

Clathrin–AP-3 association is dispensable for AP-3 vesicle budding from endosomes, which suggests that AP-3–clathrin interactions differ from those by which AP-1 and AP-2 adaptors productively engage clathrin in vesicle biogenesis.
Abstract
A role for clathrin in AP-3–dependent vesicle biogenesis has been inferred from biochemical interactions and colocalization between this adaptor and clathrin. The functionality of these molecular associations, however, is controversial. We comprehensively explore the role of clathrin in AP-3–dependent vesicle budding, using rapid chemical-genetic perturbation of clathrin function with a clathrin light chain–FKBP chimera oligomerizable by the drug AP20187. We find that AP-3 interacts and colocalizes with endogenous and recombinant FKBP chimeric clathrin polypeptides in PC12-cell endosomes. AP-3 displays, however, a divergent behavior from AP-1, AP-2, and clathrin chains. AP-3 cofractionates with clathrin-coated vesicle fractions isolated from PC12 cells even after clathrin function is acutely inhibited by AP20187. We predicted that AP20187 would inhibit AP-3 vesicle formation from endosomes after a brefeldin A block. AP-3 vesicle formation continued, however, after brefeldin A wash-out despite impairment of clathrin function by AP20187. These findings indicate that AP-3–clathrin association is dispensable for endosomal AP-3 vesicle budding and suggest that endosomal AP-3–clathrin interactions differ from those by which AP-1 and AP-2 adaptors productively engage clathrin in vesicle biogenesis.
INTRODUCTION
Exocytic and endocytic compartments exchange components and maintain their composition by means of carriers, some of which are vesicles (Bonifacino and Glick, 2004; De Matteis and Luini, 2011). The generation of vesicles and the selective loading of proteins and lipids into them require diverse monomeric and heteromeric cytosolic coats. Among the latter, heterotetrameric adaptor complexes AP-1–5 specify distinct trafficking routes. AP-1–5 adaptors comprise four subunits of various sizes: two large subunits, β1-5 and α, γ, δ, ε, or ζ; one medium-size, μ1-5 subunit; and a small, σ1-5 subunit. All of these heterotetrameric adaptor complexes share the ability to recognize specific sorting signals in the cytosolic domains of membrane proteins. Some adaptors have in common the capacity to bind phosphoinositides, GTPases, and proteins that act as modules to connect a nascent vesicle to diverse machineries. These include, but are not limited to, membrane deformation, the cytoskeleton, or the recognition of specialized cargoes. In contrast, these adaptor complexes differ in their ability to bind clathrin and/or cosediment with clathrin-coated vesicles (Kirchhausen, 2000; Bonifacino and Traub, 2003; Bonifacino and Glick, 2004; Robinson, 2004; Hirst et al., 2011; McMahon and Boucrot, 2011).
Clathrin heavy-chain and the associated clathrin light-chain polypeptides assemble into triskelia, which are the structural building blocks of clathrin cages. These cages encase and help to concentrate adaptors with their bound membrane cargoes and other associated factors in the transition from a clathrin-coated membrane pit into a clathrin-coated vesicle bud. AP-1 and AP-2 bind to the clathrin heavy chain via the appendages of β adaptins (Kirchhausen, 2000; Bonifacino and Traub, 2003; Bonifacino and Glick, 2004; Robinson, 2004; Hirst et al., 2011; McMahon and Boucrot, 2011; Willox and Royle, 2012). Extensive biochemical, structural, and functional evidence indicates that clathrin association with AP-1 and AP-2 is necessary for vesicle formation by these adaptors (Kirchhausen, 2000; Bonifacino and Traub, 2003; Bonifacino and Glick, 2004; Robinson, 2004; Hirst et al., 2011; McMahon and Boucrot, 2011; Willox and Royle, 2012). In contrast, adaptor complexes AP-4 and AP-5 do not cofractionate with clathrin-coated vesicles and may function independently of clathrin, although more research on these complexes is needed to establish certainty (Hirst et al., 1999, 2013; Borner et al., 2006, 2012). A more thoroughly studied adaptor, the AP-3 complex, has a debatable relationship with clathrin. A functional equivalence for AP-3 with AP-1 or AP-2 has been inferred based on biochemical interactions and colocalization of AP-3 with clathrin. Nonetheless, the precise functional role of clathrin in AP-3 vesicle generation has been a matter of dispute since AP-3’s discovery (Dell'Angelica et al., 1997, 1998; Faundez et al., 1997, 1998; Shi et al., 1998; Blondeau et al., 2004; Peden et al., 2004; Theos et al., 2005; Borner et al., 2006; Salazar et al., 2009; Kural et al., 2012).
AP-3 generates vesicles that traffic membrane proteins from endosomes to lysosomes, lysosome-related organelles, or synaptic vesicles. Defects in this vesicle formation machinery translate into the type 2 Hermansky–Pudlak syndrome in humans, a disease that shares a similar constellation of phenotypes with AP-3–null mice and flies, the most prominent of these being pigment dilution (Wei, 2006; Wei and Li, 2012; Danglot and Galli, 2007; Newell-Litwa et al., 2007; Raposo and Marks, 2007; Raposo et al., 2007). In this study, we comprehensively explore the role of clathrin in AP-3–dependent vesicle budding, using rapid chemical-genetic perturbation of clathrin function. We recapitulate key biochemical and colocalization findings by others. However, despite a clathrin and AP-3 interaction, AP-3–dependent budding from endosomes is resistant to rapid clathrin inhibition in whole-cell assays. We propose that the interaction between clathrin and AP-3 at endosomes fulfills functions distinct from the canonical role of clathrin in AP-1– and AP-2–dependent vesicle formation.
RESULTS
We sought to test the function of clathrin interactions with the adaptor complex AP-3, using chronic and acute chemical-genetic perturbations of clathrin function. We hypothesized that if AP-3 vesicle budding from endosomes requires clathrin, as is the case for AP-1 and AP-2, then chronic and acute clathrin perturbation should similarly affect the content of these three adaptors in clathrin-coated vesicle fractions. We used short hairpin RNA (shRNA) down-regulation of clathrin heavy chain as a mean to chronically perturb clathrin. Acute interference of clathrin function was achieved with a clathrin light-chain chimera carrying in tandem an mCherry tag and an FKBP oligomerization domain (mCh-FKBP-CLC). Induction of mCh-FKBP-CLC quaternary structures impairs clathrin function in minutes after addition of the bivalent FKBP-binding agent AP20187 by trapping clathrin polypeptides and their associated molecules on membranes (Moskowitz et al., 2003; Deborde et al., 2008; Zlatic et al., 2011). This rapid onset of effects is in contrast with clathrin heavy-chain down-regulation by shRNA, which takes several days to attain clathrin function inhibition.
Clathrin and AP-3 interactions have been documented by means of in vitro pull-downs with the hinge-ear domain of AP-3 β3 subunits, as well as by AP-3 enrichment in clathrin-coated vesicles (Dell'Angelica et al., 1998; Blondeau et al., 2004; Borner et al., 2006; Salazar et al., 2009). Under knockdown conditions, clathrin vesicle formation is impaired, leading to reduced content of proteins copurifying with clathrin-coated vesicles including AP-3 subunits (Borner et al., 2006). We revisited these criteria in HEK293 cell and PC12 cells expressing mCh-FKBP-CLC to validate this recombinant tool. We used shRNA-mediated clathrin heavy-chain knockdown in HEK cells, followed by clathrin-coated vesicle isolation, to determine whether AP-3 adaptor subunits could be considered components of clathrin-coated vesicles in cells transiently expressing mCh-FKBP-CLC. Equal protein loads from control knockdown and clathrin heavy-chain down-regulated clathrin-coated vesicle fractions were analyzed by SDS–PAGE and Western blot (Figure 1, A and B). Clathrin heavy chain was enriched in clathrin-coated vesicle fractions (Figure 1A, compare lanes 1 and 3, asterisk), and the content of clathrin heavy chain and other fraction polypeptides decreased after clathrin heavy-chain down-regulation (Figure 1A, compare lanes 3 and 4). In addition, mCh-FKBP-CLC and adaptor complexes AP-1–3 were enriched in clathrin-coated vesicle fractions (Figure 1B, compare lanes 1 and 6), and their presence in clathrin-coated vesicle enriched fractions was similarly sensitive to shRNA clathrin heavy-chain down-regulation (Figure 1B, compare lanes 6 and 12). AP-3 content in clathrin-coated vesicle enriched fractions was insensitive to down-regulation of BLOC-1 complexes, a major AP-3 interactor (Supplemental Figure S1; Di Pietro et al., 2006; Salazar et al., 2009; Gokhale et al., 2012; Lee et al., 2012). These results are compatible with the hypothesis that a subset of clathrin-coated vesicles may contain the adaptor complex AP-3.
FIGURE 1:

Cofractionation and coimmunomagnetic isolation of clathrin and AP-3 in cells expressing mCherry-FKBP-CLC. (A) Coomassie stain of homogenate (H) and clathrin-coated vesicle (CCV) fractions from HEK293 cells transiently transfected with mCh-FKBP-CLC and infected with either control shRNA (lanes 1 and 3) or shRNA directed against the clathrin heavy-chain message (lanes 2 and 4). Asterisk represent clathrin heavy-chain polypeptide. (B) Western blot of clathrin-coated vesicle fractionation from A. (C) Western blot of clathrin light-chain (lane 3) and control (lane 2) immunomagnetic isolations from HEK293 cells transiently transfected with mCherry-FKBP-CLC. Input represents 5% of load. (D) Western blot of clathrin heavy-chain (lanes 5 and 6) and control (lanes 3 and 4) immunomagnetic isolations from HEK293 cells transiently transfected with mCh-FKBP-CLC in the absence or presence of the cross-linker DSP. Input represents 5% of load. (E) Western blot of AP-3 δ (lanes 5 and 6) and AP-3 δ with peptide competition (lanes 3 and 4) in HEK293 cells transiently transfected with mCh-FKBP-CLC in the absence or presence of the cross-linker DSP. Input represents 5% of load. (F) Western blot of AP-3 δ (lane 3) and AP-3 δ with peptide competition (lane 2) in PC12 cells stably transfected with mCh-FKBP-CLC in the presence of the cross-linker DSP. Input represents 1.25% of load. Black asterisk indicates CLC signal. Double white asterisks indicate antibody signal. CCV, clathrin-coated vesicle fraction; H, homogenate; IB, immunoblot; IP, immunomagnetic isolation;P1, pellet 1; pellet 2; S1, supernatant 1; P2, S2 supernatant 2.
We further tested the possible association of AP-3 with clathrin by coimmunomagnetic precipitations. We performed cross-linking with the homo-bifunctional cross-linker dithiobis-succinimidyl propionate (DSP) in intact cells (Lomant and Fairbanks, 1976; Zlatic et al., 2010). Our rationale was to chemically stabilize putative clathrin–AP-3 associations in intact cells and use stringent isolation conditions to prevent cell-free clathrin–AP-3 associations after cell solubilization. mCh-FKBP-CLC HEK293 cells were treated in the absence or presence of DSP, and detergent cell extracts were incubated with magnetic beads containing antibody directed against clathrin light chain (Figure 1C), clathrin heavy chain (Figure 1D), or AP-3 δ (Figure 1E). Isolated immunocomplexes were analyzed by Western blot. Clathrin light-chain immunocomplexes coisolated with both clathrin heavy chain and the AP-3 adaptor subunit β3 (Figure 1C, lane 3). Similarly, isolation of clathrin heavy-chain immunocomplexes precipitated both clathrin light chain and the AP-3 adaptor subunit β3, the latter only in the presence of DSP cross-linker (Figure 1D, compare lanes 5 and 6). In addition, the association between clathrin chains and AP-3 could be detected in reverse magnetic precipitations where AP-3 was isolated with AP-3 δ antibodies, which coprecipitated AP-3 β3 and σ3 subunits, as well as both clathrin chains (Figure 1E). As before, clathrin chains coprecipitated with AP-3 only in the presence of cross-linker (Figure 1E, compare lanes 5 and 6). These clathrin–AP-3 interactions were also detected in PC12 neuroendocrine cells stably expressing mCh-FKBP-CLC (Figure 1F). We included PC12 cells in our study because of their extensively characterized AP-3 vesicle biogenesis pathway (Faundez et al., 1997, 1998; Faundez and Kelly, 2000; Shi et al., 1998). The selectivity of clathrin and AP-3 coprecipitations in HEK293 cells and PC12 cells was examined with either control isotype-matched antibodies or peptide antigen competition (Figure 1, C and D, and E and F, respectively). These results suggest that a subset of clathrin-coated vesicles contains the adaptor complex AP-3 at steady state.
Our biochemical results suggested a low extent of overlap between clathrin and AP-3 polypeptides. Our goal was to quantitatively assess the extent to which clathrin and AP-3 associate in PC12 cells expressing mCh-FKBP-CLC at steady state, in particular in early endosome compartments. Thus we performed indirect immunofluorescence experiments with high-resolution deconvolution microscopy or superresolution structured illumination microscopy (Figures 2 and 3, Supplemental Figures S2–S4, and Supplemental Movie S1). Polypeptides were detected with antibodies against AP-3 δ, clathrin light chain, or mCherry. We previously demonstrated that mCh-FKBP-CLC occupies nearly all clathrin heavy chain–positive structures in HEK293 cells (Zlatic et al., 2011). Similarly, mCh-FKBP-CLC and clathrin heavy-chain signals extensively overlapped in PC12 cells, as revealed by deconvolution microscopy (74 ± 8.6% of all clathrin heavy-chain pixels present in a cell, n = 10; data not shown), validating recombinant clathrin light chain as a marker of clathrin coats in this neuroendocrine cell. In contrast, only 10–20% of all clathrin-positive structures present in a PC12 cell overlapped with AP-3 pixels (Figures 2A and 3 and Supplemental Figure S2). Structures for which the AP-3 and clathrin overlapped in images acquired using deconvolution microscopy were imaged using superresolution structured illumination microscopy (SIM). SIM provides a theoretical doubling of spatial resolution above wide-field deconvolution (∼120 nm, x–y; 300 nm, z). Analysis of SIM images showed that the overlap of clathrin and AP-3 remained (SIM, Figure 2, B and C; Schermelleh et al., 2008).
FIGURE 2:
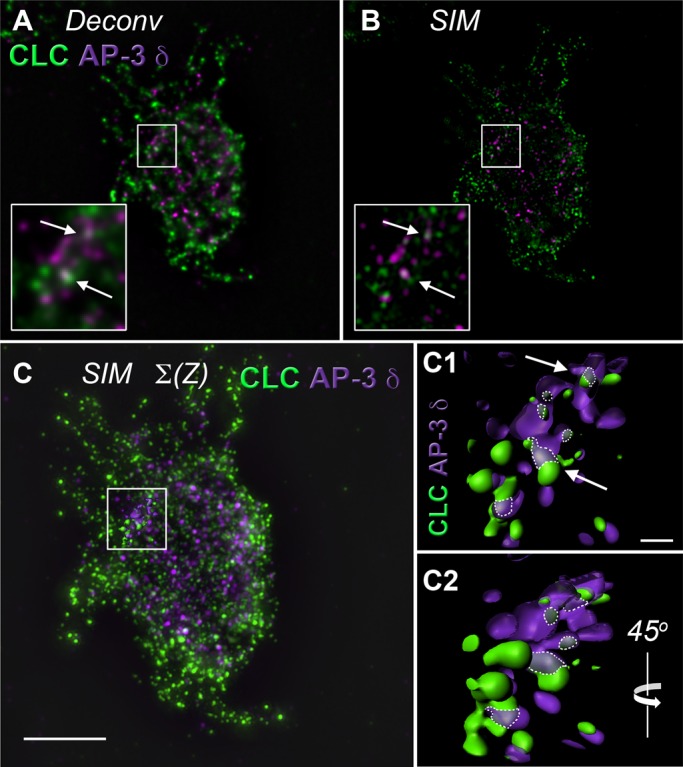
Analysis of clathrin light-chain and AP-3 δ colocalization in PC12 cells using high-resolution deconvolution microscopy and superresolution structured illumination microscopy. (A) High-resolution deconvolution microscopy done in the Huygens algorithm and (B) Superresolution SIM of a single PC12-cell z-plane. Insert, threefold enlargement of the boxed region. Arrows direct attention to discrete puncta showing colocalization between clathrin light chain (CLC) and AP-3 δ signals. (C) Isosurface rendering through 10 z-planes of SIM microscopy in B. Scale bar, 5 μm. Boxed region includes surface reconstruction enlarged in C1 and C2. (C1) Arrows draw attention to the same discrete puncta in A and B. Dotted lines outline area of colocalization. Scale bar, 0.5 μm. (C2) A 45° rotation of C1 around a vertical axis. See Supplemental Movie S1.
FIGURE 3:

Analysis of clathrin light-chain and AP-3 δ colocalization in PC12-cell early endosomes. (A, B) High-resolution deconvolution microscopy of PC12 cells incubated in the presence of vehicle or AP20187 and triple labeled with antibodies against AP-3 δ, the mCherry tag in mCh-FKBP-CLC, and EEA1. Scale bar, 5 μm. (C–E) Probability plots of the extent of pixel overlap among marker pairs (Y axis) in cells treated with vehicle (blue traces) and AP20187 (red traces). The p values were determined using the Kolmogorov–Smirnov test. Vehicle, n = 167 endosomes from 55 cells acquired from three biological replicates. AP20187, n = 203 endosomes from 68 cells acquired from three biological replicates.
In the following experiments, we specifically focused on early endosomal compartments since these endosomes bud a biochemically and pharmacologically tractable microvesicle population generated by an AP-3–dependent mechanism (Faundez et al., 1997, 1998; Faundez and Kelly, 2000; Clift-O'Grady et al., 1998; Shi et al., 1998). To examine the subcellular distribution of clathrin and AP-3 adaptor in endosomes, we determine the extent of AP-3 signal overlap with clathrin in endosomes identified with the early endosome marker EEA1 (Figure 3). We performed triple indirect immunofluorescence experiments with antibodies against AP-3 δ, the mCherry tag in mCh-FKBP-CLC, and EEA1 using high-resolution deconvolution microscopy. AP-3 and CLC colocalized in early endosomes (Figure 3). Moreover, the extent of AP-3–CLC signal overlap significantly increased from 20.7 ± 7.1% (average ± SEM) to 25.9 ± 6.6% after trapping clathrin chains on membranes by the addition of AP20187 (Figure 3, A–C). This increased overlap after AP20187 occurred at the expense of an accumulation of clathrin polypeptides on endosomes (Figure 3, A, B, and D) rather than an increase of AP-3 on these organelles (Figure 3, A, B, and E). We further tested the presence of AP-3 and clathrin overlapping domains in endosomes by transiently expressing an enhanced green fluorescent protein (EGFP)–tagged Rab5 Q79L mutant in PC12 cells stably expressing mCh-FKBP-CLC (Supplemental Figures S3 and S4). Molecules were detected in fixed cells using antibodies against EGFP, mCherry, and AP-3 δ. The EGFP-tagged Rab5 Q79L mutant allowed us to unequivocally identify early endosomes for quantitative analysis of AP-3 and clathrin subcellular overlap. Rab5 Q79L early endosomes were identified by their enlarged EGFP perimeter (Supplemental Figure S3). In these endosomes, we determined the percentage of their perimeter occupied by either clathrin or AP-3. mCh-FKBP-CLC covered 35 ± 5.2% of the EGFP-Rab5 Q79L–labeled perimeter (Supplemental Figure S4A, column 1). Similarly, AP-3 was present in 30.5 ± 5.9% of the EGFP-labeled endosome limiting membrane (Supplemental Figure S4A, column 3), the extent of which was not significantly modified by the addition of AP20187 (Supplemental Figure S4A, column 4). Of importance, a fraction of the entire AP-3 signal present in the endosome perimeter overlapped with mCh-FKBP-CLC (15.6 ± 3.8%; Supplemental Figure S4A, column 5). This result is in good agreement with previous quantitative electron immunomicroscopy data indicating that not all AP-3 budding profiles in early endosomes are decorated with clathrin in cell types other than PC12 cells (Peden et al., 2004; Theos et al., 2005). These results indicate that a pool of AP-3 overlaps with clathrin light chain in early endosomes, the site of origin for AP-3 microvesicles in PC12 cells.
PC12 cells generate clathrin-coated vesicles from the plasma membrane by a mechanism that is AP-2 and clathrin dependent yet brefeldin A insensitive (Figure 4, step 1). These clathrin-coated vesicles feed to early endosomes, where synaptic-like microvesicles (SLMVs) bud by a mechanism that instead requires AP-3 and ARF1. This AP-3 budding pathway is sensitive to brefeldin A (Figure 4, step 2; Faundez et al., 1997, 1998; Faundez and Kelly, 2000; Shi et al., 1998). Synaptic membrane proteins sorted into these SLMVs, such as SV2 or VAMP7, sequentially use these two vesicle-generation steps, whose vesicle products can be distinguished by their distinctive sedimentation properties (Figure 4). This allows selective isolation of clathrin-coated vesicles and SLMVs. We took advantage of features distinguishing these pathways to selectively test the requirement of clathrin in the budding of AP-2 and AP-3 vesicles in PC12 cells stably expressing mCh-FKBP-CLC. We first asked whether the content of adaptor subunits cosedimenting with clathrin-coated vesicles was sensitive to the acute inhibition of clathrin function in mCh-FKBP-CLC–expressing PC12 cells. Our hypothesis predicted that if AP-3 vesicle budding requires clathrin, then acute clathrin perturbation should similarly affect the content of adaptors in clathrin-coated vesicle fractions. Stably transfected mCh-FKBP-CLC PC12 cells were treated for 2 h with vehicle control or AP20187 to perturb clathrin function. We determined the global effectiveness of AP20187 treatment by the redistribution of clathrin and AP-3 toward the perinuclear area after drug incubation (Supplemental Figure S2), the accumulation of clathrin in EEA1-positive endosomes (Figure 3), and a reduction in endosome perimeter after drug addition (Supplemental Figure S4D, and data not shown; Deborde et al., 2008; Zlatic et al., 2011). Clathrin-coated vesicle–enriched fractions were prepared from vehicle and AP20187-treated cells and fraction contents analyzed by SDS–PAGE and Western blot (Figure 5A). AP20187 selectively decreased a major polypeptide of ∼180 kDa in clathrin-coated vesicle fractions resolved by SDS–PAGE. We confirmed this band as clathrin heavy chain by immunoblot (Figure 5, A and B). Concurrent with clathrin heavy chain depletion, we detected decreased content of endogenous clathrin light chain and recombinant mCh-FKBP-CLC in clathrin-coated vesicle–enriched fractions (Figure 5C, compare lanes 6 and 12). Of importance, the content of AP-1 γ adaptin and AP-2 α adaptin were similarly reduced in clathrin-coated vesicle fractions isolated from AP20187-treated cells (Figure 5, B and C, compare lanes 1 and 2, and 6 and 12, respectively, and D). Clathrin-coated vesicle cargoes synaptobrevin 2, synaptophysin, and VAMP7 (Blondeau et al., 2004; Takamori et al., 2006) were reduced after AP20187 treatment, a further indication that plasma membrane clathrin-coated vesicle formation was impaired by mCh-FKBP-CLC oligomerization (Figure 5C, compare lanes 6 and 12, and D). Strikingly, even though treatment with AP20187 impaired the formation of AP-1 and AP-2 clathrin-coated vesicles, we observed an increase in the levels of AP-3 in clathrin-coated vesicle enriched–fractions as determined by immunoblot with antibodies against β3, δ, and σ3 subunits (Figure 5, B and C, compare lanes 1 and 2, and 6 and 12, respectively, and D). These results demonstrate that clathrin-coated vesicle formation by AP-1– and AP-2–dependent mechanisms is robustly impaired by acute clathrin oligomerization in PC12 cells expressing mCh-FKBP-CLC. Our findings suggest, however, that a putative clathrin–AP-3 interaction differs from other clathrin–adaptor interactions after acute clathrin function perturbation. This differential behavior of AP-3 content in clathrin-coated vesicle fractions after AP20187 treatment raises the question of whether it also reflects a differential requirement of clathrin in endosome AP-3 vesicle generation.
FIGURE 4:
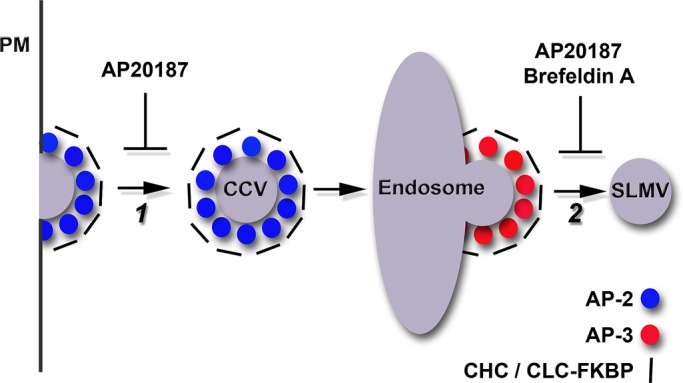
Model of acute pharmacological perturbation to clathrin and AP-3 in PC12 cells stably expressing mCh-FKBP-CLC. Step 1: AP-2 vesicles bud from the plasma membrane (PM) in a clathrin-dependent manner. Acute AP20187 treatment perturbs FKBP-CLC, and thus clathrin function, in these cells. AP-2-clathrin coated vesicles (CCV) feed into endosomal organelles. Step 2: AP-3 vesicles, presumably coated with clathrin, bud from an early endosomal compartment to form SLMVs. Acute treatment of these cells with brefeldin A disrupts AP-3 vesicle formation. We tested whether acute treatment of these cells with AP20187 similarly affects SLMV formation. We hypothesized that if clathrin is required for AP-3–derived SLMV formation, then AP20187 treatment after release of a brefeldin A block in SLMV formation should further prohibit SLMV formation.
FIGURE 5:
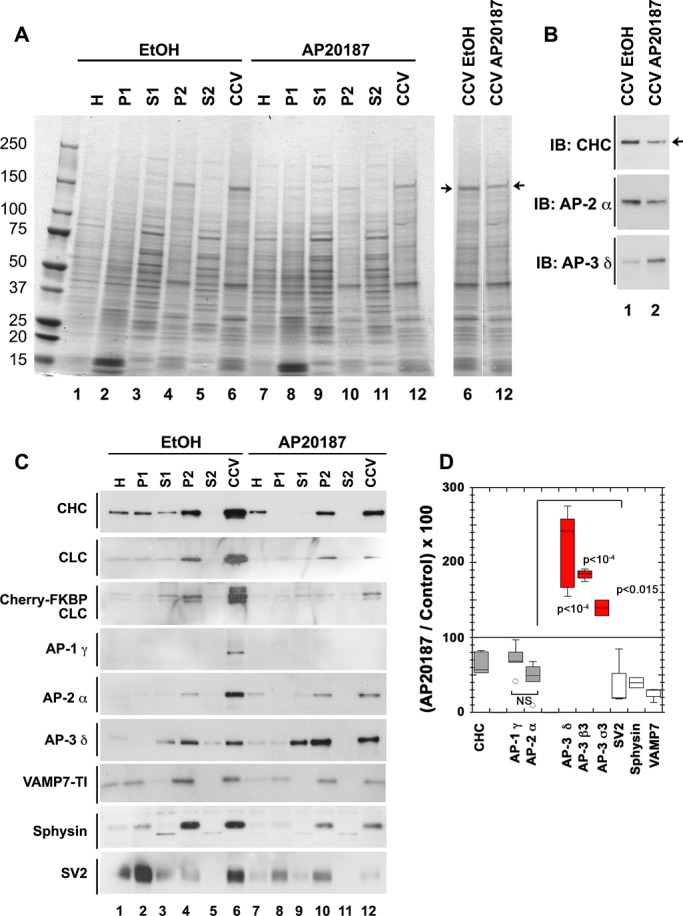
AP-3 levels increase in clathrin-coated vesicle fractions after acute clathrin function perturbation. (A) Coomassie stain of SDS–polyacrylamide gel loaded with fractions of CCVs isolated from PC12 cells stably expressing mCh-FKBP-CLC. Cells were treated for 2 h with vehicle (lanes 1–6) or the drug AP20187 to acutely perturb clathrin function (lanes 7–12). Arrows direct attention to a band that runs consistent with clathrin heavy-chain molecular weight. (B) Western blot of CCVs fraction from A. Arrow draws attention to a band in clathrin heavy-chain immunoblot that shows a decrease in the CCV fraction from cells with acute clathrin perturbation compared with vehicle control. Decrease in clathrin signal from CCV fraction of drug-treated cells indicates that acute clathrin perturbation affects clathrin vesicle formation. (C) Western blots of clathrin-coated vesicle isolation fractions from PC12 cells stably expressing mCherry-FKBP-CLC treated for 2 h with either vehicle control (lanes 1–6) or the drug AP20187 to perturb clathrin function (lanes 7–12). (D) Densitometry quantification of CCV fraction from three biological replicates each with at least two technical replicates of clathrin coated–vesicle isolations in PC12 cells stably expressing mCh-FKBP-CLC and treated for 2 h with either vehicle control or AP20187 to acutely perturb clathrin function. AP-3 subunits display significantly higher signals in CCV fractions from AP20187-treated cells compared with vehicle control–treated cells despite decreases in other adaptor subunits like AP-1 γ and AP-2 α. The extent of decrease in AP-1 γ and AP-2 α in CCV fraction treated with AP20187 compared with control is similar, as highlighted by the nonsignificance when these two are compared (not significant [NS]). CCV, clathrin-coated vesicle fraction; H, homogenate; IB, immunoblot; P1, pellet 1; P2, pellet 2; S1, supernatant 1; S2, supernatant 2; p values, analysis of variance multiple comparisons with Student–Newman–Keuls post hoc test.
To test the function of clathrin interactions with the adaptor complex AP-3, we took advantage of PC12 cells expressing mCh-FKBP-CLC and the pharmacological sensitivity of AP-3 vesicle budding to brefeldin A. The sequential organization of AP-2 and AP-3 budding steps has so far precluded a direct test of clathrin's role in endosome SLMV formation using long-term clathrin perturbations such as shRNA or expression of clathrin heavy-chain recombinant fragments (Figure 4). Therefore we used pharmacological epistasis between the brefeldin A and AP20187 blocks in mCh-FKBP-CLC–expressing PC12 cells to selectively assess a role of clathrin in AP-3 vesicle generation. Brefeldin A completely and reversibly inhibits SLMV formation by AP-3 while sparing the generation of AP-2 vesicles (Figure 4, steps 1 and 2; Faundez et al., 1997; Shi et al., 1998). If the AP-3 budding step requires clathrin, we reasoned that after a SLMV brefeldin A block, a brefeldin A washout in the presence of AP20187 should prevent de novo SLMV generation. In contrast, if clathrin is dispensable for AP-3 vesicle budding, then a brefeldin A washout in the presence of AP20187 should lead to a complete recovery of SLMV formation. We used VAMP7 and SV2 to track SLMV presence and their formation after pharmacological blockages. We selected these two transmembrane proteins because they are enriched in SLMV and mostly localize to endosome compartments at steady state in cells of the neural lineage (Newell-Litwa et al., 2009). In addition, VAMP7 is an AP-3 cargo that directly binds AP-3 (Martinez-Arca et al., 2003; Kent et al., 2012). mCh-FKBP-CLC–expressing PC12 cells were incubated in the presence of vehicle or brefeldin A for 2 h, and SLMVs were isolated by differential centrifugation followed by glycerol velocity sedimentation. Glycerol velocity gradients resolve SLMVs by size as a peak in the middle of the gradient (Figure 6, fractions 8–10; Clift-O'Grady et al., 1998). Brefeldin A decreased SLMVs as determined by VAMP7 and SV2 gradient content (Figure 6, A–C and A1–C1, open circles). This brefeldin A block was released after brefeldin A washout and incubation of cells in ethanol vehicle control for additional 2 h (Figure 6, B and B1, black circles). Of note, continuous incubation of cells in AP20187 instead of vehicle for 2 h had no effect on the reversibility of the brefeldin A washout (Figure 6, C and C1, black circles). This outcome was not due to either a reversal of the brefeldin A block after 4 h of incubation (Supplemental Figure S5) or a redistribution of mCh-FKBP-CLC chains away from endosomes after AP20187. In fact, AP20187 incubation neither modified clathrin or AP-3 nor changed AP-3-clathrin light-chain overlap in the limiting membrane of enlarged early endosomes (Supplemental Figure S4A, compare even and odd columns) over a wide range of clathrin content per endosome and endosome size (Supplemental Figure S4, B and C). This observation is consistent with the described AP20187-dependent trapping of clathrin polypeptides on membranes (Moskowitz et al., 2003; Deborde et al., 2008; Zlatic et al., 2011). These results demonstrate that the biochemical interaction between AP-3 and clathrin chains found in PC12 cells is dispensable for the budding of AP-3-derived SLMV and are in agreement with the previous contention that AP-3 complexes carrying mutations in the β3 putative clathrin binding domain are functional.
FIGURE 6:

Acute perturbation of clathrin function does not prohibit synaptic-like microvesicle formation after release of a brefeldin A block. PC12 cells stably expressing mCh-FKBP-CLC were treated for 2 h with brefeldin A (BFA) or vehicle control (EtOH) at 37°C. After this 2-h treatment, BFA-treated cells were either transferred to 4°C (BFA) or extensively washed at 4°C and then incubated with AP21087 (BFA-AP20187) or vehicle control (BFA-EtOH) for additional 2 h at 37°C. SLMVs sediment to the middle of 5–20% glycerol velocity gradients. Fractions from this gradient were loaded onto SDS–polyacrylamide gels for Western blot and densitometry quantifications. Immunoblot (IB) for SV2 and VAMP7 was used to track SLMVs. (A, A′) Densitometry quantification of fraction signal per total signal in BFA-treated cells (white circles) as compared with EtOH-only treated cells (black circles). (B, B′) Densitometry quantification of fraction signal per total signal in BFA-EtOH–treated cells (black circles). (C, C′) Densitometry quantification of fraction signal per total signal in BFA-treated cells (white circles) as compared with BFA-AP20187–treated cells (black circles). (D, E) Western blot of VAMP7 and SV2, respectively, from EtOH-, BFA-, BFA-EtOH–, and BFA-AP20187–treated cells.
The dispensability of clathrin function for AP-3–dependent SLMV formation suggests that interactions between AP-3 and clathrin detected in PC12 cells and other cells types may not follow a canonical association mechanism between the ear domain of the β subunit of the adaptor and clathrin terminal domain (Willox and Royle, 2012). To test this hypothesis, we focused on fibroblasts carrying the β3A mutation pearl (Ap3b1pe/pe). These cells carry a hypomorphic mutation in the AP-3 complex that affects the sorting of AP-3 cargoes. We used Ap3b1pe/pe cells or Ap3b1pe/pe rescued with either recombinant wild-type β3A or β3A mutations ablating putative clathrin-binding determinants (clathrin box) in the β3A hinge-ear domain to test whether an AP-3–clathrin interaction is sensitive to mutagenesis of the β3 clathrin box. These mutations included discrete changes in the β3A clathrin-binding sequence 817SLLDLD822 (β3A817AAA), a deletion of the 817SLLDLD822 clathrin box (β3AΔ807-831), and a truncation of the entire β3A ear domain (β3A807Stop). With the exception of β3A807Stop, all other β3A clathrin box mutants rescued a missorting phenotype in Ap3b1pe/pe cells (Peden et al., 2002). We performed DSP cross-linking in intact cells followed by immunomagnetic isolation of AP-3 complexes with δ antibodies. The δ adaptin antibodies precipitated σ3 subunits from Ap3b1pe/pe and notably low levels of clathrin in the presence of DSP but not BLOC-1 subunits detected with antibodies against pallidin (Figure 7, compare lanes 1 and 2). These δ-σ3 polypeptides correspond to the reported δ-σ3 adaptor hemicomplexes described in Ap3b1pe/pe cells (Peden et al., 2002). We used the BLOC-1 complex, an AP-3 interactor, and the levels of σ3 adaptin precipitated by δ antibodies as indications of reassembled AP-3 complexes with different recombinant β3A subunits (Di Pietro et al., 2006; Salazar et al., 2006, 2009; Gokhale et al., 2012). The content of clathrin precipitating with δ adaptin antibodies increased in all Ap3b1pe/pe cells where AP-3 complexes were reassembled by reexpression of recombinant β3A chains (Figure 7, compare lane 2 with lanes 4, 6, 8, and 10). Of importance, this increase in clathrin coprecipitation with reassembled AP-3 complexes was not sensitive to any of the mutations ablating putative clathrin-binding determinants in the β3A hinge-ear domain. These results indicate that the β3A hinge-ear domain is not necessary for clathrin interactions with cross-linked AP-3 complexes. Our results suggest that AP-3 diverges from the model by which AP-1 and AP-2 adaptors bind to clathrin cages for vesicle biogenesis via the C-terminus of β adaptins with the terminal domain of clathrin-heavy chain (Lemmon and Traub, 2012)
FIGURE 7:
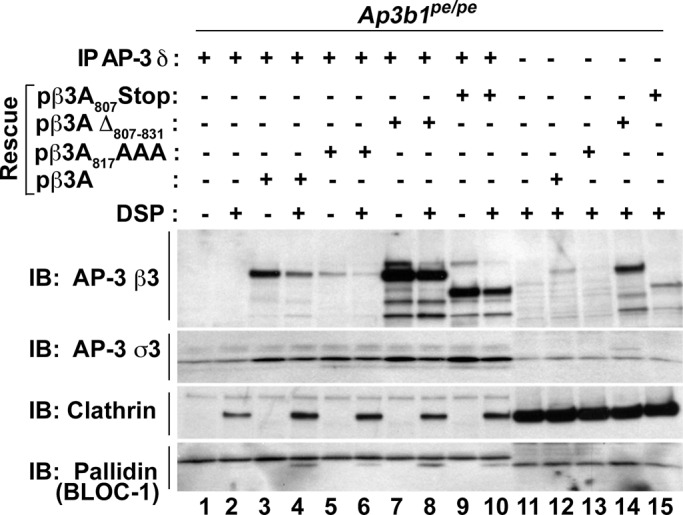
The clathrin-binding box of AP-3 β1 is dispensable for AP-3–clathrin coisolation. Pearl (Ap3b1pe/pe) fibroblasts or Pearl fibroblasts rescued with full-length Ap3b1 (pβ3A), Ap3b1 containing a triple-alanine mutation in the clathrin box (pβ3A871AAA), a deletion of the clathrin box in Ap3b1 (pβ3AΔ 807-831), or a truncation of Ap3b1 eliminating the entire ear domain containing the clathrin box (pβ3A807Stop) were treated with the cross-linker DSP. Lysates were subjected to coimmunomagnetic isolations directed against AP-3 δ (lanes 1–10). All rescue lysates from DSP-treated cells show increased coisolation of clathrin heavy chain (immunoblot [IB]; clathrin lanes 4, 6, 8, and 10) compared with unrescued lysate treated with DSP (IB, clathrin lane 2). Input represents 5% of immunomagnetic isolation load (lanes 11–15).
DISCUSSION
We explored the role of clathrin in AP-3–dependent vesicle budding by means of long-term clathrin shRNA down-regulation and rapid chemical-genetic perturbation of clathrin function. Down-regulation of clathrin heavy chain has been used to assess whether components enriched in clathrin-coated vesicle fractions truly reside in these organelles. This approach has been validated in HeLa cells using quantitative mass spectrometry alone or in combination with principal component analysis of multiple datasets (Borner et al., 2006, 2012). AP-1 and AP-2 faithfully cosegregate with clathrin-coated vesicles in both approaches. These assays provide different answers, however, as to whether AP-3 resides in clathrin-coated vesicles (Borner et al., 2006, 2012). Comparison of control and clathrin-knockdown cells by quantitative mass spectrometry suggests that AP-3 would reside in these organelles, a result that we recapitulated here. However, similar to principal component analysis of clathrin-coated vesicle fractions, we also observed a discrepancy in cosedimentation of AP-3 with clathrin-coated vesicle fractions. In our case, there is a drastic difference between chronic and acute clathrin function perturbation despite consistent behavior by AP-1 and AP-2 adaptors. This suggests that AP-3 cosediments with clathrin-coated vesicles but in membranous organelles distinct from these caged vesicles. Second, it implies that the sensitivity of AP-3 to long-term clathrin RNA interference results from indirect effects of chronic perturbations in the endocytic pathway. The “anomalous” AP-3 behavior prompted us to reevaluate whether AP-3 indeed requires clathrin to generate vesicles and whether clathrin–AP-3 interactions follow principles similar to those by adaptors AP-1 and AP-2.
Our findings in PC12 cells indicate that acute perturbation of clathrin with a mCh-FKBP-CLC chimera does not prevent the formation of AP-3 vesicles from endosomes after a brefeldin A block. We interpreted this observation as evidence of clathrin dispensability in AP-3 SLMV budding from endosomes. We examined alternative interpretations that could also explain our brefeldin A-AP20187 pharmacological epistasis results. We asked 1) whether AP-3 interacts and colocalizes with mCh-FKBP-CLC in endosomes, which gives rise to AP-3-derived vesicles; 2) whether there is biological activity for the clathrin light-chain chimera in PC12 cells, which we demonstrated by depletion of AP-1, AP-2, and synaptic vesicle cargoes from clathrin-coated vesicle fractions after acute AP20187 treatment; and 3) whether there is an alternative, “clathrin-like” molecule taking the place of clathrin heavy chain. AP-3 coprecipitates with the clathrin heavy chain encoded in human chromosome 22 (Liu et al., 2001). We estimate, however, that a role of this clathrin isoform is a remote option. Rodent cells do not express chromosome 22 clathrin, and our functional assessment of a clathrin-AP-3 interaction was conducted in rat PC12 cells (Wakeham et al., 2005). Thus a simple interpretation of our data is that the clathrin interaction with AP-3 is not required for vesicle budding from endosomes. This conclusion is in rapport with previous studies in which AP-3 function is restored in β3 mutant cells by expression of recombinant β3 chains carrying a defective clathrin-binding motif defined in vitro and presumed to be unique in AP-3 (Peden et al., 2002). The present conclusion is also consistent with our past studies in cell-free vesicle reconstitution experiments. Cell-free AP-3 budding is resistant to cytosolic clathrin depletion. Moreover, de novo vesicle formation from endosomes is reconstituted with just recombinant ARF1 and purified AP-3. A variable not controlled in these cell-free assays, however, is the contribution of donor membrane–bound clathrin to the reaction (Faundez et al., 1997, 1998; Shi et al., 1998). The whole-cell approach using mCh-FKBP-CLC oligomerization and pharmacological epistasis overcomes this limitation and provides conclusive evidence that clathrin is dispensable for AP-3 vesicle formation from PC12 endosomes.
If clathrin is unessential for AP-3 vesicle budding, then what roles may clathrin play? We postulate that an interaction between AP-3 and clathrin reflects a scaffolding role such as Hrs-clathrin patches, clathrin-sorting nexin interactions, or clathrin organization of the kinetochore microtubules (Raiborg et al., 2001; McGough and Cullen, 2012; Royle, 2012). Alternatively, clathrin binding to AP-3 could be a mechanism to deliver clathrin directionally by motors either in the absence or presence of vesicle membranes. Although we know of motors that bind to AP-1 and AP-3 adaptors directly (Nakagawa et al., 2000; Azevedo et al., 2009; Delevoye et al., 2009), there is no reported motor that binds to clathrin cages directly. In contrast, clathrin binds to dynein by means of an intervening protein, bicaudal D. This motor–clathrin association is necessary for normal neurotransmission in Drosophila (Li et al., 2010). These nonbudding models predict that the stoichiometry of AP-3 and clathrin interactions or the mode of binding between AP-3 and clathrin would differ from AP-1 and AP-2 interactions with clathrin cages. We indirectly tested the second prediction in fibroblasts carrying the β3A mutation pearl (Ap3b1pe/pe; Peden et al., 2002). These Ap3b1pe/pe cells possess low levels of δ-σ3 adaptor hemicomplexes that we found coprecipitated clathrin. This association is specific, as experiments in mocha Ap3d1mh/mh cells, which lack δ adaptin, are free of clathrin in these cross-linked precipitates, thus excluding spurious binding of clathrin to antibody–magnetic beads complexes. An equally striking finding is the observation that Ap3b1pe/pe rescued with a recombinant β3A carrying a truncation of the entire β3A hinge-ear domain (β3A807Stop) still coprecipitates clathrin. These observations are consistent with a model by which either δ-σ3 adaptor hemicomplexes or AP-3 complexes lacking the ear domain associate to clathrin, directly or indirectly. This would represent a departure of the model by which canonical AP-1 and AP-2 adaptors productively engage clathrin cages for vesicle biogenesis via the C-terminus of β adaptins with the terminal domain of clathrin heavy chain (Edeling et al., 2006; Knuehl et al., 2006; Schmid et al., 2006; Lemmon and Traub, 2012). Although clathrin binds preponderantly to the C-terminus of β2 adaptin, two-hybrid evidence suggests a putative interaction between clathrin and the C-terminal domain of α adaptin (Knuehl et al., 2006). However, the functional relevance of the appendage domain of α adaptin has been tested, and it is dispensable (Motley et al., 2006). In fact, the α adaptin down-regulation phenotype can be fully rescued by expression of α adaptin lacking its appendage domain (Motley et al., 2006). In contrast, the δ allele Ap3d1mh2j/mh2j, which lacks the appendage domain of δ adaptin, possesses phenotypes indicating that the δ appendage is not dispensable (Kantheti et al., 2003). Collectively, the evidence supports our contention that clathrin-AP-3 interactions differ from those between clathrin and the adaptor complexes AP-1 and AP-2.
A question that arises from our data concerns the extent to which the functionality of a clathrin–AP-3 association observed in PC12 SLMV budding from endosomes reflects other putative AP-3–dependent budding events. Nearly 40% of all AP-3 immunoreactivity in cells is present in endosomes (Peden et al., 2004; Theos et al., 2005). A similar amount of AP-3, however, associates to tubules/vesicles not identified as endosomes, and 4–9% associates with the trans-Golgi network (Peden et al., 2004; Theos et al., 2005). This leaves open the possibility that there may be additional modalities of AP-3-dependent budding distinct from vesicle generation from endosomes, such as the formation of large, dense secretory vesicles from the Golgi complex (Asensio et al., 2010), where AP-3 and clathrin may interact in a productive manner.
MATERIALS AND METHODS
Antibodies and constructs
Anti–clathrin heavy chain was purchased from EMD Millipore (Billerica, MA; CP45, clone: X22) and BD Transduction Laboratories (Pasadena, CA; 610499). Anti–clathrin light chain was purchased from EMD Millipore (AB9884) and Covance (Berkeley, CA; MMS-423P, clone: CON.1). Anti–adaptin α (610501), anti–adaptin γ (610385), and EEA1 (610456) were purchased from BD Transduction Laboratories. The AP3δ SA4, c-myc 9E10, and SV2 10H antibodies, developed by Andrew Peden, J. Michael Bishop, and Kathleen M. Buckley, respectively, were obtained from the Developmental Studies Hybridoma Bank (University of Iowa, Department of Iowa City, IA). Anti-synaptophysin was purchased from EMD Millipore (MAB5258, clone: SY38). Anti-AP3β1 (13384-1-AP) and anti-pallidin (10891-1-AP) were purchased from Protein Tech Group (Chicago, IL). Anti-mCherry/DsRed was purchased from Clontech (Mountain View, CA; 632496). Anti-GFP was purchased from AVES Labs (Tigard, OR; GFP-1020). Antibodies directed against anti–muted, a kind gift from E. C. Dell'Angelica, AP-3 β-NAP, AP-3 σ, and VAMP7 have been described (Larimore et al., 2011; Zlatic et al., 2011; Gokhale et al., 2012). Horseradish peroxidase–conjugated anti-mouse and anti-rabbit (A10668 and G21234) and Alexa Fluor 488– and 555–conjugated anti-chicken (A11039), anti-mouse (A21121 and A21127), and anti-rabbit (A11034 and A21429) were purchased from Invitrogen (Carlsbad, CA).
Peptide directed against the AP3 δ SA4 antibody antigen binding site (AQQVDIVTEEMPENALPSDEDDKDPNDPYRA) was obtained from the Emory University Microchemical Facility (Atlanta, GA) and Invitrogen (EvoQuest Team, Carlsbad, CA).
Clathrin heavy chain (RHS39799577067) and muted (RHS397998822549) pLKO.1 shRNA constructs were purchased from Open Biosystems (Huntsville, AL). The constructs mCherry-FKBP-CLC and EGFP-Rab5-Q79L have been described (Craige et al., 2008; Zlatic et al., 2011).
Cell culture, transfection, and infection
HEK293 and SH-SY5Y cells (American Type Culture Collection, Manassas, VA) cultured in DMEM (HyClone, Logan, UT) containing 10% fetal bovine serum (HyClone) and 100 μg/ml penicillin and streptomycin (HyClone) and PC12 cells (American Type Culture Collection) cultured in DMEM containing 10% donor equine serum (HyClone), 5% fetal bovine serum, and 100 μg/ml penicillin and streptomycin were incubated at 37°C and 10% CO2. Generation and culture of Ap3b1pe/pe cells or Ap3b1pe/pe rescued with either recombinant wild-type β3A or β3A mutations ablating putative clathrin-binding determinants was as described (Peden et al., 2002).
For transient transfections, HEK293 cells in wells of six-well plates were incubated with 1 μg of DNA in OptiMEM (Life Technologies, Grand Island, NY) containing 0.5% Lipofectamine 2000 (Invitrogen) for 4 h, followed by incubation with culture media. For stable transfections, PC12 cells were transfected as in transient transfections, followed by 0.2 mg/ml G418 (Invitrogen) drug selection. Selected cells were serially diluted, and clonal populations were maintained in PC12 culture media supplemented with 0.1 mg/ml G418.
For lentiviral infection, lentivirus containing shRNA constructs were prepared by the Emory University Viral Vector Core. Antibiotic-free culture medium containing 1 μl of high-titer lentivirus was incubated with HEK293 or SH-SY5Y cells for 24 h, followed by incubation for 6 d with culture medium supplemented with 0.1 mg/ml selection drug, hygromycin B (Invitrogen), in 10-cm culture plates.
Acute clathrin perturbation and clathrin-coated vesicle fractionation
HEK293 or PC12 cells expressing the mCh-FKBP-CLC construct were grown to confluence. Cells were incubated for 2 h at culture conditions in medium supplemented with 50 nM AP20187 (B/B Homodimerizer 635060; Clontech) or 0.05% ethanol vehicle control (Zlatic et al., 2011). HEK293 cells infected with lentivirus containing scramble or clathrin heavy-chain shRNA constructs and transiently expressing mCh-FKBP-CLC, SH-SY5Y cells infected with lentivirus containing scramble or muted shRNA constructs, and PC12 cells stably expressing mCh-FKBP-CLC with acute clathrin perturbation were prepared for clathrin-coated vesicle fractionation as previously described (Girard et al., 2004). Fractions were analyzed by either Western blot or Coomassie staining of 4–20% Criterion polyacrylamide gels (Bio-Rad, Hercules, CA).
DSP cross-linking and immunomagnetic precipitation
HEK293 and PC12 cells expressing mCh-FKBP-CLC were prepared for DSP cross-linking, followed by immunomagnetic precipitation as previously described (Salazar et al., 2009; Zlatic et al., 2010, 2011; Gokhale et al., 2012; Perez-Cornejo et al., 2012). The resulting isolated proteins were analyzed by Western blot of 4–20% Criterion polyacrylamide gels.
Brefeldin A/AP20187 treatment followed by synaptic-like microvesicle fractionation
PC12 cells expressing mCh-FKBP-CLC were grown to confluence in 10-cm plates, moved to an ice bath at 4°C, and washed three times in ice-cold phosphate-buffered saline (PBS)/1 mM MgCl2/0.1 mM CaCl2. Sequential drug–treated cells were incubated with warm DMEM supplemented with 10 mg/ml brefeldin A (Epicentre Biotechnologies, Madison, WI) for 2 h at 37°C. Plates were then moved to an ice bath at 4°C and washed for 5 min with ice-cold DMEM three times. Cells were then incubated with ice-cold DMEM supplemented with either 50 nM AP20187 or 0.05% ethanol vehicle control for 15 min on an ice bath at 4°C to allow for drug equilibration. Cells were then moved to 37°C water bath for 10 min to quickly warm media and then to 37°C incubator for 2 h. Single drug–treated cells were incubated with warm DMEM supplemented with 10 mg/ml brefeldin A or 1% ethanol vehicle control for 2 or 4 h at 37°C. After incubation, cells were placed directly on an ice bath at 4°C and washed two times with ice-cold PBS/1 mM MgCl2/0.1 mM CaCl2 with subsequent synaptic-like microvesicle fractionation.
Synaptic-like microvesicle fractionation was performed as previously described (Faundez et al., 1997; Faundez and Kelly, 2000; Clift-O'Grady et al., 1998). Briefly, cells were lifted in intracellular buffer (38 mM potassium aspartate, 38 mM potassium glutamate, 38 mM potassium gluconate, 20 mM 3-(N-morpholino)propanesulfonic acid–KOH, pH 7.2, 5 mM reduced glutathione, 5 mM sodium carbonate, 2.5 mM magnesium sulfate, 2 mM ethylene glycol tetraacetic acid) and sedimented for 5 min at 800 × g at 4°C in a table-top centrifuge. Cell pellet was suspended in 300 μl of Intracellular buffer supplemented with Complete antiprotease (Roche, Indianapolis, IN). Homogenate was prepared with a 12-μm clearance cell cracker with intracellular buffer supplemented with Complete antiprotease at 4°C. Equal protein load from homogenate was centrifuged at 1000 × g for 5 min at 4°C in a table-top centrifuge, generating the first supernatant (S1) and pellet (P1). S1 was centrifuged at 27,000 × g for 35 min at 4°C in a Sorvall SS34 rotor (Thermo Scientific, Waltham, MA) to generate the second supernatant (S2) and pellet (P2). Equal volumes of the S2 were overlaid on a 5–25% glycerol gradient prepared in intracellular buffer generated by a Biocomp Gradient Master (Biocomp, Fredericton, Canada). Gradients were then centrifuged at 218,000 × g for 75 min at 4°C in a Beckman SW55 rotor (Beckman Coulter, Palo Alto, CA). Aliquots of the glycerol gradient after centrifugation were analyzed by Western blot on 4–20% Criterion polyacrylamide gels (Bio-Rad).
Immunofluorescence, microscopy, and analysis
Coverslips were prepared as previously described (Craige et al., 2008; Zlatic et al., 2011). Cells were seated to Matrigel-coated (BD Bioscience, San Jose, CA) coverslips. Cells were washed twice in PBS/1 mM MgCl2/0.1 mM CaCl2 and fixed with 4% paraformaldehyde in PBS. Cells were blocked, permeabilized, and probed with primary and secondary antibody in block buffer (PBS containing 15% donor equine serum [HyClone], 2% bovine serum albumin [HyClone], 1% fish gelatin [Sigma-Aldrich, St. Louis, MO], and 0.02% saponin [Sigma-Aldrich]). After secondary antibody probing, cells were washed twice in block buffer and once with PBS supplemented with 1 mM MgCl2 and 0.1 mM CaCl2 and mounted on slides with Gelvatol.
Deconvolution microscopy was carried out as previously described on a 200M inverted microscope (Carl Zeiss, Thornwood, NY) and Sedat filter set (Chroma Technology, Rockingham, UT) run with a multiwavelength, sequential capture, wide-field, three-dimensional microscopy system from SlideBook 4.0 OS X software (Intelligent Imaging Innovations, Denver, CO; Craige et al., 2008; Newell-Litwa et al., 2009; Zlatic et al., 2011). Samples were imaged with a 100×/1.40 numerical aperture (NA) oil differential interference contrast objective (Carl Zeiss) and captured with a scientific-grade, cooled, charge-coupled device (CCD) CoolSnap HQ camera (Photometrics, Tucson, AZ) run with an ORCA-ER chip (Hamamatsu Photonics, Hamamatsu, Japan). Out-of-focus light was removed using a nearest-neighbor constrained iterative deconvolution algorithm with Gaussian smoothing.
SIM microscopy was carried out using a Nikon N-SIM microscopy system on an Eclipse Ti inverted microscope run with Nikon Elements software (Nikon, Melville, NY). The samples were imaged with a 100–/1.49 NA objective and an iXon DU897 electron-multiplying CCD camera (Andor Technology, Belfast, Northern Ireland). Wide-field images were acquired with a mercury lamp and the appropriate filters: 480/30-nm excitation, 535/40-nm emission (Alexa 488) or 540/25-nm excitation, 692/68-nm emission (Alexa 555). Wide-field images were deconvolved with Huygens software (Scientific Volume Imaging, Hilversum, Netherlands). SIM images were acquired with laser excitation and emission filters, 488-nm excitation, 520/40-nm emission (Alexa 488) and 561-nm excitation, 640/40-nm emission (Alexa 555). Images were acquired in three-dimensional SIM mode (for each SIM image 15 images with five different phases of three different angular orientations of illumination were collected), and z-stacks were collected for each image. SIM images were processed with Nikon Elements software.
Three-dimensional surface reconstruction was carried out with Imaris 6.3.1 software (Bitplane, St. Paul, MN) from SIM and deconvolution image z-stacks. Whole-cell and profile colocalization, profile perimeter, and total fluorophore pixel content were determined from three z-plane focal slices per region of interest and carried out with MetaMorph 6.1 software (Universal Imaging, Sunnyvale, CA) with manual thresholding.
Statistical analysis
Experimental conditions were compared using Synergy KaleidaGraph, version 4.1.3 (Reading, PA), or StatPlus Mac Built5.6.0pre/Universal (AnalystSoft, Vancouver, Canada). Data are presented as boxplots displaying the four quartiles of the data, with the “box” comprising the two middle quartiles, separated by the median. The upper and lower quartiles are represented by the single lines extending from the box.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM077569 and NS42599) and the Emory University Research Committee to V.F. P.V.R. was supported by National Research Service Award Fellowship F31NS0765. This work was supported in part by the National Institute of Environmental Health Sciences of the National Institutes of Health under Awards P01ES016731 (V.F.) and T32ES012870 (S.Z.). This research project was supported in part by the Emory University Integrated Cellular Imaging Microscopy Core and Viral Cores of the Emory Neuroscience National Institute of Neurological Disorders and Stroke Core Facilities Grant P30NS055077
Abbreviations used:
- AP-3
adaptor protein complex 3
- BLOC
biogenesis of liposome-related organelles complex
- CHC
clathrin heavy chain
- CLC
clathrin light chain
- DSP
dithiobis(succinimidylpropionate)
- EEA1
early endosome antigen 1
- FKBP
FK506-binding protein
- GFP
green fluorescent protein
- PBS
phosphate-buffered saline
- shRNA
short hairpin RNA
- siRNA
small interfering RNA
- SIM
structured illumination microscopy
- SLMV
synaptic-like microvesicle
- SV2
synaptic vesicle glycoprotein 2
- VAMP7
vesicle-associated membrane protein 7
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-12-0860) on June 12, 2013.
REFERENCES
- Asensio CS, Sirkis DW, Edwards RH. RNAi screen identifies a role for adaptor protein AP-3 in sorting to the regulated secretory pathway. J Cell Biol. 2010;191:1173–1187. doi: 10.1083/jcb.201006131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo C, Burton A, Ruiz-Mateos E, Marsh M, Saiardi A. Inositol pyrophosphate mediated pyrophosphorylation of AP3B1 regulates HIV-1 Gag release. Proc Natl Acad Sci USA. 2009;106:21161–21166. doi: 10.1073/pnas.0909176106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondeau F, et al. Tandem MS analysis of brain clathrin-coated vesicles reveals their critical involvement in synaptic vesicle recycling. Proc Natl Acad Sci USA. 2004;101:3833–3838. doi: 10.1073/pnas.0308186101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Borner GH, Antrobus R, Hirst J, Bhumbra GS, Kozik P, Jackson LP, Sahlender DA, Robinson MS. Multivariate proteomic profiling identifies novel accessory proteins of coated vesicles. J Cell Biol. 2012;197:141–160. doi: 10.1083/jcb.201111049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borner GH, Harbour M, Hester S, Lilley KS, Robinson MS. Comparative proteomics of clathrin-coated vesicles. J Cell Biol. 2006;175:571–578. doi: 10.1083/jcb.200607164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clift-O'Grady L, Desnos C, Lichtenstein Y, Faundez V, Horng JT, Kelly RB. Reconstitution of synaptic vesicle biogenesis from PC12 cell membranes. Methods. 1998;16:150–159. doi: 10.1006/meth.1998.0662. [DOI] [PubMed] [Google Scholar]
- Craige B, Salazar G, Faundez V. Phosphatidylinositol-4-kinase type ii alpha contains an AP-3 sorting motif and a kinase domain that are both required for endosome traffic. Mol Biol Cell. 2008;19:1415–1426. doi: 10.1091/mbc.E07-12-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danglot L, Galli T. What is the function of neuronal AP-3. Biol Cell. 2007;99:349–361. doi: 10.1042/BC20070029. [DOI] [PubMed] [Google Scholar]
- Deborde S, Perret E, Gravotta D, Deora A, Salvarezza S, Schreiner R, Rodriguez-Boulan E. Clathrin is a key regulator of basolateral polarity. Nature. 2008;452:719–723. doi: 10.1038/nature06828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delevoye C, et al. AP-1 and KIF13A coordinate endosomal sorting and positioning during melanosome biogenesis. J Cell Biol. 2009;187:247–264. doi: 10.1083/jcb.200907122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Angelica EC, Klumperman J, Stoorvogel W, Bonifacino JS. Association of the AP-3 adaptor complex with clathrin. Science. 1998;280:431–434. doi: 10.1126/science.280.5362.431. [DOI] [PubMed] [Google Scholar]
- Dell'Angelica EC, Ohno H, Ooi CE, Rabinovich E, Roche KW, Bonifacino JS. AP-3: an adaptor-like protein complex with ubiquitous expression. EMBO J. 1997;16:917–928. doi: 10.1093/emboj/16.5.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Matteis MA, Luini A. Mendelian disorders of membrane trafficking. N Engl J Med. 2011;365:927–938. doi: 10.1056/NEJMra0910494. [DOI] [PubMed] [Google Scholar]
- Di Pietro SM, Falcon-Perez JM, Tenza D, Setty SR, Marks MS, Raposo G, Dell'Angelica EC. BLOC-1 interacts with BLOC-2 and the AP-3 complex to facilitate protein trafficking on endosomes. Mol Biol Cell. 2006;17:4027–4038. doi: 10.1091/mbc.E06-05-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edeling MA, Mishra SK, Keyel PA, Steinhauser AL, Collins BM, Roth R, Heuser JE, Owen DJ, Traub LM. Molecular switches involving the AP-2 beta2 appendage regulate endocytic cargo selection and clathrin coat assembly. Dev Cell. 2006;10:329–342. doi: 10.1016/j.devcel.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Faundez V, Horng JT, Kelly RB. ADP ribosylation factor 1 is required for synaptic vesicle budding in PC12 cells. J Cell Biol. 1997;138:505–515. doi: 10.1083/jcb.138.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faundez V, Horng JT, Kelly RB. A function for the AP3 coat complex in synaptic vesicle formation from endosomes. Cell. 1998;93:423–432. doi: 10.1016/s0092-8674(00)81170-8. [DOI] [PubMed] [Google Scholar]
- Faundez V, Kelly RB. The AP-3 complex required for endosomal synaptic vesicle biogenesis is associated with a casein kinase ialpha-like isoform. Mol Biol Cell. 2000;11:2591–2604. doi: 10.1091/mbc.11.8.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard M, Allaire PD, Blondeau F, McPherson PS. Isolation of clathrin-coated vesicles. In: Bonifacino JS, Dasso M, Harford J, Lippincott-Schwartz J, Yamada K, editors. Current Protocols in Cell Biology. Vol. 1. Hoboken, NJ: John Wiley & Sons; 20042004. pp. 3.13.11–13.13.31. [Google Scholar]
- Gokhale A, Larimore J, Werner E, So L, Moreno-De-Luca A, Lese-Martin C, Lupashin VV, Smith Y, Faundez V. Quantitative proteomic and genetic analyses of the schizophrenia susceptibility factor dysbindin identify novel roles of the biogenesis of lysosome-related organelles complex 1. J Neurosci. 2012;32:3697–3711. doi: 10.1523/JNEUROSCI.5640-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, Barlow LD, Francisco GC, Sahlender DA, Seaman MN, Dacks JB, Robinson MS. The fifth adaptor protein complex. PLoS Biol. 2011;9:e1001170. doi: 10.1371/journal.pbio.1001170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, Bright NA, Rous B, Robinson MS. Characterization of a fourth adaptor-related protein complex. Mol Biol Cell. 1999;10:2787–2802. doi: 10.1091/mbc.10.8.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, Irving C, Borner GH. Adaptor protein complexes AP-4 and AP-5: new players in endosomal trafficking and progressive spastic paraplegia. Traffic. 2013;14:153–164. doi: 10.1111/tra.12028. [DOI] [PubMed] [Google Scholar]
- Kantheti P, Diaz ME, Peden AE, Seong EE, Dolan DF, Robinson MS, Noebels JL, Burmeister ML. Genetic and phenotypic analysis of the mouse mutant mh(2J), an Ap3d allele caused by IAP element insertion. Mamm Genome. 2003;14:157–167. doi: 10.1007/s00335-002-2238-8. [DOI] [PubMed] [Google Scholar]
- Kent HM, Evans PR, Schafer IB, Gray SR, Sanderson CM, Luzio JP, Peden AA, Owen DJ. Structural basis of the intracellular sorting of the SNARE VAMP7 by the AP3 adaptor complex. Dev Cell. 2012;22:979–988. doi: 10.1016/j.devcel.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhausen T. Clathrin. Annu Rev Biochem. 2000;69:699–727. doi: 10.1146/annurev.biochem.69.1.699. [DOI] [PubMed] [Google Scholar]
- Knuehl C, Chen CY, Manalo V, Hwang PK, Ota N, Brodsky FM. Novel binding sites on clathrin and adaptors regulate distinct aspects of coat assembly. Traffic. 2006;7:1688–1700. doi: 10.1111/j.1600-0854.2006.00499.x. [DOI] [PubMed] [Google Scholar]
- Kural C, Tacheva-Grigorova SK, Boulant S, Cocucci E, Baust T, Duarte D, Kirchhausen T. Dynamics of intracellular clathrin/AP1- and clathrin/AP3-containing carriers. Cell Rep. 2012;2:1111–1119. doi: 10.1016/j.celrep.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larimore J, et al. The schizophrenia susceptibility factor dysbindin and its associated complex sort cargoes from cell bodies to the synapse. Mol Biol Cell. 2011;22:4854–4867. doi: 10.1091/mbc.E11-07-0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Nemecek D, Schindler C, Smith WJ, Ghirlando R, Steven AC, Bonifacino JS, Hurley JH. Assembly and architecture of the biogenesis of lysosome-related organelles complex-1 (BLOC-1) J Biol Chem. 2012;287:5882–5890. doi: 10.1074/jbc.M111.325746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon SK, Traub LM. Getting in touch with the clathrin terminal domain. Traffic. 2012;13:511–519. doi: 10.1111/j.1600-0854.2011.01321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Kuromi H, Briggs L, Green DB, Rocha JJ, Sweeney ST, Bullock SL. Bicaudal-D binds clathrin heavy chain to promote its transport and augments synaptic vesicle recycling. EMBO J. 2010;29:992–1006. doi: 10.1038/emboj.2009.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SH, Towler MC, Chen E, Chen CY, Song W, Apodaca G, Brodsky FM. A novel clathrin homolog that co-distributes with cytoskeletal components functions in the trans-Golgi network. EMBO J. 2001;20:272–284. doi: 10.1093/emboj/20.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomant AJ, Fairbanks G. Chemical probes of extended biological structures: synthesis and properties of the cleavable protein cross-linking reagent [35S]dithiobis(succinimidyl propionate) J Mol Biol. 1976;104:243–261. doi: 10.1016/0022-2836(76)90011-5. [DOI] [PubMed] [Google Scholar]
- Martinez-Arca S, et al. A dual mechanism controlling the localization and function of exocytic v-SNAREs. Proc Natl Acad Sci USA. 2003;100:9011–9016. doi: 10.1073/pnas.1431910100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGough IJ, Cullen PJ. Clathrin is not required for SNX-BAR-retromer mediated carrier formation. J Cell Sci. 2012;126:45–52. doi: 10.1242/jcs.112904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2011;12:517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- Moskowitz HS, Heuser J, McGraw TE, Ryan TA. Targeted chemical disruption of clathrin function in living cells. Mol Biol Cell. 2003;14:4437–4447. doi: 10.1091/mbc.E03-04-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley AM, Berg N, Taylor MJ, Sahlender DA, Hirst J, Owen DJ, Robinson MS. Functional analysis of AP-2 alpha and mu2 subunits. Mol Biol Cell. 2006;17:5298–5308. doi: 10.1091/mbc.E06-05-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Setou M, Seog D, Ogasawara K, Dohmae N, Takio K, Hirokawa N. A novel motor, KIF13A, transports mannose-6-phosphate receptor to plasma membrane through direct interaction with AP-1 complex. Cell. 2000;103:569–581. doi: 10.1016/s0092-8674(00)00161-6. [DOI] [PubMed] [Google Scholar]
- Newell-Litwa K, Salazar G, Smith Y, Faundez V. Roles of BLOC-1 and AP-3 complexes in cargo sorting to synaptic vesicles. Mol Biol Cell. 2009;20:1441–1453. doi: 10.1091/mbc.E08-05-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell-Litwa K, Seong E, Burmeister M, Faundez V. Neuronal and non-neuronal functions of the AP-3 sorting machinery. J Cell Sci. 2007;120:531–541. doi: 10.1242/jcs.03365. [DOI] [PubMed] [Google Scholar]
- Peden AA, Oorschot V, Hesser BA, Austin CD, Scheller RH, Klumperman J. Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J Cell Biol. 2004;164:1065–1076. doi: 10.1083/jcb.200311064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peden AA, Rudge RE, Lui WW, Robinson MS. Assembly and function of AP-3 complexes in cells expressing mutant subunits. J Cell Biol. 2002;156:327–336. doi: 10.1083/jcb.200107140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Cornejo P, Gokhale A, Duran C, Cui Y, Xiao Q, Hartzell HC, Faundez V. Anoctamin 1 (Tmem16A) Ca2+-activated chloride channel stoichiometrically interacts with an ezrin-radixin-moesin network. Proc Natl Acad Sci USA. 2012;109:10376–10381. doi: 10.1073/pnas.1200174109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiborg C, Bache KG, Mehlum A, Stang E, Stenmark H. Hrs recruits clathrin to early endosomes. EMBO J. 2001;20:5008–5021. doi: 10.1093/emboj/20.17.5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, Marks MS. Melanosomes—dark organelles enlighten endosomal membrane transport. Nat Rev Mol Cell Biol. 2007;8:786–797. doi: 10.1038/nrm2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, Marks MS, Cutler DF. Lysosome-related organelles: driving post-Golgi compartments into specialisation. Curr Opin Cell Biol. 2007;19:394–401. doi: 10.1016/j.ceb.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MS. Adaptable adaptors for coated vesicles. Trends Cell Biol. 2004;14:167–174. doi: 10.1016/j.tcb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Royle SJ. The role of clathrin in mitotic spindle organisation. J Cell Sci. 2012;125:19–28. doi: 10.1242/jcs.094607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar G, et al. BLOC-1 complex deficiency alters the targeting of adaptor protein complex-3 cargoes. Mol Biol Cell. 2006;17:4014–4026. doi: 10.1091/mbc.E06-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar G, Zlatic S, Craige B, Peden AA, Pohl J, Faundez V. Hermansky-Pudlak syndrome protein complexes associate with phosphatidylinositol 4-kinase type II alpha in neuronal and non-neuronal cells. J Biol Chem. 2009;284:1790–1802. doi: 10.1074/jbc.M805991200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schermelleh L, et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science. 2008;320:1332–1336. doi: 10.1126/science.1156947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid EM, Ford MG, Burtey A, Praefcke GJ, Peak-Chew SY, Mills IG, Benmerah A, McMahon HT. Role of the AP2 beta-appendage hub in recruiting partners for clathrin-coated vesicle assembly. PLoS Biol. 2006;4:e262. doi: 10.1371/journal.pbio.0040262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G, Faundez V, Roos J, Dell'Angelica EC, Kelly RB. Neuroendocrine synaptic vesicles are formed in vitro by both clathrin- dependent and clathrin-independent pathways. J Cell Biol. 1998;143:947–955. doi: 10.1083/jcb.143.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, et al. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Theos AC, et al. Functions of adaptor protein (AP)-3 and AP-1 in tyrosinase sorting from endosomes to melanosomes. Mol Biol Cell. 2005;16:5356–5372. doi: 10.1091/mbc.E05-07-0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeham DE, Abi-Rached L, Towler MC, Wilbur JD, Parham P, Brodsky FM. Clathrin heavy and light chain isoforms originated by independent mechanisms of gene duplication during chordate evolution. Proc Natl Acad Sci USA. 2005;102:7209–7214. doi: 10.1073/pnas.0502058102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei ML. Hermansky-Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res. 2006;19:19–42. doi: 10.1111/j.1600-0749.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- Wei AH, Li W. Hermansky-Pudlak syndrome: pigmentary and non-pigmentary defects and their pathogenesis. Pigment Cell Melanoma Res. 2012;26:176–192. doi: 10.1111/pcmr.12051. [DOI] [PubMed] [Google Scholar]
- Willox AK, Royle SJ. Functional analysis of interaction sites on the N-terminal domain of clathrin heavy chain. Traffic. 2012;13:70–81. doi: 10.1111/j.1600-0854.2011.01289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatic SA, Ryder PV, Salazar G, Faundez V. Isolation of labile multi-protein complexes by in vivo controlled cellular cross-linking and immuno-magnetic affinity chromatography. J Vis Exp. 2010;1855 doi: 10.3791/1855. 1810.3791/1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatic SA, Tornieri K, L'Hernault SW, Faundez V. Clathrin-dependent mechanisms modulate the subcellular distribution of class C Vps/HOPS tether subunits in polarized and nonpolarized cells. Mol Biol Cell. 2011;22:1699–1715. doi: 10.1091/mbc.E10-10-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.