actin disassembly in the presence of cofilin, coronin, and Aip1.
The second caveat is that actin filaments depolymerize much faster in cells than in pure solution, implying the existence of cellular factors that accelerate actin disassembly (Zigmond, 1993). These factors could in principle accelerate pointed-end dissociation to enhance treadmilling, but treadmilling need not be an essential feature of actin filaments based on evidence from the world of bacterial actin filaments. Prokaryotic ParM, for example, is structurally related to actin (Carballido-Lopez, 2006), but ParM uses the energy from ATP hydrolysis to drive dynamic instability, not treadmilling (Garner et al., 2004). In principle, then, if cellular factors were to help actin capture more of the energy from ATP hydrolysis, they could alter the mechanism to any of those depicted in Figure 1.
The discrepancy between in vitro and in vivo actin disassembly rates motivated the search for actin disassembly factors, and members of the cofilin family of small actin-binding proteins were the leading candidates (Bamburg, 1999). Lappalainen and Drubin (1997) combined yeast genetics with imaging and a small-molecule inhibitor of actin assembly to prove that cofilin is essential for actin depolymerization in cells. Cofilin severs actin filaments in pure solution but does not appear to accelerate the rate at which actin subunits dissociate from filament ends (Andrianantoandro and Pollard, 2006), and cofilin-mediated severing of actively growing filaments has been detected in vitro (Michelot et al., 2007). Cofilin-mediated actin filament severing, presumably followed by slow depolymerization from the pointed end, is now the prevailing model for actin disassembly in cells. Bundles of actin filaments can be seen fragmenting in neuronal growth cones of Aplysia (Medeiros et al., 2006). We do not know the fate of such severed filaments, however, or how they might depolymerize down to monomer. Nor is it even clear whether actin-filament severing is the dominant mode of disassembly operating in cells. Fluorescence speckle microscopy of actin in lamellipodia, for example, suggests that fast actin depolymerization extends all the way to the tip of the leading edge (Watanabe and Mitchison, 2002), but the short filaments we expect to see from frequent severing events are not found in electron tomograms of lamellipodia (Urban et al., 2010). In fact, a number of nagging observations of actin filament turnover in cells are inconsistent with the hypothesis that cofilin-mediated actin-filament severing is the primary mode of disassembly driving actin dynamics in cells.
CAN SEVERING AND TREADMILLING ACCOUNT FOR ACTIN DISASSEMBLY IN CELLS?
Several years ago Hao Yuan Kueh, Tim Mitchison, and I compared the effects of two drugs, latrunculin B and cytochalasin D, on the dynamics of Listeria actin comet tails (Kueh et al., 2008). Cytochalasin D is a drug that caps the barbed ends of actin filaments, preventing it from growing or shrinking. Latrunculin, on the other hand, binds to actin monomer and prevents it from incorporating into polymer. Listeria is a bacterial pathogen that harnesses the energy of actin polymerization on its surface to propel it through the host's cytoplasm (Tilney and Portnoy, 1989). As the bacterium is propelled forward, it leaves an actin comet tail behind it that rapidly depolymerizes with exponential kinetics (Theriot et al., 1992). Cofilin is necessary for comet tail disassembly (Rosenblatt et al., 1997). Both latrunculin and cytochalasin D stopped Listeria propulsion, which was expected. However, cytochalasin D also blocked comet tail disassembly, which was unexpected. The cytochalasin D result is inconsistent with treadmilling—why should capping the barbed end block disassembly at the pointed end? The result is also inconsistent with severing—cytochalasin D should, if anything, enhance severing by blocking filament reannealing. This is not an isolated case unique to Listeria, because cytochalasin D also blocked actin depolymerization in protruding lamellipodia (Kueh et al., 2008). Either we have more to learn about cytochalasin D or, just maybe, actin filaments in cells depolymerize from their barbed ends.
One might be tempted to brush aside the cytochalasin D result as some effect of the drug other than capping of filament barbed ends (Cooper, 1987). Nevertheless, cofilin-mediated actin filament severing remains an unsatisfactory model for actin filament turnover in cells because it cannot explain the morphogenesis of the Listeria actin comet tail. Comet tail assembly is restricted to the bacterial surface, whereas the rest of the comet tail only depolymerizes (Theriot et al., 1992; Kueh et al., 2010). Although severing will produce more pointed ends that shrink, severing will also create more barbed ends that grow. Actin-filament barbed ends produced by cofilin-mediated severing serve as sites for actin assembly in vitro (Ichetovkin et al., 2002). Furthermore, acute activation of cofilin at the leading edge of migrating cells results in a burst of actin assembly, not disassembly, presumably due to the abrupt formation of new barbed ends created by a round of severing (Ghosh et al., 2004). Disassembly dominated by severing cannot be operating in the Listeria actin comet tail because no new actin assembly can be detected along the length of the dissolving comet tail (Kueh et al., 2010). Finally, loss of polymer mass from comet tails via severing alone would require two cuts before the resulting filament could diffuse away from the comet tail, which is inconsistent with their exponential decay profile (Kueh et al., 2010).
AUXILIARY FACTORS COULD ALTER THE MECHANISM OF DEPOLYMERIZATION
Thus cofilin-mediated filament severing alone cannot account for actin disassembly dynamics detected in cells. The implication is that cells express additional factors that augment cofilin function and alter the mechanism of depolymerization. In fact, whereas cofilin is necessary for actin depolymerization, it does not appear to be sufficient under conditions normally found inside cells, and additional factors appear to be required to help cofilin overcome intracellular obstacles that would otherwise inhibit actin disassembly. Cells contain high concentrations of polymerizable actin monomer, which is used to drive fast actin assembly, and depolymerization must occur against this thermodynamic barrier. There is also a stoichiometric problem caused by the high concentration of actin polymer in cells that is in large excess of the depolymerizers. A variety of auxiliary factors have been identified that augment cofilin function, including Aip1, coronin, and CAP (Ono, 2007). The importance of these factors in actin turnover dynamics has been demonstrated in multiple organisms (Ono, 2007), and some of them help cofilin to overcome inhibition of disassembly by excess monomer and polymer (Brieher et al., 2006; Normoyle and Brieher, 2012). The key question is whether these auxiliary factors simply accelerate cofilin-mediated severing or alter the mechanism.
In many cases the auxiliary factors enhance cofilin-mediated severing (Ono et al., 2004; Ono, 2007; Moseley et al., 2006; Gandhi et al., 2009; Normoyle and Brieher, 2012; Chaudhry et al., 2013). In contrast, the combination of cofilin, coronin, and Aip1 altered the mechanism of depolymerization to one in which long stretches of actin polymer appeared to disassemble catastrophically in a single step (Kueh et al., 2008). This mechanism has been referred to alternatively as bursting or whole-filament destabilization to reflect either the abruptness of the disassembly event or the possibility that the filament is disassembling under these conditions through every imaginable pathway (severing, pointed-end loss, barbed-end loss, unraveling of the protofilaments). The molecular mechanism underlying whole-filament destabilization is not known, but it is presumably a highly cooperative process. One possibility is that the combination of cofilin and coronin drives highly cooperative cofilin binding to F-actin to create long stretches of polymer coated with cofilin and possibly coronin. These coated segments of polymer are envisioned to be highly unstable in the presence of Aip1. Although this model is highly speculative, cofilin binds cooperatively to F-actin. The cooperativity is weak (Andrianantoandro and Pollard, 2006), but it might be strengthened by coronin, which enhances cofilin binding to Listeria actin comet tails as well as F-actin in pure solution (Brieher et al., 2006)—this, despite the fact that coronin occludes the cofilin-binding site on F-actin (Galkin et al., 2008) and the two proteins compete for binding to the filament (Cai et al., 2007). Such stretches of cofilin-coronin–laden polymer are then envisioned to be superior substrates for Aip1-mediated disassembly. Our understanding of the mechanism of disassembly under these conditions is still in its infancy and arguably too murky to justify the names that I helped assign to it. Nevertheless, despite what we call it, severing does not suffice to describe filament disassembly in the presence of these three factors, and the triple mixture of cofilin, coronin, and Aip1 is the only depolymerization cocktail that we know of that is blocked by cytochalasin D, which is the situation in cells (Kueh et al., 2008). Catastrophic disassembly that removes entire filaments in a single step could also account for exponential decay of Listeria actin comet tails (Kueh et al., 2010). Determining whether this mode of actin disassembly is operating in cells and understanding why cytochalasin D blocks it will require a detailed understanding of the underlying mechanism driving bursting/whole-filament destabilization.
POSSIBLE IMPLICATIONS FOR ACTIN-DEPENDENT CELL BIOLOGY
The number of auxiliary factors, in conjunction with their potential ability to alter the mechanism of actin depolymerization, could have important consequences for actin-dependent cell biology. Cells could conceivably control the activities of the disassembly factors to tune the mechanism of depolymerization to meet physiological demands. Whole-filament destabilization, for example, could be advantageous when cells needs to remove an actin array quickly and completely, such as during phagosome maturation (Bengtsson et al., 1993). A severing-dominated regime, on the other hand, could allow cells to deform an actin array without losing much polymer mass. Semaphorin 3A, for example, induces neuronal growth cone collapse (Fan et al., 1993). The process requires both cofilin and myosin II (Aizawa et al., 2001; Gallo, 2006). Although actin polymer mass is lost upon exposure to Semaphorin 3A, some F-actin remains, and myosin II pulls on it to retract the neurite (Gallo, 2006). Severing might weaken the array, allowing it to flow more freely in response to myosin II–dependent contractile forces.
Different auxiliary factors could also be used to selectively control the stability of different actin arrays in cells. The stability of actin within different networks can vary from seconds to hours (Zigmond, 1993; Kueh and Mitchison, 2009). How cells manage to simultaneously maintain actin arrays of such varying stabilities in a shared cytosol is not known. Selectively stabilizing certain actin arrays with tropomyosin appears to be one mechanism (Kuhn and Bamburg, 2008). Selectively destabilizing different actin arrays with different combinations of cofilin and supplementary factors could be a complementary mechanism. Selective destabilization of a particular actin array might be operating in Drosophila nurse cells, in which CAP specifically controls the amount of actin that assembles at apical cell–cell junctions, whereas cofilin controls the amount of actin throughout the entire cell (Baum and Perrimon, 2001). The extent to which other disassembly factors help control the amount and stability of F-actin in other actin arrays is an interesting question for the future.
FUTURE DIRECTIONS
Identifying which mechanisms of actin depolymerization are operating in cells is the obvious goal, but directly visualizing depolymerization of single filaments in cells is still beyond the resolution of light microscopy. As an alternative, we can analyze actin depolymerization in vitro under defined conditions in greater detail to better distinguish between different mechanisms of disassembly. This information could then be used to help better detect the mechanism of actin depolymerization operating within an actin array in vivo. In the process, we might uncover new roles for actin depolymerization in controlling the morphogenesis of the actin cytoskeleton, with potential consequences for our understanding of actin-dependent processes such as cell motility.
Acknowledgments
I thank colleagues and mentors for sharing and discussing their ideas with me on actin depolymerization. I thank Ambika Nadkarni and Vivian Tang for commenting on the manuscript.
Footnotes
REFERENCES
- Aizawa H, et al. Phosphorylation of cofilin by LIM-kinase is necessary for semaphorin 3A-induced growth cone collapse. Nat Neurosci. 2001;4:367–373. doi: 10.1038/86011. [DOI] [PubMed] [Google Scholar]
- Andrianantoandro E, Pollard TD. Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol Cell. 2006;24:13–23. doi: 10.1016/j.molcel.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- Baum B, Perrimon N. Spatial control of the actin cytoskeleton in Drosophila epithelial cells. Nat Cell Biol. 2001;3:883–890. doi: 10.1038/ncb1001-883. [DOI] [PubMed] [Google Scholar]
- Bengtsson T, Jaconi ME, Gustafson M, Magnusson KE, Theler JM, Lew DP, Stendahl O. Actin dynamics in human neutrophils during adhesion and phagocytosis is controlled by changes in intracellular free calcium. Eur J Cell Biol. 1993;62:49–58. [PubMed] [Google Scholar]
- Brieher WM, Kueh HY, Ballif BA, Mitchison TJ. Rapid actin monomer-insensitive depolymerization of Listeria actin comet tails by cofilin, coronin, and Aip1. J Cell Biol. 2006;175:315–324. doi: 10.1083/jcb.200603149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Makhov AM, Bear JE. F-actin binding is essential for coronin 1B function in vivo. J Cell Sci. 2007;120:1779–1790. doi: 10.1242/jcs.007641. [DOI] [PubMed] [Google Scholar]
- Carballido-Lopez R. The bacterial actin-like cytoskeleton. Microbiol Mol Biol Rev. 2006;70:888–909. doi: 10.1128/MMBR.00014-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry F, Breitsprecher D, Little K, Sharov G, Sokolova O, Goode BL. Srv2/cyclase-associated protein forms hexameric shurikens that directly catalyze actin filament severing by cofilin. Mol Biol Cell. 2013;24:31–41. doi: 10.1091/mbc.E12-08-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA. Effects of cytochalasin and phalloidin on actin. J Cell Biol. 1987;105:1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Mansfield SG, Redmond T, Gordon-Weeks PR, Raper JA. The organization of F-actin and microtubules in growth cones exposed to a brain-derived collapsing factor. J Cell Biol. 1993;121:867–878. doi: 10.1083/jcb.121.4.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkin VE, Orlova A, Brieher W, Kueh HY, Mitchison TJ, Egelman EH. Coronin-1A stabilizes F-actin by bridging adjacent actin protomers and stapling opposite strands of the actin filament. J Mol Biol. 2008;376:607–613. doi: 10.1016/j.jmb.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo G. RhoA-kinase coordinates F-actin organization and myosin II activity during semaphorin-3A-induced axon retraction. J Cell Sci. 2006;119:3413–3423. doi: 10.1242/jcs.03084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi M, Achard V, Blanchoin L, Goode BL. Coronin switches roles in actin disassembly depending on the nucleotide state of actin. Mol Cell. 2009;34:364–374. doi: 10.1016/j.molcel.2009.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner EC, Campbell CS, Mullins RD. Dynamic instability in a DNA-segregating prokaryotic actin homolog. Science. 2004;306:1021–1025. doi: 10.1126/science.1101313. [DOI] [PubMed] [Google Scholar]
- Ghosh M, Song X, Mouneimne G, Sidani M, Lawrence DS, Condeelis JS. Cofilin promotes actin polymerization and defines the direction of cell motility. Science. 2004;304:743–746. doi: 10.1126/science.1094561. [DOI] [PubMed] [Google Scholar]
- Ichetovkin I, Grant W, Condeelis J. Cofilin produces newly polymerized actin filaments that are preferred for dendritic nucleation by the Arp2/3 complex. Curr Biol. 2002;12:79–84. doi: 10.1016/s0960-9822(01)00629-7. [DOI] [PubMed] [Google Scholar]
- Kueh HY, Brieher WM, Mitchison TJ. Quantitative analysis of actin turnover in Listeria comet tails: evidence for catastrophic filament turnover. Biophys J. 2010;99:2153–2162. doi: 10.1016/j.bpj.2010.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kueh HY, Charras GT, Mitchison TJ, Brieher WM. Actin disassembly by cofilin, coronin, and Aip1 occurs in bursts and is inhibited by barbed-end cappers. J Cell Biol. 2008;182:341–353. doi: 10.1083/jcb.200801027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kueh HY, Mitchison TJ. Structural plasticity in actin and tubulin polymer dynamics. Science. 2009;325:960–963. doi: 10.1126/science.1168823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn TB, Bamburg JR. Tropomyosin and ADF/cofilin as collaborators and competitors. Adv Exp Med Biol. 2008;644:232–249. doi: 10.1007/978-0-387-85766-4_18. [DOI] [PubMed] [Google Scholar]
- Lappalainen P, Drubin DG. Cofilin promotes rapid actin filament turnover in vivo. Nature. 1997;388:78–82. doi: 10.1038/40418. [DOI] [PubMed] [Google Scholar]
- Medeiros NA, Burnette DT, Forscher P. Myosin II functions in actin-bundle turnover in neuronal growth cones. Nat Cell Biol. 2006;8:215–226. doi: 10.1038/ncb1367. [DOI] [PubMed] [Google Scholar]
- Michelot A, Berro J, Guerin C, Boujemaa-Paterski R, Staiger CJ, Martiel JL, Blanchoin L. Actin-filament stochastic dynamics mediated by ADF/cofilin. Curr Biol. 2007;17:825–833. doi: 10.1016/j.cub.2007.04.037. [DOI] [PubMed] [Google Scholar]
- Moseley JB, Okada K, Balcer HI, Kovar DR, Pollard TD, Goode BL. Twinfilin is an actin-filament-severing protein and promotes rapid turnover of actin structures in vivo. J Cell Sci. 2006;119:1547–1557. doi: 10.1242/jcs.02860. [DOI] [PubMed] [Google Scholar]
- Normoyle KP, Brieher WM. Cyclase-associated protein (CAP) acts directly on F-actin to accelerate cofilin-mediated actin severing across the range of physiological pH. J Biol Chem. 2012;287:35722–35732. doi: 10.1074/jbc.M112.396051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono S. Mechanism of depolymerization and severing of actin filaments and its significance in cytoskeletal dynamics. Int Rev Cytol. 2007;258:1–82. doi: 10.1016/S0074-7696(07)58001-0. [DOI] [PubMed] [Google Scholar]
- Ono S, Mohri K, Ono K. Microscopic evidence that actin-interacting protein 1 actively disassembles actin-depolymerizing factor/Cofilin-bound actin filaments. J Biol Chem. 2004;279:14207–14212. doi: 10.1074/jbc.M313418200. [DOI] [PubMed] [Google Scholar]
- Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- Ponti A, Vallotton P, Salmon WC, Waterman-Storer CM, Danuser G. Computational analysis of F-actin turnover in cortical actin meshworks using fluorescent speckle microscopy. Biophys J. 2003;84:3336–3352. doi: 10.1016/S0006-3495(03)70058-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt J, Agnew BJ, Abe H, Bamburg JR, Mitchison TJ. Xenopus actin depolymerizing factor/cofilin (XAC) is responsible for the turnover of actin filaments in Listeria monocytogenes tails. J Cell Biol. 1997;136:1323–1332. doi: 10.1083/jcb.136.6.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot JA, Mitchison TJ, Tilney LG, Portnoy DA. The rate of actin-based motility of intracellular Listeria monocytogenes equals the rate of actin polymerization. Nature. 1992;357:257–260. doi: 10.1038/357257a0. [DOI] [PubMed] [Google Scholar]
- Tilney LG, Portnoy DA. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J Cell Biol. 1989;109:1597–1608. doi: 10.1083/jcb.109.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban E, Jacob S, Nemethova M, Resch GP, Small JV. Electron tomography reveals unbranched networks of actin filaments in lamellipodia. Nat Cell Biol. 2010;12:429–435. doi: 10.1038/ncb2044. [DOI] [PubMed] [Google Scholar]
- Wang YL. Exchange of actin subunits at the leading edge of living fibroblasts: possible role of treadmilling. J Cell Biol. 1985;101:597–602. doi: 10.1083/jcb.101.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Mitchison TJ. Single-molecule speckle analysis of actin filament turnover in lamellipodia. Science. 2002;295:1083–1086. doi: 10.1126/science.1067470. [DOI] [PubMed] [Google Scholar]
- Wegner A. Treadmilling of actin at physiological salt concentrations. An analysis of the critical concentrations of actin filaments. J Mol Biol. 1982;161:607–615. doi: 10.1016/0022-2836(82)90411-9. [DOI] [PubMed] [Google Scholar]
- Zigmond SH. Recent quantitative studies of actin filament turnover during cell locomotion. Cell Motil Cytoskeleton. 1993;25:309–316. doi: 10.1002/cm.970250402. [DOI] [PubMed] [Google Scholar]