

Insulin stimulates glucose transport by delivering GLUT4 from a specialized storage compartment to the plasma membrane. Subcellular fractionation and an in vitro assay for GLUT4-storage vesicle formation show that the Sec1/Munc18 protein mVps45 is required to correctly sort GLUT4 into this compartment.
Abstract
Insulin stimulates glucose transport in fat and muscle cells by regulating delivery of the facilitative glucose transporter, glucose transporter isoform 4 (GLUT4), to the plasma membrane. In the absence of insulin, GLUT4 is sequestered away from the general recycling endosomal pathway into specialized vesicles, referred to as GLUT4-storage vesicles. Understanding the sorting of GLUT4 into this store is a major challenge. Here we examine the role of the Sec1/Munc18 protein mVps45 in GLUT4 trafficking. We show that mVps45 is up-regulated upon differentiation of 3T3-L1 fibroblasts into adipocytes and is expressed at stoichiometric levels with its cognate target–soluble N-ethylmaleimide–sensitive factor attachment protein receptor, syntaxin 16. Depletion of mVps45 in 3T3-L1 adipocytes results in decreased GLUT4 levels and impaired insulin-stimulated glucose transport. Using subcellular fractionation and an in vitro assay for GLUT4-storage vesicle formation, we show that mVps45 is required to correctly traffic GLUT4 into this compartment. Collectively our data reveal a crucial role for mVps45 in the delivery of GLUT4 into its specialized, insulin-regulated compartment.
INTRODUCTION
The binding of insulin to its receptor on the surface of adipose and muscle cells initiates a signaling cascade that alters the trafficking itinerary of the facilitative glucose transporter, glucose transporter isoform 4 (GLUT4), releasing it from intracellular stores and delivering it to the cell surface (Bryant et al., 2002; Stockli et al., 2011). In the absence of insulin, ∼95% of GLUT4 is confined to intracellular compartment(s). Insulin stimulation results in GLUT4 redistribution from these intracellular stores to the plasma membrane via alterations in membrane trafficking (Bryant et al., 2002; Stockli et al., 2011). This insulin-stimulated translocation of GLUT4 from intracellular sites to the plasma membrane is defective in individuals with insulin resistance and type 2 diabetes, providing an impetus to understand how this trafficking pathway is controlled (Graham and Kahn, 2007; Bogan, 2012).
Models suggest that intracellular GLUT4 traffics via two interlinked endosomal cycles (Bryant et al., 2002; Bryant and Gould, 2011). The first operates between the plasma membrane and early endosomes and serves to efficiently internalize GLUT4 in the absence of insulin (Piper et al., 1992). GLUT4 is subsequently sorted into a slowly recycling pathway, encompassing recycling endosomes and the trans-Golgi network (TGN). It is from this cycle that GLUT4 is delivered to the cell surface in response to insulin, and hence the compartments from which this delivery occurs are referred to as insulin-sensitive GLUT4-storage vesicles (GSVs; Livingstone et al., 1996; Bryant et al., 2002; Bryant and Gould, 2011).
All eukaryotic membrane trafficking events require the assembly of specific soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) complexes (Hong, 2005; Cai et al., 2007). The formation of complexes between members of the target (t) family of SNARE proteins and their cognate vesicle (v) SNAREs localized on the appropriate donor membrane is sufficient to drive bilayer fusion (Weber et al., 1998; Hu et al., 2003) and impart a degree of specificity to membrane traffic (Scales et al., 2000). SNARE proteins therefore provide the cell with a mechanism by which to regulate membrane traffic, and recent studies identified a number of important SNAREs in the trafficking itinerary of GLUT4 (Bryant and Gould, 2011). Studies from this laboratory identified a role for the intracellular syntaxin, Sx6, in GLUT4 sorting. Overexpression of a mutant form of Sx6 lacking the transmembrane anchor (and thus interfering with the function of endogenous Sx6) delayed reinternalization of GLUT4 from the cell surface upon insulin removal (Perera et al., 2003). Although the locus of action of Sx6 remains to be defined, studies in other systems suggest that this SNARE acts at the TGN, consistent with a role for Sx6 in sorting into GSVs (Bock et al., 1997; Wendler et al., 2001; Murray et al., 2005). Consistent with this, recent studies showed that recycling of the insulin-responsive aminopeptidase (IRAP; a GSV-resident protein) from the cell surface back to GSVs requires Sx6 (Watson and Pessin, 2008). Syntaxin 16 (Sx16) forms a t-SNARE complex with Sx6 and has also been implicated in GLUT4 sorting. Sx16 and GLUT4 exhibit a marked level of colocalization, and GLUT4 recycles through a subdomain of the TGN enriched in both Sx16 and Sx6 (Shewan et al., 2003). Studies involving either depletion of endogenous Sx16 or overexpression of a dominant-negative mutant led us to propose that Sx16 facilitates sorting of GLUT4 into GSVs and that disruption of this pathway results in mistargeting of GLUT4 (Proctor et al., 2006).
The Sec1/Munc18 (SM) family of proteins are key regulators of SNARE complex formation and function (Sudhof and Rothman, 2009). It is well established that the SM protein that binds to Sx16 is mVps45 (Tellam et al., 1997; Bryant and James, 2003; Carpp et al., 2006; Struthers et al., 2009). We therefore hypothesized that mVps45 may play an important role in GLUT4 trafficking, and here we describe an analysis of the localization and function of mVps45 in GLUT4 trafficking. Consistent with an important role for this SM protein in GLUT4 trafficking, we report that mVps45 levels increase during adipocyte differentiation concomitant with those of Sx16 and the acquisition of insulin-sensitive glucose transport. Depletion of mVps45 significantly reduces total cell GLUT4 levels, abrogates insulin-stimulated glucose transport and GLUT4 translocation, and results in missorting of GLUT4 such that levels within a GSV-enriched fraction are markedly reduced. Furthermore, an in vitro budding assay that recapitulates the formation of GSVs from donor membranes indicates that this sorting step requires mVps45. We therefore propose that mVps45 is an important regulatory molecule in GLUT4 sorting, acting in concert with Sx16 to allow entry of GLUT4 into GSVs.
RESULTS
mVps45 levels in adipocytes
Previous studies showed that molecules involved in insulin-stimulated GLUT4 translocation are often up-regulated during the differentiation of 3T3-L1 fibroblasts into adipocytes (El-Jack et al., 1999). Consistent with this, we observed that levels of mVps45 were elevated in adipocytes compared with fibroblasts, as previously reported for other molecules involved in GLUT4 sorting, including Sx16 and GLUT4 (Figure 1A; El-Jack et al., 1999; Shewan et al., 2003). By comparative immunoblotting using bacterially expressed recombinant Sx16 or mVps45, we quantified the absolute levels of these proteins in 3T3-L1 fibroblasts (unpublished data) and adipocytes (Figure 1B). Quantification of such data (together with that observed in primary rat adipocytes) is shown in Table 1. As shown, levels of Sx16 and mVps45 on a per-cell basis were similar in 3T3-L1 adipocytes (635,000 vs. 459,000 copies/cell, respectively), and levels of both proteins increased significantly during the fibroblast-to-adipocyte conversion. These levels are similar to those for syntaxin 4/Munc18c in 3T3-L1 adipocytes (374,000 vs. 452,000 copies/cell, respectively) and compare favorably with other estimates of 280,000 copies of GLUT4 per cell (Hickson et al., 2000). Sx16 and mVps45 also were found to be expressed at roughly stoichiometric levels in rat adipocytes (3.6 × 1012 vs. 2.8 × 1012 copies/μg lysate, respectively).
FIGURE 1:
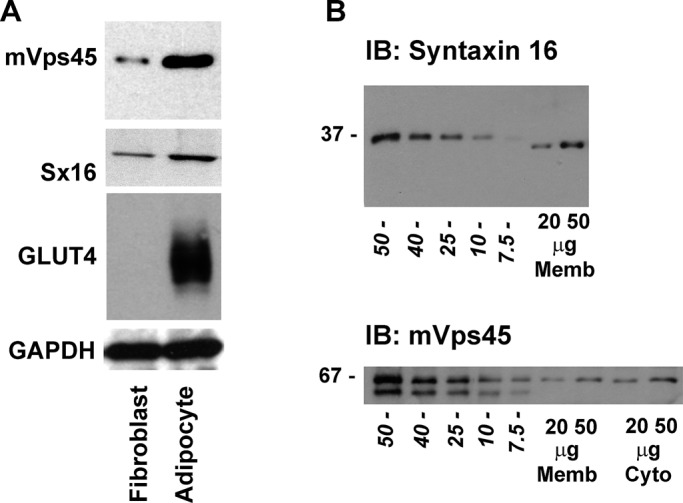
mVps45 and syntaxin 16 levels in 3T3-L1 adipocytes. (A) Representative immunoblot of 50 μg of lysate prepared from either 3T3-L1 fibroblasts or adipocytes (as labeled) and probed with anti-mVps45, anti-Sx16, anti-GLUT4, or anti-GAPDH. Data from a representative experiment is shown, repeated three times with qualitatively similar results. Table 1 shows quantification of levels of Sx16 and mVps45 in these cells. (B) Representative immunoblot of 3T3-L1 adipocyte membranes (20- and 50-μg membranes for Sx16) or membranes and cytosol (20- and 50-μg membranes and cytosol for mVps45) compared with known amounts of recombinant Sx16 or mVps45 expressed and purified from bacteria. Figures in italics are nanograms of recombinant protein, and the position of molecular weight markers is shown. Data are from a representative experiment repeated at least four times.
TABLE 1:
Quantification of syntaxin 16 and mVps45 levels.
| Syntaxin 16 | mVps45 | |
|---|---|---|
| 3T3-L1 fibroblasts (copies/cell × 103) | 116 ± 21 | 45 ± 5.8 |
| 3T3-L1 adipocytes | ||
| Total (copies/cell × 103) | 635 ± 59 | 459 ± 61 |
| Membrane (copies/cell × 103) | 635 ± 59 | 203 ± 35 |
| Cytosol (copies/cell × 103) | Not detected | 247 ± 21 |
| 3T3-L1 adipocytes (copies/mg) | 1.52 ± 0.2 × 1012 | 1.10 ± 0.3 × 1012 |
| Rat adipocytes (copies/mg) | 3.6 ± 0.5 × 1012 | 2.8 ± 0.45 × 1012 |
Levels of mVps45 and Sx16 were determined in 3T3-L1 fibroblasts, adipocytes, or rat adipocytes as described in Materials and Methods. Shown are the mean values (±SD) of at least three determinations, using at least three different preparations of lysates or membrane fractions.
Knockdown of mVps45 impairs insulin-stimulated glucose transport and GLUT4 translocation
To understand the functional role of mVps45 in GLUT4 sorting, we engineered four different short hairpin RNAs (shRNAs) designed to target murine mVps45. These were delivered into 3T3-L1 fibroblasts using retroviruses, and the cells were then grown and differentiated. Figure 2A shows the result of a typical experiment comparing cells infected with the four mVps45 shRNA targets, a virus delivering random shRNA (as a non-mVps45–targeted control), and cells not infected with any virus. Two of the constructs, shRNAs #1 and #2, effectively reduced mVps45 levels (by 77.9 ± 8 and 41.1 ± 11%, p < 0.01 and 0.02, respectively); glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is shown as a control. We therefore performed a series of immunoblot analyses examining the effect of depletion of mVps45 using shRNAs #1 and #2 on a range of membrane trafficking proteins, some of which are shown in Figure 2B and quantified in Figure 2C (see also Supplemental Table S1). Knockdown of mVps45 significantly reduced levels of expression of its cognate Qa-SNARE Sx16, as expected based on other studies that revealed an important role of SM proteins in the maintenance of SNARE stability (∼82% reduction with shRNA #1, p < 0.02; Figure 2, B and C; Bryant and James, 2001). By contrast, other SNAREs, including Vti1a (Figure 2B), Sx6, Vti1b, Sx4, Sx12/13, and SNAP23, were unaffected by mVps45 depletion (Supplemental Table S1). Sx6 and Vti1a form a functional SNARE complex with Sx16 that is involved in endosome–TGN trafficking (Simonsen et al., 1998). Note that knockdown of Sx16 in 3T3-L1 adipocytes also had no effect on Sx6 (Proctor et al., 2006) or Vti1a levels (our unpublished data). This presumably reflects the involvement of these t-SNAREs in multiple SNARE complexes (Hong, 2005). This is further supported by observations that deletion of yeast Vps45 reduced levels of Tlg2p (Sx16 orthologue) but not those of Vti1p or Tlg1p (Sx6 orthologue; Bryant and James, 2001). We also examined levels of VAMP3 and VAMP4, which have been proposed to play a role in intracellular GLUT4 traffic (Williams and Pessin, 2008). Levels of VAMP3 did not significantly change upon mVps45 knockdown (Supplemental Table S1). By contrast, levels of VAMP4 were significantly reduced (by 41 ± 12%, p = 0.04; see Supplemental Table S1), suggesting that this v-SNARE functions in a complex regulated by mVps45, consistent with published work on other cell types (Tran et al., 2007). Levels of endosomal proteins (EEA1, transferrin receptor [TfR], and Rab5) were unaffected (Supplemental Table S1). Levels of Rabenosyn-5 were reduced upon mVps45 knockdown (Figure 2, B and C), consistent with other reports (Morrison et al., 2008; Rahajeng et al., 2009). Furthermore, we observed that the total cellular levels of tubulin (Figure 2, B and C), actin, and GAPDH (Figure 2A) or the adipocyte marker proteins PPAR-γ (Figure 2, B and C), FAS, and C/EBPα were unaffected by mVps45 knockdown (Supplemental Table S1). Such observations suggest that the differentiation process was not impaired by the absence of mVps45 (this conclusion is also supported by similar levels of fat droplet accumulation in these cells; data not shown). By contrast, there were significant reductions in the levels of GLUT4 (76 ± 12% with shRNA #1, p = 0.01) and IRAP (75 ± 15% with shRNA #1, p = 0.01) upon depletion of mVps45 (Figure 2B and Supplemental Table S1), suggesting that mVps45 is required for the maintenance of normal cellular GLUT4 homeostasis.
FIGURE 2:
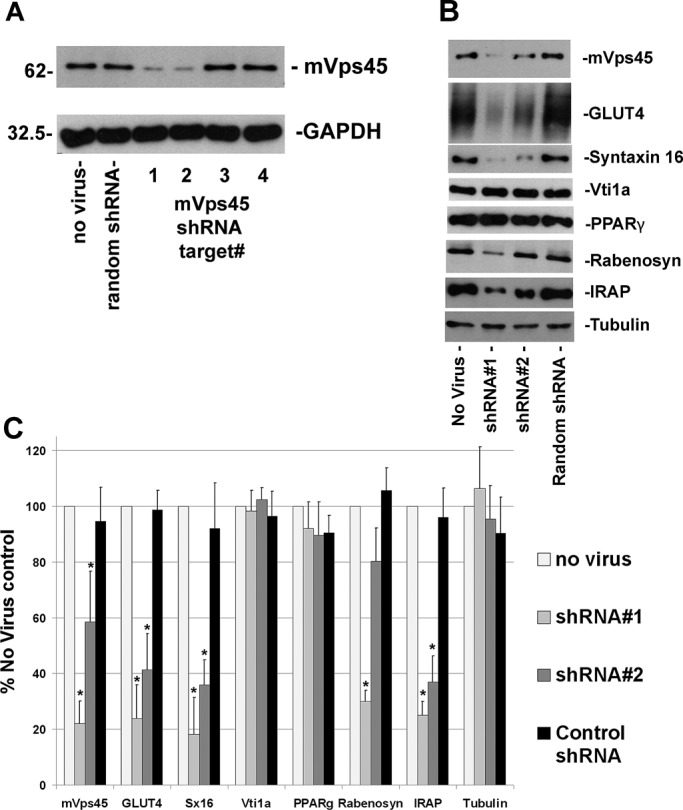
mVps45 knockdown perturbs GLUT4 levels. (A) Whole-cell lysates of 3T3-L1 adipocytes infected with four different retroviruses harboring shRNA species designed to knock down mVps45, a random shRNA, or control (noninfected) cells immunoblotted with anti-mVps45 or GAPDH as a loading control. For each lane 50 μg of protein was loaded, and the position of molecular weight markers is shown. Data from a typical experiment are presented. (B) Whole-cell lysates from noninfected cells or cells infected with shRNA #1, shRNA #2, or random shRNA, immunoblotted with the antibodies indicated. Data from a representative experiment is shown; all were repeated a minimum of three times using different batches of cells infected with at least two different batches of the indicated virus. (C) Quantification of data of this type, in which the extent of knockdown is quantified. Data shown are mean ± SD with the data normalized against control cells. Asterisk indicates a statistically significant decrease in expression (see text for details; p ≤ 0.05 for each case).
Consistent with the foregoing, we observed that cells depleted of mVps45 exhibit a significant reduction in insulin-stimulated deoxyglucose uptake compared with either noninfected or control-infected cells (Figure 3A). Depletion of mVps45, despite resulting in a reduced magnitude of insulin-stimulated glucose transport, did not significantly alter the insulin dose–response curve (Figure 3B). Consistent with this, we observed no impairment of insulin-stimulated Akt phosphorylation (Figure 3C) or AS160 phosphorylation (data not shown) upon depletion of mVps45. We next used a green fluorescent protein (GFP)–tagged GLUT4 species to assess insulin-stimulated translocation in cells depleted of mVps45 (Figure 3D). In cells expressing control shRNA, insulin stimulated a robust translocation of GLUT4 to the plasma membrane, such that the majority of cells exhibited a characteristic ring of GFP-GLUT4 at the cell surface (>95% in 50 cells from three separate experiments). In cells depleted of mVps45, insulin-stimulated GLUT4 translocation was significantly impaired (fluorescent rings of GFP-GLUT4 were evident in <15% of cells from three separate experiments of this type). Finally, analysis of GLUT4 distribution in subcellular fractions isolated using differential centrifugation revealed a marked impairment of insulin-stimulated translocation of endogenous GLUT4 to the plasma membrane fraction in mVps45-depleted cells (Figure 3E).
FIGURE 3:

mVps45 knockdown diminishes insulin-stimulated glucose transport and GLUT4 translocation. (A) Basal and insulin-stimulated 2-deoxy-d-glucose (deGlc) transport was assayed in cells infected with either mVps45 shRNA #1 or #2 retroviruses and compared with random shRNA virus or noninfected controls. Cells were incubated with or without 100 nM insulin for 30 min before assay of deGlc uptake (50 μM for 3 min), and uptake values were corrected for nonspecific association of deGlc with the cells by performing parallel assays in the presence of 10 μM cytochalasin B. Shown are data from a typical experiment, repeated three times with qualitatively similar results, in which each point is the mean of triplicate determinations (±SD) and the deGlc uptake is expressed as percentage of the basal rate in control cells. The reduction in insulin-stimulated DeGlc uptake in cells infected with mVps45 shRNA #1 and #2 retrovirus was statistically significant in all experiments, and in the experiment shown, p < 0.05 for both. (B) Insulin dose–response curve for insulin-stimulated deGlc transport, assayed as described in A. The IC50 for insulin did not differ significantly between control and mVps45-knockdown cells. Shown are data from a typical experiment, repeated three times with similar results. (C) 3T3-L1 adipocytes infected with either random shRNA or mVps45 shRNA #1 were incubated in serum-free media for 2 h and then incubated with 100 nM insulin for the times shown. Lysates were prepared and 50 μg immunoblotted for mVps45, phosphor-Akt, or total Akt. Data from a typical experiment are shown. (D) 3T3-L1 adipocytes expressing HA-GLUT4-GFP were infected with retroviruses carrying random shRNA or mVps45 shRNA #1 as described. On the day of experiment, cells were incubated in serum-free medium for 2 h, 100 nM insulin was added to half of the cells for 30 min, and then cells were fixed and mounted. Shown are representative fields of cells. Insulin-stimulated GLUT4 translocation was consistently reduced in mVps45-knockdown cells (note that higher gain levels were used in the mVps45-depeleted cells, as levels of HA-GLUT4-GFP expressed were lower). (E) Subcellular fractionation of 3T3-L1 adipocyte into PM, LDM, high-density microsome (HDM), and cytosolic fractions using a well-characterized protocol. In control cells, insulin stimulates movement of GLUT4 from the LDM into the PM fraction. In mVps45 cells, GLUT4 levels are reduced, and translocation to the plasma membrane is not observed. In this experiment, representative of at least three, 25 μg of each fraction was loaded. Note that the immunoblot for mVps45-depleted cells is exposed for a longer time in this case to reveal a clear signal (cf. Figure 2), and hence the levels of GLUT4 in the different fractions cannot be directly compared with those observed in control cells.
Locus of action of mVps45 on membrane traffic
These data reveal a requirement for mVps45 for the proper maintenance of insulin-stimulated glucose transport and GLUT4 levels. We therefore next sought to define the locus of action of mVps45. SM proteins cycle between membrane-bound and cytosolic fractions (Tellam et al., 1997; Sudhof and Rothman, 2009), and hence our first objective was to establish whether insulin modulated this or the distribution of mVps45 among cell compartments. Figure 4A shows that insulin had no effect on either distribution of mVps45 between subcellular fractions or the membrane/cytosol ratio of mVps45.
FIGURE 4:
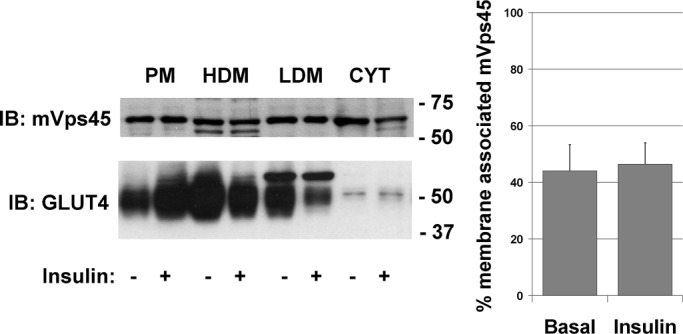
Insulin does not alter the distribution of mVps45. Noninfected 3T3-L1 adipocytes were incubated with or without 100 nM insulin for 30 min and then fractionated into PM, LDM, HDM, and cytosolic fractions, and 25 μg of each fraction was separated by SDS–PAGE. Immunoblot analysis with anti-GLUT4 and anti-mVps45 is shown. The endogenous mVps45 does not appear to exhibit any marked redistribution between fractions. Analysis of three experiments of this type revealed that the membrane/cytosol distribution of mVps45 does not change in response to acute insulin treatment (values are mean ± SD of three determinations).
In an effort to determine whether lack of mVps45 inhibited the sorting of GLUT4 into GSVs, we first used a simple fractionation procedure to examine the distribution of GLUT4 between the pellet and supernatant fractions obtained after fractionation of a 3T3-L1 adipocyte homogenate at 16,000 × g (Lamb et al., 2010). Previous studies from this and other groups established that GLUT4 in the supernatant from this simple centrifugation represents a highly enriched GLUT4-containing GSV fraction (Lamb et al., 2010). We therefore compared the pellet and supernatant fractions obtained using this procedure from control and mVps45-depleted cells (Figure 5A; note that for the GLUT4 blot only, we loaded 3× the sample of the control in order to reveal a clear GLUT4 signal due to decreased expression of GLUT4 in mVps45-knockdown cells). The data show that the fraction of GLUT4 in the 16k supernatant is decreased from 61 ± 12 to 34 ± 12% upon mVps45 knockdown (p < 0.05; Figure 5B). Note that the distribution of endosomal markers (e.g., TfR) was not impaired by mVps45 depletion. We further fractionated the low-density microsome (LDM) fraction of control and mVps45-knockdown cells using iodixanol gradient centrifugation to resolve GLUT4 in GSV-enriched fractions and GLUT4 in endosomal fractions (Hashiramoto and James, 2000; Perera et al., 2003). Figure 5, C and D, shows that infection of 3T3-L1 adipocytes with virus containing mVps45 shRNA #1 decreased the fraction of intracellular GLUT4 localized within the GSV peaks. These data are consistent with the hypothesis that mVps45 is required for sorting of GLUT4 into GSVs and that its depletion selectively abrogates this process.
FIGURE 5:
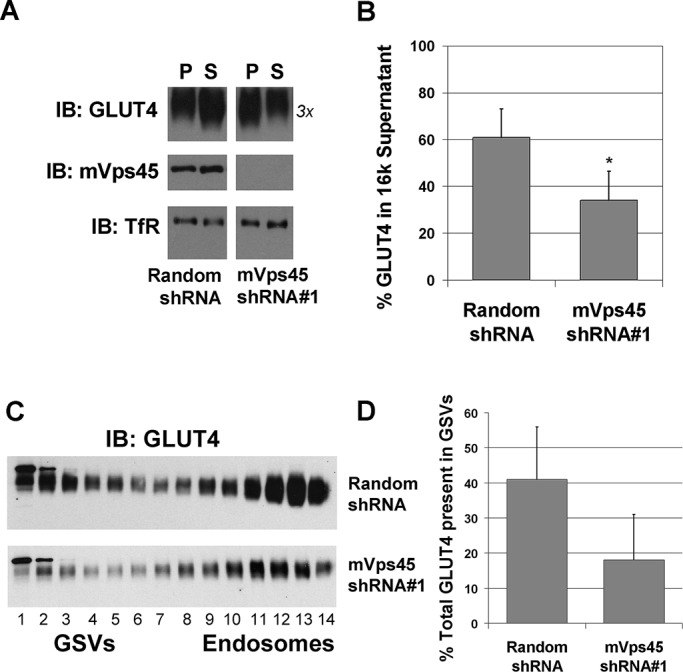
GLUT4 sorting into GSVs is impaired in mVps45 knockdown cells. (A) Control and mVps45-depleted 3T3-L1 adipocytes incubated in serum-free medium for 2 h in the absence of insulin were subjected to a simple 16,000 × g subcellular fractionation to prepare a pellet (P) and a supernatant (S) fraction, the latter being enriched in GSVs. Shown is a representative experiment of this type in which these fractions are immunoblotted for GLUT4, mVps45, or TfR (as a loading control). For the P fractions, 5 μg of protein was loaded per lane; for the S fractions, 20 μg. Note that as GLUT4 levels are decreased upon mVps45 knockdown, we loaded 3× the amount of protein in this specific case. The percentage GLUT4 in the supernatant was quantified from three experiments of this type, and the result (mean ± SD) is shown in B. Asterisk indicates a statistically significant difference, p = 0.05. (C) LDM fractions from cells infected with retrovirus delivering either random shRNA or mVps45 shRNA #1 were prepared and then further fractionated using iodixanol gradients as described. Fractions were collected from the bottom (fraction 1) to top (fraction 14) of the tubes and analyzed for GLUT4 levels by immunoblotting. Data from a typical experiment are shown, repeated with similar results. Note that fractions 1–5 are enriched in GSVs, and fractions 10–14 are enriched in markers for recycling endosomes (see text for details). Note that the two blots are not identical exposures, as GLUT4 levels are reduced in mVps45 knockdown cells. (D) Levels of GLUT4 in GSVs (fractions 1–5) expressed as percentage of the total across all fractions.
To further test this hypothesis, we used a well-characterized in vitro budding assay to recapitulate the formation of GSVs (Shi et al., 2008). In brief, a membrane fraction (donor membranes) is prepared from a 16,000 × g centrifugation of a homogenate of 3T3-L1 adipocytes. After washing, this fraction is incubated at 37°C with ATP and cytosol from 3T3-L1 adipocytes. After incubation, the donor membranes are resedimented, and any GSVs that are formed remain in the supernatant. We first reproduced this assay and showed that the formation of GSVs is both cytosol and ATP dependent (Figure 6A). We repeated this assay using donor membranes or cytosol from control (random shRNA-treated) cells or mVps45-depleted cells. Using donor membranes and cytosol from control cells resulted in the formation of GLUT4-containing vesicles. Substitution of the control cytosol with cytosol from mVps45-depleted cells consistently reduced the incorporation of GLUT4 into GSVs but did not completely inhibit this process (Figure 6B), presumably reflecting the presence of mVps45 in the donor membranes. The use of donor membranes from mVps45-depleted cells, however, completely abrogated GLUT4 sorting into GSVs (Figure 6B). These data are consistent with mVps45 functioning in trafficking step(s) that culminate with the sorting of GLUT4 into GSVs.
FIGURE 6:
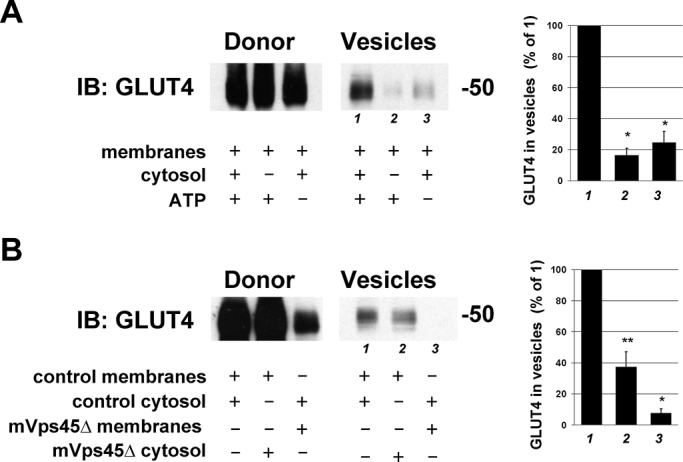
Impaired GLUT4 trafficking revealed using an in vitro assay. (A) Budding assays were performed as described in Materials and Methods first using control cells. Donor membranes from control cells were incubated in the presence of cytosol and ATP (or certain components omitted as indicated on the figure) and incubated at 37°C for 20 min before centrifugation to repellet the donor membranes. GLUT4 levels in the donor and supernatant (Vesicle) fractions were assayed by immunoblotting. Data from a typical experiment are shown, repeated three times with similar results. GLUT4 levels in the vesicles (lanes 1–3) were quantified by image analysis and expressed as percentage of the value for lane 1. Values of mean ± SD are shown in the graph to the right. Asterisk indicates a significant reduction compared with lane 1, p = 0.01. (B) The same assay, this time using combinations of donor and cytosol fractions isolated from random shRNA-treated cells or mVps45-depleted cells. The indicated fractions were incubated in the presence of an ATP-regenerating system at 37C for 20 min and the donor and vesicle fractions immunoblotted for GLUT4. The blot shown is from a representative experiment repeated five times with quantitatively similar results. GLUT4 levels in the vesicles (lanes 1–3) were quantified by image analysis and expressed as percentage of the value for lane 1. Values of mean ± SD are shown in the graph to the right. Statistically significant reductions are indicated by *p = 0.01 and **p = 0.05.
To determine whether the defective sorting observed for GLUT4 was specific or reflected generalized disruption of endosomal traffic, we examined the trafficking of TfRs using a fluorescence-based assay. Assay of cell surface TfR levels in the presence and absence of insulin revealed that the modest insulin-stimulated increase in plasma membrane (PM) TfR levels reported by us and others (Tanner and Lienhard, 1987; Foran et al., 1999; Perera et al., 2003) remained intact upon mVps45 depletion (Figure 7A). However, the absolute levels of TfR at the PM appeared lower in mVps45-knockdown cells (29 ± 4% decrease over four experiments of this type, p < 0.05). Analysis of TfR recycling revealed that mVps45 depletion appeared to slightly reduce the recycling rate, but this did not reach statistical significance in every experiment; Figure 7B). Hence we conclude that mVps45 depletion has minimal effect on TfR recycling and insulin responsiveness.
FIGURE 7:
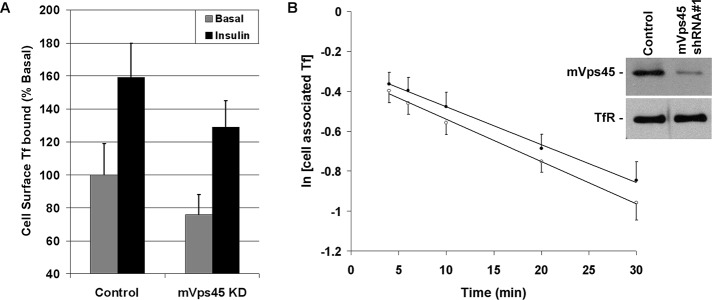
TfR trafficking is not significantly perturbed by mVps45 knockdown. (A) Data from a representative experiment assaying levels of cell surface Texas red Tf binding under basal or insulin-stimulated (100 nM for 30 min) conditions in control or mVps45-knockdown cells. Data are presented as percentage of the signal observed in basal control cells; each point is the mean of triplicate determinations at each condition. The experiment was repeated a further two occasions with similar results. The lower cell surface binding values observed in knockdown cells were significant in all experiments. (B) Recycling of Texas red Tf in control (open circles) and mVps45-knockdown (filled circles) cells. Briefly, cells were incubated with Texas red Tf for 3 h, surface-associated Tf was removed by a brief acid wash, and then cells were incubated in recycle buffer for the times shown. At each time, the Texas red Tf released into the media and that remaining cell associated were quantified in quadruplicate. The data from a typical experiment of this type are shown, in which the logarithm of the cell-associated Tf is plotted as a function of time. Inset, immunoblots of lysates of the same cells probed with anti-mVps45 or anti-TfR. Four experiments of this type revealed no statistically significant difference between the two conditions.
DISCUSSION
Intracellular retention of GLUT4 is a key feature of this facilitative glucose transporter. By sorting GLUT4 into insulin-responsive GSVs, adipocytes and muscle are able to quickly respond to insulin by delivering GLUT4 to the plasma membrane (Bryant et al., 2002; Bryant and Gould, 2011; Stockli et al., 2011). Hence the ability to sequester GLUT4 away from recycling endosomes represents an important regulatory step. Many of the molecular details of this process, however, are unclear. Recent studies from this group revealed a role for the intracellular t-SNAREs Sx6 and Sx16 in GLUT4 sorting. Depletion of Sx16 results in a diminution of insulin-stimulated glucose transport and a reduction in cellular GLUT4 levels (Proctor et al., 2006). We postulated that Sx16 is therefore required for the correct intracellular trafficking of GLUT4 and that depletion of Sx16 results in missorting of GLUT4 into a degradative pathway. Consistent with this, others have shown that intracellular GLUT4 sorting involves a Sx16-positive subdomain of the trans-Golgi network (Shewan et al., 2003). Sx16 is a Qa-SNARE that interacts with the Sec1/Munc18 protein mVps45 (Tellam et al., 1997). Because SM proteins act to regulate the activity and function of SNARE-dependent trafficking events, we sought to determine whether mVps45 regulates GLUT4 sorting and, if so, at what step(s) this SM protein may act.
We first quantified the expression of Sx16 and mVps45 in cells using quantitative immunoblotting with recombinant Sx16 or mVps45 as standards to determine an approximate level of expression of these proteins. As shown in Supplemental Table S1, expression of both Sx16 and mVps45 increases as 3T3-L1 fibroblasts differentiate into adipocytes, with levels of mVps45 increasing ∼10-fold and Sx16 ∼6-fold. In both 3T3-L1 adipocytes and freshly isolated rat adipocytes, Sx16 and mVps45 are expressed at approximately stoichiometric levels, as would be predicted from studies in other systems.
We chose to use shRNA, delivered into growing 3T3-L1 fibroblasts using a retrovirus, to knock down mVps45 levels in an attempt to ascertain the function of this SM protein in GLUT4 traffic. As shown in Figure 2, we were able to identify two shRNA species that effectively knocked down mVps45 levels in cells subsequently differentiated into adipocytes. As shown (Figure 2), depletion of mVps45 resulted in a marked decrease in cellular GLUT4 levels. In contrast, depletion of mVps45 had minimal effect on markers for the general endosomal system, other SNAREs, or phenotypic markers for adipocyte differentiation (Figure 2 and Supplemental Table S1). As would be expected, the reduction in GLUT4 levels was accompanied by reduced insulin-stimulated glucose transport and GLUT4 translocation despite apparently normal insulin signaling to Akt (Figure 3). These data make a compelling case for a requirement for mVps45 in the maintenance of intracellular GLUT4 homeostasis, presumably involving mVps45 regulating one (or more) GLUT4-specific intracellular sorting steps. We propose that knockdown of mVps45 changed the trafficking itinerary of GLUT4 so that it enters a degradative pathway. Future analysis of the half-life of GLUT4 in Sx16- or mVps45-depleted cells will be informative in this regard.
Given that GLUT4 has been reported to cycle through a Sx16-positive subdomain of the trans-Golgi network (Shewan et al., 2003), we reasoned that mVps45 may act to control the sorting of GLUT4 from the TGN to GSVs. Using a simple subcellular fractionation procedure, we found that the amount of total GLUT4 found in a GSV-enriched fraction is markedly decreased in mVps45-depleted cells (Figure 5; Lamb et al., 2010). Although total levels of GLUT4 are lower in mVps45-depleted cells, it is clear from this analysis that the sorting of the remaining GLUT4 is also impaired. This impairment appears selective for GLUT4, as TfR levels in the respective fractions are unchanged by mVps45 depletion (Figure 5). Iodixanol gradient analysis supported this contention, with a clear reduction in the fraction of intracellular GLUT4 localized within GSV-enriched fractions (Figure 5).
We sought to confirm these finding using an in vitro budding assay for GSV formation first described by the Kandror laboratory (Shi et al., 2008). We compared the effect of using cytosol from control or mVps45-depleted cells on the budding of GSVs from control or mVps45-depleted donor membranes. As shown in Figure 6B, cytosol lacking mVps45 resulted in a significant impairment of GLUT4 sorting into GSVs and is consistent with our model that mVps45 functions to regulate a step involved in the sorting or trafficking of GLUT4 into GSVs. It is important to note that the sorting of GLUT4 into GSVs reported by this assay likely involves several budding/fusion steps (e.g., between recycling endosomes and the TGN, TGN to GSVs, etc.). Although we cannot define precisely the step(s) regulated by mVps45, our data clearly reveal a requirement for this SM protein in the trafficking pathway leading to GSV formation. This experiment also supports the view that SM proteins cycle on and off membranes during membrane trafficking (Bryant and James, 2003), as we postulate that low levels of mVps45 in the cytosolic fraction of mVps45-knockdown cells are inhibitory for GLUT4 sorting into GSVs. This action of mVps45 would appear to be selective for GLUT4 and does not reflect a general effect on endosomes, since TfR trafficking is not significantly impaired upon mVps45 knockdown (Figures 5 and 7) and cellular levels of other endosomal proteins are not perturbed upon mVps45 knockdown (Figure 2 and Supplemental Table S1).
In sum, we show that mVps45 levels increase during adipocyte differentiation and that mVps45 and Sx16 are expressed in stoichiometric amounts. Depletion of mVps45 significantly reduced total cell GLUT4 levels, abrogated insulin-stimulated glucose transport and GLUT4 translocation, and resulted in missorting of GLUT4 such that levels within a GSV-enriched fraction were markedly reduced. Furthermore, the sorting of GLUT4 into GSVs is markedly impaired in an in vitro assay. We therefore propose that mVps45 is an important regulatory molecule in GLUT4 sorting, acting in concert with Sx16 to control entry of GLUT4 into GSVs.
MATERIALS AND METHODS
Cells, antibodies, and plasmid constructs
3T3-L1 fibroblasts were obtained from the American Tissue Culture Collection (Manassas, VA) and grown and differentiated into adipocytes as outlined in Perera et al. (2003) and Proctor et al. (2006). Anti-GLUT4, anti-mVps45, anti-VAMP3, anti-VAMP4, and syntaxin 16 antibodies were from Synaptic Systems (Göttingen, Germany), anti-SNAP23 and anti-syntaxin 4 were from Abcam (Cambridge, MA), and anti-Vti1a and 1b, anti-tubulin, and anti-Rab4 were from BD Biosciences (San Diego, CA). Anti–PPAR-γ was from Santa Cruz Biotechnology (Santa Cruz, CA), anti–Rabenosyn 5 was from Sigma-Aldrich (Poole, United Kingdom), anti-akt and anti–phospho-Akt S473 were from Cell Signaling (Beverly, MA), and phospho-AS160 T642 and anti-AS160 were from Millipore (Billerica, MA). All other antibodies were as described in Proctor et al. (2006). HA-GLUT4-GFP in the pRRL-PGK plasmid was from Cynthia Mastick (University of Nevada, Reno), and rat cDNA encoding mVps45 was from Robert Piper (University of Iowa, Iowa City). Hexahistidine-tagged mVps45 was generated using PCR, fully sequenced on both strands, and cloned into pQE30 for expression in bacteria. All other cDNAs were as previously outlined (Brandie et al., 2008; Aran et al., 2009). Insulin was from Sigma-Aldrich. Rat epididymal adipocytes were prepared as described in Collison et al. (2000).
Preparation and characterization of shRNA viruses to knock down mVps45
shRNA oligonucleotides were designed to knock down mouse vesicular transport protein mVps45 using a retroviral vector system (Clontech, Mountain View, CA). We chose 19–base pair sequences that were not near the start codon or in untranslated regions and had between 40 and 60% GC content. Four sets of target sequences were generated using these criteria and checked for secondary structures and long base runs. For each target, two complementary oligonucleotides were synthesized with a 5′-BamHI restriction site overhang on the top strand and a 5′-EcoRI restriction site overhang on the bottom strand. The target sense sequence had an added G at the 5′ end and a nucleotide hairpin loop sequence (5′-TTCAAGAGA-3′). The target antisense sequence had a terminator sequence of a five- to six-nucleotide (nt) poly(T) tract. Each target sequence then consisted of a restriction site (five or six bases) at the 5′ end, 19 bases of sense strand, seven to nine bases of hairpin loop, 19 bases of antisense strand, six bases of terminator poly(T), and six bases of restriction site at the 3′ end (Clontech). Four such targets were generated, separated from one another by at least 200 base pairs. The oligonucleotides were synthesized by (Integrated DNA Technologies, Munich, Germany) and ligated into RNAi-Ready pSIREN-RetroQ vector (Clontech). The 19-nt sequences were as follows: shRNA #1, GGTATAGTGAGTATGGTCT; shRNA #2, CGGTGAATCCACAATTTGT; shRNA #3, CAGTAATGTGATCAGCAAG; and shRNA #4, AGTGGACCTCAGGAGTAAA. pSIREN-RetroQ containing a scrambled shRNA sequence was used as a control.
EcoPack2-293 cells (American Tissue Culture Collection) were grown on collagen-coated T150 flasks before being subcultured into six-well plates at a density of 30,000 cells/well in complete medium (DMEM, 10% fetal calf serum, 1 mM sodium pyruvate, 1% [vol/vol] 10,000 U/ml penicillin/streptomycin). shRNA constructs were transfected into these viral packaging cells using Lipofectamine-2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. After 24 h, medium was removed from the cells and stored, and fresh complete medium was applied for a further 24 h. The conditioned medium was centrifuged at 2000 × g for 5 min, and the supernatant was collected and stored at 4°C. After 48 h, the second addition of complete media was similarly processed. It was then pooled with the previous supernatant, 1 mg/ml Polybrene was added, and it was stored at 4°C. Viral titers were determined in NIH 3T3 cells (Clontech) as described by the manufacturer.
3T3-L1 fibroblasts were grown in 10-cm plates and infected with 5 ml/plate of 2 × 106/ml colony-forming units of virus per target in growth medium (10% newborn calf serum/DMEM). Cells were incubated with virus for 24 h, and then the medium was changed to growth medium containing 2.5 μg/ml puromycin (Sigma-Aldrich). Cells were subsequently grown to confluence and differentiated as described for noninfected cells and used for experiments between days 8 and 11 postdifferentiation. In all experiments, cells infected with specific virus (shRNAs #1 and #2 were used routinely) were compared with cells infected with scrambled shRNA or noninfected cells.
Hemagglutinin-GLUT4-GFP cells
Hemagglutinin (HA)-GLUT4-GFP lentivirus was prepared using the ViraPower Lentiviral system (Invitrogen) starting from a plasmid provided by Cynthia Mastick (Muretta et al., 2008). 293T cells (American Tissue Culture Collection) were cotransfected with the HA-GLUT4-GFP plasmid and ViraPower packaging mix using Lipofectamine 2000. Lentivirus-containing supernatants were harvested 72 h after transfection and used to infect 3T3-L1 fibroblasts as outlined in Muretta et al. (2008). In experiments using retroviruses to knock down mVps45, this knockdown was performed in 3T3-L1 fibroblasts previously infected with HA-GLUT4-GFP lentivirus.
Recombinant protein production
Syntaxin 16 lacking its transmembrane domain was subcloned in-frame with a protein A tag and purified on immunoglobulin G–Sepharose (GE Healthcare, Piscataway, NJ) as outlined in Brandie et al. (2008). The entire coding sequence of rat mVps45 was amplified by PCR and ligated into the hexahistidine tag vector pQE30 and fully sequenced on both strands. Histidine- tagged mVps45 was produced in Escherichia coli BL-21 (DE3) after induction with isopropyl-β-d-thiogalactoside at 22°C overnight. The next day, bacteria were harvested and lysed, and mVps45 was isolated using nickel–nitriloacetic acid beads (Qiagen, Valencia, CA) according to the manufacturer's instructions. Protein levels were determined using Micro-BCA kits (Pierce, Loughborough, United Kingdom).
Subcellular fractionation
For quantification of Sx16 and mVps45 levels of expression in membrane and cytosol fraction, 10-cm plates of 3T3-L1 adipocytes were washed in ice-cold HES buffer (25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.4, 250 mM sucrose, 1 mM EDTA) and then scraped in the same buffer containing proteinase inhibitors. The cells were homogenized using a Balch-type homogenizer (16-μm clearance) and the homogenate centrifuged at 1000 × g for 5 min to pellet nuclei and insoluble material. The supernatant from this spin was then further sedimented at 100,000 × g for 1 h to pellet all membranes. The supernatant from this step was collected (cytosolic fraction) and precipitated using trichloroacetic acid. Protein levels in these fractions was determined using the micro bicinchoninic acid (BCA) method.
To determine the distribution of GLUT4 at a crude level, 10-cm plates of cells were washed and homogenized as outlined and centrifuged at 1000 × g for 5 min to pellet nuclei and insoluble material. The supernatant from this step was subsequently centrifuged at 16,000 × g and the pellet and supernatant fractions screened by SDS–PAGE and immunoblotting. The supernatant from this step is enriched for GSVs (Lamb et al., 2010).
Subcellular fractionation into plasma membrane, high- and low-density membranes, and cytosolic fractions was performed using a well-characterized procedure (Simpson et al., 1983; Perera et al., 2003). Iodixanol gradient centrifugation analysis of freshly isolated LDM fractions from control or mVps45-knockdown cells was performed as described (Hashiramoto and James, 2000; Perera et al., 2003). To quantify the fraction of GLUT4 within the GSV fractions, the immunoblot signal derived from lanes 1–5 was expressed as a fraction of the total GLUT4 signal across all fractions.
Budding assays
The formation of GSVs can be assayed in vitro using a budding assay (Shi et al., 2008). A donor membrane fraction is incubated with cytosol and an ATP-regenerating system; GSVs form in a cytosol- and ATP-dependent manner that can be readily separated from the donor membrane fraction. In brief, on day 8 postdifferentiation, 3T3-L1 adipocytes previously infected with either random shRNA virus or shRNA #1 virus were serum starved for 2 h, washed twice in cold budding buffer (38 mM potassium aspartate, 38 mM potassium glutamate, 20 mM 3-(N-morpholino)propanesulfonic acid, 5 mM sodium carbonate, 2.5 mM magnesium sulfate, 5 mM reduced glutathione, and a protease inhibitor tablet, pH 7.2), scraped into budding buffer, and passed through a 26-gauge needle 10 times and centrifuged at 1000 × g for 5 min at 4°C. The supernatants from this spin were transferred to fresh tubes and further sedimented at 16,000 × g for 20 min at 4°C. The pellets were retained, twice washed in budding buffer, and then resuspended in budding buffer and kept on ice (hereafter this material is referred to as the “donor membrane fraction”). During this procedure, parallel plates of adipocytes were scraped and homogenized as described, but after the 1000 × g spin, the supernatant was sedimented at 100,000 × g for 1 h at 4°C. The supernatant from this step is referred to as the “cytosol fraction.” Both membrane and cytosol fractions were analyzed for protein content using the micro BCA method.
An ATP-regeneration system (800 mM creatine phosphate, 500U/ml creatine kinase, 100 mM ATP; all from Sigma-Aldrich) was combined with the cytosol and membrane fractions in a reaction consisting of 250 μg of membrane, 2 mg/ml cytosol, and 0.3% (vol/vol) ATP regeneration system. The reactions were incubated at 37°C for 20 min before being centrifuged at 16,000 × g for 20 min at 4°C. The resultant pellets (the “donor fraction”) were then resuspended in Laemmli sample buffer. The supernatants (which contain newly formed GSVs budding from the donor membrane fraction) were recentrifuged at 100,000 × g for 1 h at 4°C and the pellets (the “vesicle fraction”) resuspended in Laemmli sample buffer. Donor and vesicle fractions were then subjected to SDS–PAGE and immunoblot analysis.
Transferrin recycling assays
Relative transferrin (Tf) levels at the cell surface were determined using Texas red Tf (Molecular Probes, Eugene, OR) binding. Briefly, cells were incubated in serum-free medium for 2 h with or without 100 nM insulin for the final 30 min. Thereafter, cells were washed three times in ice-cold 20 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM KCl, and 1 mM MgCl2 and incubated in the same buffer containing Texas red Tf (250 μg/ml) or Texas red Tf (250 μg/ml) plus holo-Tf (Sigma-Aldrich; 25 mg/ml) on ice for 3 h. Cells were washed five times in ice-cold buffer and the fluorescence associated with each well determined using a FLUOstar OPTIMA (BMG Labtech, Ortenberg, Germany). For transferrin-recycling experiments, cells were incubated in serum-free medium containing Texas red Tf (250 μg/ml) or Texas red Tf (250 μg/ml) plus holo-Tf (25 mg/ml) for 6 h at 37°C. Cells were quickly washed twice in 20 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM KCl, and 1 mM MgCl2 and then acid washed with ice-cold buffer (200 mM NaCl, 50 mM 2-(N-morpholino)ethanesulfonic acid, pH 5). Cells were then rapidly rewarmed into recycle buffer (220 mM NaHCO3, 20 mM HEPES, pH 7.4, 250 μg/ml holotransferrin, and 100 μM desferrioxamine [Sigma-Aldrich]) and incubated for the times shown in the figure legends. At the end of each incubation, Texas red Tf in the media was assayed, together with levels of Texas red Tf in the cell lysates. All experimental points were performed in quadruplicate.
Other methods
SDS–PAGE and immunoblotting were performed as described (Perera et al., 2003; Proctor et al., 2006). Immunoblot signals were quantified using ImageJ (National Institutes of Health, Bethesda, MD) software from multiple exposures of x-ray film to ensure linearity of signal used for quantification. Deoxyglucose transport assays were performed exactly as outlined (Proctor et al., 2006). Confocal microscopy of HA-GLUT4-GFP–expressing cells was performed on a Zeiss Exciter equipped with a 63× lens (Carl Zeiss, Jena, Germany). Images were captured and processed using Zeiss software.
Supplementary Material
Acknowledgments
This work was supported by a Colin MacArthur Studentship award (for J.R.), the Diabetes UK Arthur and Sadie Pethybridge Studentship (for J.B.A.S.), and grants from Diabetes UK to G.W.G. and N.J.B. N.J.B. is a Prize Fellow of the Lister Institute of Preventive Medicine. We thank Rob Piper for the mVps45 cDNA, Cynthia Mastick for the HA-GLUT4-GFP lentiviral plasmid, Silvia Bijland for assistance with the phospho-Akt/AS160 blots, and Claire Miller for technical assistance.
Abbreviations used:
- GLUT4
glucose transporter isoform 4
- GSV
GLUT4 storage vesicles
- PM
plasma membrane
- SM
Sec1/Munc18
- SNARE
soluble N-ethylmaleimide–sensitive factor attachment protein receptor
- Sx
syntaxin
- Tf
transferrin
- TfR
transferrin receptor
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-01-0011) on June 5, 2013.
REFERENCES
- Aran V, Brandie FM, Boyd AR, Kantidakis T, Rideout EJ, Kelly SM, Gould GW, Bryant NJ. Characterisation of two distinct binding modes between syntaxin 4 and Munc18c. Biochem J. 2009;419:655–660. doi: 10.1042/BJ20082293. [DOI] [PubMed] [Google Scholar]
- Bock JB, Klumperman J, Davanger S, Scheller RH. Syntaxin 6 functions in trans-Golgi network vesicle trafficking. Mol Biol Cell. 1997;8:1261–1271. doi: 10.1091/mbc.8.7.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan JS. Regulation of glucose transporter translocation in health and diabetes. Annu Rev Biochem. 2012;81:507–532. doi: 10.1146/annurev-biochem-060109-094246. [DOI] [PubMed] [Google Scholar]
- Brandie FM, Aran V, Verma A, McNew JA, Bryant NJ, Gould GW. Negative regulation of syntaxin4/SNAP-23/VAMP2-mediated membrane fusion by Munc18c in vitro. PLoS ONE. 2008;3:e4074. doi: 10.1371/journal.pone.0004074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant NJ, Gould GW. SNARE proteins underpin insulin-regulated GLUT4 traffic. Traffic 12, 657–664. 2011 doi: 10.1111/j.1600-0854.2011.01163.x. [DOI] [PubMed] [Google Scholar]
- Bryant NJ, Govers R, James DE. Regulated trafficking of the glucose transporter, Glut4. Nat Rev Mol Cell Biol. 2002;3:267–277. doi: 10.1038/nrm782. [DOI] [PubMed] [Google Scholar]
- Bryant NJ, James DE. Vps45p stabilizes the syntaxin homologue Tlg2p and positively regulates SNARE complex formation. EMBO J. 2001;20:3380–3388. doi: 10.1093/emboj/20.13.3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant NJ, James DE. The Sec1p/Munc18 (SM) protein, Vps45p, cycles on and off membranes during vesicle transport. J Cell Biol. 2003;161:691–696. doi: 10.1083/jcb.200212078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Reinsisch K, Ferro-Novick S. Coats, tethers, Rabs and SNAREs work together to mediate the intracellular destination of a transport vesicle. Dev Cell. 2007;12:671–682. doi: 10.1016/j.devcel.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Carpp LN, Ciufo LF, Shanks SG, Boyd A, Bryant NJ. The Sec1p/Munc18 protein Vps45p binds its cognate SNARE proteins via two distinct modes. J Cell Biol. 2006;173:927–936. doi: 10.1083/jcb.200512024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collison M, Glazier AM, Aitman TJ, Scott J, Graham D, Morton JJ, Dominiczak MH, Connell JMC, Gould GW, Dominiczak AF. Cd36 and molecular mechanisms on insulin resistance in the stroke-prone spontaneously hypertensive rat. Diabetes. 2000;49:2222–2226. doi: 10.2337/diabetes.49.12.2222. [DOI] [PubMed] [Google Scholar]
- El-Jack AK, Kandror K, Pilch PF. The formation of an insulin-responsive vesicular cargo compartment is an early event in 3T3-L1 adipocyte differentiation. Mol Biol Cell. 1999;10:1581–1594. doi: 10.1091/mbc.10.5.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foran PGP, Fletcher LM, Oatey PB, Mohammed N, Dolly JO, Tavaré JM. Protein kinase B stimulates the translocation of Glut4 but not Glut1 or transferrin receptors in 3T3-L1 adipocytes by a pathway involving SNAP-23, synaptobrevin-2, and/or cellubrevin. J Biol Chem. 1999;274:28087–28095. doi: 10.1074/jbc.274.40.28087. [DOI] [PubMed] [Google Scholar]
- Graham TE, Kahn BB. Tissue-specific alterations of glucose transport and molecular mechanisms of intertissue communication in obesity and type 2 diabetes. Horm Metab Res. 2007;39:717–721. doi: 10.1055/s-2007-985879. [DOI] [PubMed] [Google Scholar]
- Hashiramoto M, James DE. Characterization of insulin-responsive GLUT4 storage vesicles isolated from 3T3-L1 adipocytes. Mol Cell Biol. 2000;20:416–427. doi: 10.1128/mcb.20.1.416-427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson GRX, Chamberlain LH, Maier VH, Gould GW. Quantification of SNARE protein levels in 3T3-L1 adipocytes: implications for insulin-stimulated glucose transport. Biochem Biophys Res Commun. 2000;270:841–845. doi: 10.1006/bbrc.2000.2525. [DOI] [PubMed] [Google Scholar]
- Hong W. SNAREs and traffic. Biochim Biophys Acta. 2005;1744:493–517. [PubMed] [Google Scholar]
- Hu C, Ahmed M, Melia TJ, Sollner TH, Mayer T, Rothman JE. Fusion of cells by flipped SNAREs. Science. 2003;300:1745–1749. doi: 10.1126/science.1084909. [DOI] [PubMed] [Google Scholar]
- Lamb CA, McCann RK, Stockli J, James DE, Bryant NJ. Insulin-regulated trafficking of GLUT4 requires ubiquitination. Traffic. 2010;11:1445–1454. doi: 10.1111/j.1600-0854.2010.01113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingstone C, James DE, Rice JE, Hanpeter D, Gould GW. Compartment ablation analysis of the insulin responsive glucose transporter, GLUT4, in 3T3-L1 adipocytes. Biochem J. 1996;315:487–495. doi: 10.1042/bj3150487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison HA, Dionne H, Rusten TE, Brech A, Fisher WW, Pfeiffer BD, Celniker SE, Stenmark H, Bilder D. Regulation of early endosomal entry by the Drosophila tumor suppressors Rabenosyn and Vps45. Mol Biol Cell. 2008;19:4167–4176. doi: 10.1091/mbc.E08-07-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muretta JM, Romenskaia I, Mastick CC. Insulin releases Glut4 from static storage compartments into cycling endosomes and increases the rate constant for Glut4 exocytosis. J Biol Chem. 2008;283:311–323. doi: 10.1074/jbc.M705756200. [DOI] [PubMed] [Google Scholar]
- Murray RZ, Wylie FG, Khromykh T, Hume DA, Stow JL. Syntaxin 6 and Vti1b Form a novel SNARE complex, which is up-regulated in activated macrophages to facilitate exocytosis of tumor necrosis factor-{alpha} J Biol Chem. 2005;280:10478–10483. doi: 10.1074/jbc.M414420200. [DOI] [PubMed] [Google Scholar]
- Perera HKI, Clark M, Morris NJ, Hong W, Chamberlain LH, Gould GW. Syntaxin 6 regulates Glut4 trafficking in 3T3-L1 adipocytes. Mol Biol Cell. 2003;14:2946–2958. doi: 10.1091/mbc.E02-11-0722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper RC, Tai C, Slot JW, Hahn CS, Rice CM, Huang H, James DE. The efficient intracellular sequestration of the insulin-regulatable glucose transporter (GLUT-4) is conferred by the NH2 terminus. J Cell Biol. 1992;117:729–743. doi: 10.1083/jcb.117.4.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor KM, Miller SCM, Bryant NJ, Gould GW. Syntaxin 16 controls the intracellular sequestration of GLUT4 in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2006;347:433–438. doi: 10.1016/j.bbrc.2006.06.135. [DOI] [PubMed] [Google Scholar]
- Rahajeng J, Caplan S, Naslavsky N. Common and distinct roles for the binding partners Rabenosyn-5 and Vps45 in the regulation of endocytic trafficking in mammalian cells. Exp Cell Res. 2009;316:859–874. doi: 10.1016/j.yexcr.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scales SJ, Chen YA, Yoo BY, Patel SM, Doung Y-C, Scheller RH. SNAREs contribute to the specificity of membrane fusion. Neuron. 2000;26:457–464. doi: 10.1016/s0896-6273(00)81177-0. [DOI] [PubMed] [Google Scholar]
- Shewan AM, van Dam EM, Martin S, Luen TB, Hong W, Bryant NJ, James DE. GLUT4 recycles via a trans-Golgi network (TGN) subdomain enriched in syntaxins 6 and 16 but not TGN38: Involvement of an acidic targeting motif. Mol Biol Cell. 2003;14:973–986. doi: 10.1091/mbc.E02-06-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Huang G, Kandror KV. Self-assembly of Glut4 storage vesicles during differentiation of 3T3-L1 adipocytes. J Biol Chem. 2008;283:30311–30321. doi: 10.1074/jbc.M805182200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen A, Bremnes B, Ronning E, Aasland R, Stenmark H. Syntaxin-16, a putative Golgi t-SNARE. Eur J Cell Biol. 1998;75:223–231. doi: 10.1016/S0171-9335(98)80116-7. [DOI] [PubMed] [Google Scholar]
- Simpson IA, Yver DR, Hissin PJ, Wardzala LJ, Karnieli E, Salans LB, Cushman SC. Insulin-stimulated translocation of glucose transporters in isolated rat adipose cells: characterisation of subcellular fractions. Biochim Biophys Acta. 1983;763:393–407. doi: 10.1016/0167-4889(83)90101-5. [DOI] [PubMed] [Google Scholar]
- Stockli J, Fazakerley DJ, James DE. GLUT4 exocytosis. J Cell Sci. 2011;124:4147–4159. doi: 10.1242/jcs.097063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struthers MS, Shanks SG, MacDonald C, Carpp LN, Drozdowska AM, Kioumourtzoglou D, Furgason ML, Munson M, Bryant NJ. Functional homology of mammalian syntaxin 16 and yeast Tlg2p reveals a conserved regulatory mechanism. J Cell Sci. 2009;122:2292–2299. doi: 10.1242/jcs.046441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner LI, Lienhard GE. Insulin elicits a redistribution of transferrin receptors in 3T3-L1 adipocytes through an increase in the rate constant for receptor externalisation. J Biol Chem. 1987;262:8975–8980. [PubMed] [Google Scholar]
- Tellam JT, James DE, Stevens TH, Piper RC. Identification of a mammalian Golgi Sec1p-like protein, mVps45. J Biol Chem. 1997;272:6187–6193. doi: 10.1074/jbc.272.10.6187. [DOI] [PubMed] [Google Scholar]
- Tran THT, Zeng Q, Hong W. VAMP4 cycles from the cell surface to the trans-Golgi network via sorting and recycling endosomes. J Cell Sci. 2007;120:1028–1041. doi: 10.1242/jcs.03387. [DOI] [PubMed] [Google Scholar]
- Watson RT, Pessin JE. Recycling of IRAP from the plasma membrane back to the insulin-responsive compartment requires the Q-SNARE syntaxin 6 but not the GGA clathrin adaptors. J Cell Sci. 2008;121:1243–1251. doi: 10.1242/jcs.017517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- Wendler F, Page L, Urbe S, Tooze SA. Homotypic fusion of immature secretory granules during maturation requires syntaxin 6. Mol Biol Cell. 2001;12:1699–1709. doi: 10.1091/mbc.12.6.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D, Pessin JE. Mapping of R-SNARE function at distinct intracellular GLUT4 trafficking steps in adipocytes. J Cell Biol. 2008;180:375–387. doi: 10.1083/jcb.200709108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.