

In normal aging, nuclear sirtuin levels decline and specific histone acetylation increases. In diabetic mice, Sirt1 and Sirt6 levels increase, which may induce gluconeogenesis through the PGC-1α pathway and DNA repair of the oxidative damage caused by hyperglycemia.
Abstract
Hyperglycemia, like aging, induces chromatin remodeling in mouse hepatocytes in comparison to normoglycemia and younger age, respectively. Changes in glucose metabolism also affect the action and expression of sirtuins, promoting changes in chromatin conformation and dynamics. Here we investigate the abundance and activity of the nuclear sirtuins Sirt1, Sirt6, and Sirt7 in mouse hepatocytes in association with specific histone acetylation, DNA damage, and the activation of nucleolar organizing regions (NORs) in hyperglycemic nonobese diabetic (NOD) and old normoglycemic BALB/c mouse strains. Higher levels of Sirt1 and PGC-1α and increased expression of gluconeogenesis pathway genes are found in the hyperglycemic NOD mice. Increased Sirt6 abundance is found in the hyperglycemic NOD mice, which might increase DNA damage repair. With aging, lower Sirt1 abundance and activity, increased acetylated histone modifications and Sirt7 levels, and NOR methylation are found. Thus, whereas in normal aging cell metabolism is reduced, in the diabetic mice a compensatory mechanism may elevate Sirt1 and Sirt6 levels, increasing gluconeogenesis and DNA repair from the oxidative damage caused by hyperglycemia. Therefore understanding the regulation of epigenetic factors in diabetes and aging is crucial for the development of new therapeutic approaches that could prevent diseases and improve quality of life.
INTRODUCTION
Type 1 diabetes mellitus (T1DM) is an autoimmune disease caused by lymphocyte infiltration in the endocrine pancreas, which leads to the destruction of β-cells and consequently to hyperglycemia (Makino et al., 1980). This type of diabetes occurs mainly in childhood and puberty, and management requires daily insulin replacement therapy in addition to dietary measures and physical activity. According to the International Diabetes Federation, ∼480,000 children <15 yr of age and a similar number of individuals aged 15–25 yr have T1DM. In the liver, the absence of insulin signaling promotes deregulation in glucose and lipid metabolism, such as increased ketogenesis, gluconeogenesis, and decreased glycolysis, which result in a severe hyperglycemic state (Berg et al., 2002; Nelson and Cox, 2004).
Furthermore, changes in cell metabolism may affect chromatin organization. Mononucleated hepatocytes of severe nonobese diabetic (NOD) mice, for instance, can attain high levels of polyploidy and undergo chromatin remodeling (Mello et al., 2009; Ghiraldini et al., 2012), suggesting that hyperglycemia affects the chromatin organization of hepatocytes in a similar, but not identical, manner as found in aged mice (Moraes et al., 2007; Mello et al., 2009; Ghiraldini et al., 2012). Because increased hyperplasia and apoptosis have been reported in hepatocytes of diabetic rats (Herrman et al., 1999; Ahmed, 2005), it has been hypothesized that hyperglycemic animals might suffer from early aging (Blazer et al., 2005; Mello et al., 2009).
In aging, a number of modifications in gene expression and epigenetic changes are observed, such as loss of DNA methylation, chromatin remodeling, and histone modifications that facilitate this process (Cao et al., 2001; Sarg et al., 2002; Mehta et al., 2007; Moraes et al., 2007; Sedivy et al., 2007; Nakamura et al., 2010; Shin et al., 2011). In hepatocytes particularly, there is an increase in chromatin accessibility to micrococcal nuclease digestion and a global loss of DNA methylation with aging (Mehta et al., 2007; Ghiraldini et al., 2012).
Studies on NAD+-dependent deacetylase Sirt1, a mammalian orthologue of Sir2 in yeast, show that this protein is involved in aging and caloric restriction, increasing lifespan (Blander and Guarente, 2004; Haigis and Guarente, 2006). Although the sirtuin family comprises seven proteins, only Sirt1, Sirt6, and Sirt7 are nuclear residents (Schwer and Verdin, 2008). Because of the dependence of sirtuins on the cofactor NAD+, they serve as metabolic sensors, and changes in [NAD+]/[NADH] ratio affect their activity and, ultimately, chromatin structure and gene expression (Leibiger and Berggren, 2006; Imai, 2009).
Sirt1 deacetylates specific histone sites and nonhistone proteins, such as FoxO1 and PGC-1α. In the liver, the transcription factor PGC-1α is deacetylated, working as a bridge between metabolic changes and gene expression, increasing the expression of gluconeogenic and decreasing the expression of glycolytic genes (Rodgers et al., 2008). Sirt6 is another NAD-dependent deacetylase and is involved not only in cell metabolism and metabolic diseases such as diabetes, but also in stress resistance, lifespan, aging, and inflammation (Pfluger et al., 2008; Van Gool et al., 2009; Xiao et al., 2010). Despite its preferably ADP-ribosyltransferase activity, this sirtuin can deacetylate H3K9 and H3K56 sites, thus controlling the expression of multiple glycolytic genes (Zhong et al., 2010). Sirt6 is also involved in the DNA-damage repair system, or base excision repair, and is usually highly abundant in the presence of high amounts of reactive oxygen species (Mostoslavsky et al., 2006; Lombard et al., 2008; McCord et al., 2009; Zhong et al., 2010). Sirt6-deficient mice present DNA-damage hypersensitivity and genome instability, with progressive hypoglycemia culminating in an aged-like phenotype and subsequent death (Mostoslavsky et al., 2006).
Sirt7, on the other hand, located in nucleoli, is mostly abundant in the liver (Ford et al., 2006). This protein is associated with transcriptionally active rRNA genes and increases RNA polymerase I activity (Ford et al., 2006; Murayama et al., 2008; Grob et al., 2009, Tsai et al., 2012). In the liver, changes in the NAD+/NADH ratio can regulate Sirt7 to couple changes in energy status with levels of rRNA synthesis and ribosome production (Ford et al., 2006). During aging, an increase in ribosomal DNA (rDNA) detachment from the nuclear matrix of mouse hepatocytes could indicate decreased rDNA transcription, reflecting the cellular metabolism conditions (Moraes et al., 2010).
If chromatin organization is affected by hyperglycemia and aging in mouse hepatocytes, the same may be true for the abundance and activity of nuclear sirtuins, as well as their primary targets. Moreover, given the close relationship of Sirt1, Sirt6, and Sirt7 with cell metabolism, glucose homeostasis, and DNA integrity, it is important to address the consequences for their actions under a metabolic disorder context. Therefore the aim of this study is to compare the effects of hyperglycemia and those of aging on nuclear sirtuin abundance and activity, histone modification, DNA fragmentation, and nucleolar organization in mouse hepatocytes.
RESULTS
Hyperglycemia and aging affect Sirt1 abundance and activity differently
Sirt1 abundance and mRNA expression were increased in hepatocytes of severely hyperglycemic mice in comparison to normoglycemic controls, whereas old, normoglycemic animals presented a lower Sirt1 abundance than young-adult controls (Figure 1, A and D, and Supplemental Figure S1A).
FIGURE 1:

Sirt1 is differently affected in hepatocytes of diabetic adults and normoglycemic old mice. (A) Sirt1 relative abundance in young-adult BALB/c mouse hepatocyte nuclei as determined by Western blotting. (B) Sirt1 relative activity in hepatocyte nuclei compared with young-adult BALB/c, determined by the Sirt1 Activity Assay Kit. Positive control, resveratrol (12.5 mM). (C) NAD+/NADH ratio in mouse hepatocytes. (D) Sirt1 representative image of the immunoblotting. Image was chosen to specifically highlight the differences shown in the graph. Histone H3 was used as loading control. B, BALB/c; N, NOD; +, hyperglycemia; –, normoglycemia. *Difference significant at p < 0.05 by ANOVA. †Difference significant at p < 0.05 by Student's t test.
Sirt1 activity levels, assessed in order to determine whether Sirt1 abundance has an effect on protein activity, revealed no difference between the hepatocytes of the diabetic mice and the normoglycemic BALB/c and NOD controls. Old mice, however, presented a decrease in Sirt1 activity (Figure 1B). Because Sirt1 activity did not correspond to the increased abundance of this protein in the diabetic animals, the NAD+/NADH ratio was calculated to evaluate the availability of this molecule. In hepatocytes from diabetic mice, the NAD+/NADH ratio did not differ from that in normoglycemic animals, whereas in old, normoglycemic animals this ratio was decreased compared with young-adult controls, similar to results for Sirt1 activity (Figure 1C). In all experimental conditions, Sirt1 was identified especially in euchromatic and central regions of the nuclei (Supplemental Figure S2).
Abundance of PGC-1α and Sirt1-histone targets in hepatocytes of diabetic and normoglycemic old mice
Both diabetic and normoglycemic old mice presented the same PGC-1α abundance pattern in hepatocytes as that found for Sirt1; increased and decreased PGC-1α levels in diabetic and old mice, respectively, were found only in comparison to their controls (Figure 2, A and B). Like Sirt1, PGC-1α was observed in euchromatic and central regions of the hepatocyte nuclei in all experimental conditions (Supplemental Figure S2). mRNA of target molecules of PGC-1α was also analyzed to evaluate whether increased PGC-1α abundance reflected changes in gene expression. Pck1 and Foxo1 are positively regulated by PGC-1α (Puigserver et al., 2003; Herzog et al., 2005). As expected, their expression in hyperglycemic mice increased in comparison to that in normoglycemic animals. In old animals there was no change in the expression of both genes in comparison with young-adult BALB/c mice (Figure 2, C and D).
FIGURE 2:

PGC-1α presents the same abundance pattern as Sirt1 and activates its targets in hyperglycemic animals. (A) PGC-1α relative abundance, determined by Western blotting. *Difference significant at p < 0.05 by ANOVA. †Difference significant at p < 0.05 by Student's t test. (C) PGC-1α representative image of the immunoblotting. Image was chosen to specifically highlight the differences shown in the graph. B, BALB/c; N, NOD. Histone H3 was used as the loading control. +, Hyperglycemia; –, normoglycemia. (C) Pck1 and (D) Foxo1 expression, determined by quantitative PCR. *Difference significant in comparison to normoglycemic NOD mice at p < 0.05 by Student's t test.
The abundance of acetylated lysine 26 at histone H1 (H1K26Ac), lysine 16 at histone H4 (H4K16Ac), lysine 14 at histone H3 (H3K14ac), and lysine 9 at histone H3 (H3K9Ac), Sirt1-histone targets, was also evaluated (Figure 3, A–D and F). Acetylated lysine 56 at histone H3 (H3K56Ac), the Sirt6 target, was also analyzed (Figure 3, E and F). Except for H3K9Ac, no difference in histone modification abundance was found in the hepatocyte chromatin of hyperglycemic compared with normoglycemic mice (Figure 3, C and F). This epigenetic marker was more abundant in the hepatocytes of hyperglycemic mice. H3K14Ac was highly abundant in hyperglycemic mice compared with BALB/c normoglycemic controls but did not differ from NOD normoglycemic controls (Figure 3, D and F). With the exception of H1K26Ac, old mice presented increased abundance of all histone modifications analyzed in comparison to young-adult controls (Figure 3, A–F). To determine whether the position of these epigenetic marks in different chromatin areas differed between experimental conditions, we performed immunofluorescence analyses. The histone modifications selected were H3K9Ac and H4K16Ac because of their established position and high availability in hepatocyte nuclei. Both marks were located in euchromatic regions throughout the nuclei in all experimental conditions (Supplemental Figure S2).
FIGURE 3:

Abundance of Sirt1 histone targets in diabetic and old-mouse hepatocytes. (A) H1K26Ac, (B) H4K16Ac, (C) H3K9Ac, (D) H3K14Ac, and (E) H3K56Ac. The abundance of Sirt1 and Sirt6 histone targets was determined in Western blotting using young-adult BALB/c hepatocyte nuclei as control. (F) Representative image of the immunoblotting of H1K26Ac, H4K16Ac, H3K9Ac, H3K14Ac, and H3K56Ac. Image was chosen to specifically highlight the differences shown in the graph. B, BALB/c; N, NOD. Histone H3 was used as the loading control for histone H4 and H1 modifications, and H4 was used as the loading control for histone H3 modifications. +, Hyperglycemia; –, normoglycemia. *Difference significant at p < 0.05 by ANOVA. †Difference significant at p < 0.05 by Student's t test.
Hyperglycemia, but not aging, induces increase in Sirt6 abundance and DNA damage in mouse hepatocytes
Sirt6 abundance and expression increased in the hepatocytes of hyperglycemic mice but were less abundant in the hepatocytes of normoglycemic old mice in comparison to normoglycemic and young-adult animals, respectively (Figure 4, A and C). To establish a possible connection between Sirt6 abundance and DNA damage, we performed a comet assay. Higher levels of DNA damage and number of damaged hepatocytes were found in diabetic mice in comparison with normoglycemic mice of both strains (Table 1). However, no differences were found in normoglycemic aged mice compared with young adults for either parameter. Treatment with hydrogen peroxide (H2O2), used as positive control for the comet assay, induced increased DNA damage (>300%) and a higher percentage of damaged cells in all cases (>200%), except in hyperglycemic mice, which presented a nonsignificant increase in both parameters (Table 1).
FIGURE 4:
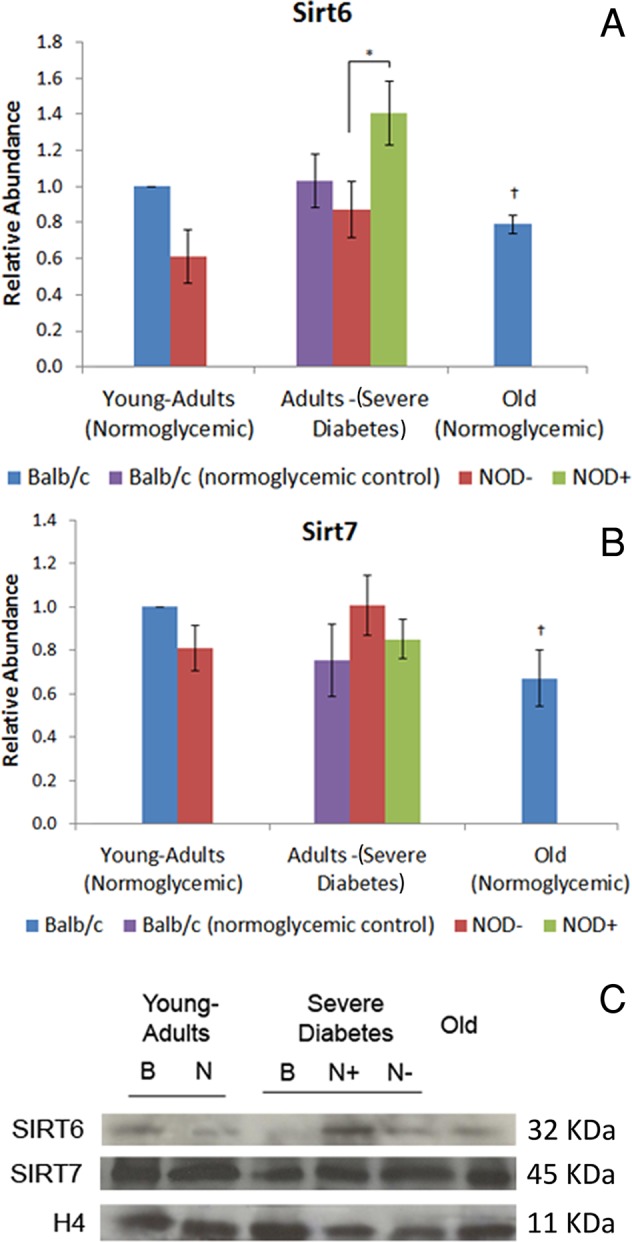
Abundance of Sirt6 and Sirt7 in diabetic and old-mouse hepatocytes. (A) Sirt6, (B) Sirt7. Sirtuin abundance was calculated in Western blotting using young-adult BALB/c hepatocyte nuclei as control. (C) Sirt6 and Sirt7 representative image of the immunoblotting. Image was chosen to specifically highlight the differences shown in the graph. Histone H4 was used as loading control. B, BALB/c; N, NOD; +, hyperglycemia; –, normoglycemia. *Difference significant at p < 0.05 by ANOVA. †Difference significant at p < 0.05 by Student's t test.
TABLE 1:
Percentage of damaged cells and DNA damage index in mouse hepatocytes after H2O2 treatment.
| Damaged cells (%) | DNA damage index | ||||
|---|---|---|---|---|---|
| Animals | H2O2 | Arithmetic mean | SE | Arithmetic mean | SE |
| Young-adult BALB/c | No | 21.38a | 04.7 | 034.72a | 08.60 |
| Yes | 53.75* | 14.6 | 111.97* | 29.50 | |
| Normoglycemic BALB/c | No | 20.67a | 03.0 | 037.58a | 06.11 |
| Yes | 53.40* | 12.4 | 105.09* | 24.30 | |
| Normoglycemic NOD | No | 22.58a | 04.1 | 040.89a | 09.32 |
| Yes | 56.30** | 11.5 | 128.72* | 31.50 | |
| Hyperglycemic NOD | No | 43.78b | 03.8 | 084.23b | 09.97 |
| Yes | 61.57* | 13.8 | 148.77* | 38.50 | |
| Old BALB/c | No | 25.46a | 02.6 | 048.34a | 06.27 |
| Yes | 63.09* | 14.5 | 159.75* | 30.60 | |
*Difference from H2O2 treatment significant at P < 0.05 (Student's t test); different subscript letters a and b in the same column indicate statistical differences significant at p < 0.05 (ANOVA).
Both hyperglycemia and aging induce a decrease in silver-stained nucleolar organizer region–positive area/nuclear area ratio, but only aging promotes decrease in Sirt7 abundance and increase in rDNA methylation
Sirt7 abundance and expression in the hepatocytes of diabetic mice did not differ from results in normoglycemic controls but was decreased in normoglycemic old mice compared with young adults (Figure 4, B and C). To establish a possible connection between Sirt7 and nucleolar activity, we performed a silver-stained nucleolar organizer region (AgNOR) assay to stain specific nucleolar proteins.
Both diabetic adults and normoglycemic old animals presented an increase in nuclear area in comparison with their respective controls, but this increase was more evident in the hepatocytes of the hyperglycemic mice (Figure 5, A and D). These mice, however, presented no differences in AgNOR-positive areas compared with normoglycemic animals, whereas normoglycemic NOD and BALB/c mice differed with regard to this parameter (Figure 5, B and D). Because the hepatocytes of old mice presented a decrease in AgNOR-positive areas as compared with young-adult animals, the AgNOR-positive area/nuclear area ratio decreased for both diabetic and old mice compared with normoglycemic and young adult animals, respectively (Figure 5, C and D).
FIGURE 5:

Nuclear and AgNOR-positive areas in diabetic and old-mouse hepatocytes. (A) Nuclear area. (B) AgNOR-positive area in imprinted mouse hepatocytes. (C) AgNOR area relative to nuclear area. +, Hyperglycemia; –, normoglycemia. *Difference significant at p < 0.05 by ANOVA. †Difference significant at p < 0.05 by Student's t test. (D) AgNOR-stained hepatocyte: 1, young-adult BALB/c; 2, old BALB/c; 3, hyperglycemic NOD. Bars, 10 μm.
To examine whether the decrease in AgNOR-positive areas in the hepatocytes of old mice was related to DNA methylation, we performed a native-chromatin immunoprecipitation (ChIP) assay to identify the abundance of DNA methylation in the 18S rDNA region. Diabetic animals did not show any difference in DNA methylation in the 18S rDNA region as compared with normoglycemic animals (Figure 6). Old normoglycemic BALB/c mice, however, showed a significant increase in this epigenetic mark in comparison to young-adult normoglycemic mice (Figure 6).
FIGURE 6:
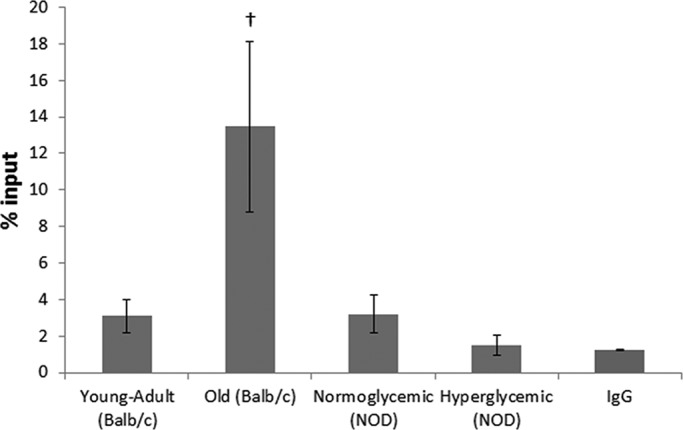
Native ChIP from mouse hepatocytes as percentage of input. IgG was used as mock control. †Difference significant at p < 0.05 by Student's t test.
DISCUSSION
Hyperglycemia and aging promote chromatin remodeling (Moraes et al., 2007; Mello et al., 2009). Although changes in polyploidy and chromatin packing states are observed in both cases, their magnitude is different (Ghiraldini et al., 2012). The results of the present study indicate that, among other epigenetic proteins involved in the modulation of chromatin functions, sirtuins play a role in coordinating changes in chromatin organization and function in the hepatocytes of diabetic and old mice.
Sirt1, in association with the transcription factor PGC-1α, modulates the gluconeogenesis and glycolysis pathway in the liver, acting as a nutritional sensor (Rodgers et al., 2008). PGC-1α binds either to transcriptor factors, like FoxO1, which activate genes involved in gluconeogenesis in hepatocytes (Puigserver et al., 2003), or to gene sequences responsible for transcription of key proteins of gluconeogenesis, like the enzyme phosphoenolpyruvate carboxykinase 1 (Pck1; Herzog et al., 2005). In the present study, Sirt1 and PGC-1α were more abundant in hyperglycemic mice than in normoglycemic controls and, as expected, PGC-1α targets were also more expressed in hyperglycemic than in normoglycemic mice. Although Sirt1 activity level was not as increased as expected when compared with protein abundance, analysis of the NAD+/NADH ratio indicated that this molecule could be limiting Sirt1 activity. In diabetic animals, the limitation of the NAD+ levels could be due to the higher rate of β-oxidation that occurs in hepatocytes in the absence of insulin signaling (Berg et al., 2002). In untreated cases of T1DM, the absence of insulin signaling could be interpreted by the cell as a fasting state (Nelson and Cox, 2004) despite glucose availability, leading to an increase in sirtuin abundance but not in its activity. In the liver, especially, glucose in excess could reduce this protein activity, as well as glycolysis (Chen et al., 2008). In old animals, the decrease in Sirt1 abundance and activity and NAD+/NADH ratio levels could indicate a general decrease in cell metabolism (Gonzalo, 2010; Shin et al., 2011). Other authors (Spindler et al., 2003; Braidy et al., 2011) also reported a decrease in gluconeogenesis and ketogenesis accompanied by a decrease in NAD+/NADH with aging, similar to results found here.
The histone modification H3K9Ac was the only Sirt1-histone target highly abundant in both diabetic and old-mice hepatocytes in comparison with normoglycemic animals from both strains and young adults, respectively. H3K9Ac, specifically, is a hallmark for gene expression activation (Nakamura et al., 2010). The abundance of this histone modification in diabetic mice could indicate a differentiated gene expression pattern that enables the organism to adapt to long periods of insulin absence. All of the histone modifications analyzed, with the exception of H1K26Ac, were more abundant in old animals, indicating that the decrease in Sirt1 abundance and activity might be related to an increase in the remaining acetylated histone sites, which might contribute to more unraveling of the chromatin state.
Like Sirt1, Sirt6 is a nuclear protein whose target is also the histone site H3K9Ac, besides the H3K56Ac modification (Michishita et al., 2009; Yang et al., 2009). Of interest, Sirt6 abundance in diabetic and old mice was similar to that observed for Sirt1, probably because Sirt1 and FoxO3a act directly on the Sirt6 gene, modulating its expression and consequently glycolysis, triglyceride synthesis, and fat metabolism rates (Kim et al., 2010). Sirt6-knockout mice present hypoglycemia by failing to repress the transcription factor Hifα, increasing the glucose capture and glycolysis rate (Zhong et al., 2010). In the present work, Sirt6 was found to be highly increased in hyperglycemic animals, a finding that could be related to the decreased glycolysis rate in diabetes. In diabetes, misbalance in ketogenesis leads to an accumulation of high amounts of reactive oxygen species, which could generate DNA damage (Berg et al., 2002), and it is known that Sirt6 might play a role in the base excision repair machinery (Mostoslavsky et al., 2006). Indeed, in hyperglycemic animals a great abundance of Sirt6 and a high percentage of damaged cells and elevated DNA damage index were found, as high as those in the positive control. Sirt6 abundance was lower in the old mice, similar to Sirt1. Reduction in Sirt6 abundance, followed by increase of its histone targets, H3K9Ac and H3K56Ac, is in accordance with the literature, given that Sirt6 has been related to early aging in knockout mice (Tennen and Chua, 2011).
Sirt7 is also a nuclear sirtuin occurring mostly in nucleoli and is involved with the activation of RNA polymerase I (Ford et al., 2006). There is no report, however, on the relative abundance of Sirt7 and NOR activation in the context of diabetes or aging. In diabetic animals, Sirt7 abundance was not different from that in normoglycemic animals, whereas in old mice it was decreased. Active NORs are usually observed in cycling cells, and this could be identified by the AgNOR technique due to nucleolar protein retention (McKeown and Schaw, 2009). In old-mouse hepatocytes, rDNA inactivation occurs, mainly because of the detachment of these DNA sequences from the nuclear matrix (Moraes et al., 2010). The decrease in the AgNOR-positive/nuclear area ratio that we observed in both diabetic and old animals was promoted by different factors: whereas in diabetic mice there was a significant increase in nuclear area, in old mice there were an increase in nuclear area and a decrease in AgNOR-positive areas. It has been established that diabetic and old animals present an increase in nuclear area, mainly because of an increase in ploidy and a decrease in total RNA levels (Moraes et al., 2007; Mello et al., 2009). The decrease in AgNOR-positive areas in old-mouse hepatocytes might be related not only to diminishment in cellular metabolism, but also to increase in 18S rDNA methylation, as observed in this work and in others (Swisshelm et al., 1990; Machwe et al., 2000).
In conclusion, there is a generalized decrease in liver cell metabolism with aging that affects the gluconeogenesis and glycolysis pathways, leading to a decrease in the production of NAD+ and rRNA. We assume that this reduced NAD+ production could induce a decrease in sirtuin abundance. In T1DM, the early-age phenotype presented by hyperglycemic animals is a response to the absence of insulin signaling from diverse metabolic processes. In the liver, it could activate the Sirt1–PGC-1α pathway, increasing the expression of genes related to the gluconeogenesis pathway, besides the DNA repair machinery induced by Sirt6. Therefore future studies specifically on epigenetic regulation in diabetes and aging are crucial for a better understanding of complex diseases such as diabetes mellitus and the discovery of new therapeutic approaches that might prevent diseases of aging, such as Alzheimer's, thus improving quality of life.
MATERIALS AND METHODS
Animals
Female NOD/Unib and BALB/cAnUnib mice were obtained from the Multidisciplinary Center of Biological Investigation of the University of Campinas. The animals were reared under standard controlled conditions, fed extruded chow (Nuvital, Colombo, Brazil), and provided water ad libitum.
The glycemia levels of the NOD and BALB/c mice were measured once a week, up to 24 h before they were killed. Blood samples were obtained by caudal puncture and analyzed using an automatic Accu-Check Active glucose meter (Roche Diagnostica do Brasil, Jaguare, Brazil), which measures glycemia in the range of 10–600 mg/dl or 0.6–33.3 mmol/l. Glycemia levels within the range 90–100 mg/dl (5.00–5.55 mmol/l) were considered normal; glycemia levels >500 mg/dl (27.5 mmol/l) were considered indicative of severe hyperglycemia. A histological evaluation of the pancreas of NOD mice was performed to confirm their hyperglycemic status.
Four mouse groups were used in this study: 1) normoglycemic young (8-wk-old) NOD adults, 2) normoglycemic young (8-wk-old) BALB/c adults, 3) severe hyperglycemic NOD adults, and 4) normoglycemic 56-wk-old BALB/cAnUnib mice. Normoglycemic BALB/c and NOD mice matched for age for both diabetic groups were used as controls. NOD mice are commonly used as a model to study type I diabetes mellitus because 80% of the females of this strain develop type I diabetes spontaneously. Therefore the normoglycemic NOD mice used as a control in this study consisted of those 20% of the female individuals that failed to develop the disease.
The protocols involving animal care and use were approved by the Committee for Ethics in Animal Use of the University of Campinas (registration no. 1608-1) and meet the guidelines of the Canadian Council on Animal Care.
Livers for the molecular assays were frozen in liquid nitrogen and stored at −80°C in MNase until use.
Isolation of nuclei
Liver cell nuclei were isolated according to previously described procedures (Blobel and Potter, 1966; Matunis, 2006). Briefly, the livers were homogenized manually in cold TKM buffer (0.05 M Tris-HCl, pH 7.5, 15 mM KCl, 5 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride [PMSF], 2 mM dithiothreitol, 10 mM sodium butyrate) with 0.25 M sucrose, and a sucrose gradient was produced with a 2.3 M sucrose TKM buffer. The samples were ultracentrifuged (Beckman Coulter, Brea, CA) at 105,000 × g and 4°C for 30 min. The nuclei were then resuspended in 0.25 M sucrose TKM buffer supplemented with 2% Triton X-100 and centrifuged at 800 × g for 5 min to eliminate endoplasmic reticulum remnants. The nuclei were kept in 0.25 M sucrose TKM buffer supplemented with 30% glycerol at −20°C until use. Nucleus integrity and purity were checked by phase contrast microscopy and by measuring the absorbance ratio at 260/280 nm in a UV Diode Array Spectrophotometer (HP, Palo Alto, CA), respectively.
Western blotting
After quantification by the Bradford protein assay, equal amounts of proteins were used to prepare the samples. Briefly, nuclei were lysed in 1× SDS sample buffer (60 mM Tris-HCl, pH 6.75, 2% SDS, 5% β-mercaptoethanol, 10% glycerol, and bromophenol blue) and sonicated. Protein was fractionated on a 12% SDS–polyacrylamide gel for Sirt1, Sirt6, Sirt7, and PGC-1α and on a 17% gel for histone modification analysis. All blots were run with a standard molecular weight to confirm and identify the target-protein weight. Proteins were transferred to a nitrocellulose membrane (Millipore, Billerica, MA) by electroblotting. The membranes were probed with the following antibodies: anti-Sirt1 (H300; Santa Cruz Biotechnology, Santa Cruz, CA, and Millipore), anti-Sirt6 (Abcam, Cambridge, United Kingdom, and Millipore), anti-Sirt7 (Abcam and Millipore), anti-PGC-1 (Millipore), anti–acetyl-histone H3 (Lys-9; Millipore), anti–acetyl-histone H3 (Lys-14; Millipore), anti–acetyl-histone H1.4 (Lys-26; Sigma-Aldrich, St. Louis, MO), anti–acetyl-histone H3 (Lys-56; Millipore), and anti–acetyl-histone H4 (Lys-16; Abcam). As loading controls, anti–histone H4 and anti–histone H3 antibodies (both from Millipore) were used. Detection and enhancement were performed by enhanced chemiluminescence (Amersham, Pittsburgh, PA). The membranes were exposed to Hyperfilms ECL (Amersham), and the resulting images were quantified by densitometry with ImageJ software (National Institutes of Health, Bethesda, MD). The results were normalized with the loading control bands for each gel and expressed as abundance relative to the BALB/c young-adult group.
Immunofluorescence
Immediately after decapitation, the livers were removed, placed in cold 0.9% NaCl solution, and sliced. Imprints on glass slides freshly prepared from the liver slices were fixed in cold 4% paraformaldehyde for 10 min and rinsed three times in cold phosphate-buffered saline (PBS) for 5 min each. The samples were permeabilized with 0.2% Triton X-100 in PBS buffer and blocked with 5% fetal bovine serum in PBS. The following primary antibodies were used: anti–PGC-1 (Millipore), anti-Sirt1 (Santa Cruz Biotechnology), anti–acetyl-histone H3 (Lys-9; Abcam), and anti–acetyl-histone H4 (Lys-16; Abcam). Sheep anti–rabbit immunoglobulin G (IgG) fluorescein–conjugated and goat anti–mouse IgG rhodamine–conjugated secondary antibodies were purchased from Millipore. Slides were counterstained with 4′,6-diamidino-2-phenylindole (Sigma-Aldrich) and mounted in Vectashield medium (Vector Labs, Burlingame, CA). The images were observed under a Zeiss Axiophot 2 microscope (Carl Zeiss, Oberkochen, Germany) equipped for epifluorescence with an HBO-100W stabilized mercury lamp as light source.
Sirt1 deacetylase activity assay
Equal amounts of total protein from liver nuclei were used for each experimental condition. Positive (resveratrol) control was included for young-adult BALB/c samples using reagents from the SIRT1 Assay Kit (CS1040; Sigma-Aldrich) according to the manufacturer's instructions. Sirt1 deacetylase activity was measured using fluorescence intensity signals at 460 nm (excitation, 365 nm) as captured in a Fusion microplate fluorimeter (Packard, Meridien, MS). Experimental values were represented as activity relative to BALB/c young-adult mice.
NAD+/NADH assay
Twenty milligrams of liver tissue was homogenized, and total NAD+ and NADH levels were determined according to the protocol provided in the NAD+/NADH Assay Kit (Abcam). Following the manufacturer's instructions, total NAD (NADt) and NADH were detected in a 96-well plate, and color was developed and read at 450 nm in an ELISA plate reader (VersaMax, Sunnyvale, TX). The NAD/NADH ratio was calculated according to [NADt – NADH]/NADH.
Comet assay
The alkaline procedure described by Singh et al. (1988) was used, with modifications required by the material. Fresh livers were minced in Hank's buffer (0.137 mM NaCl, 5.4 mM KCl, 0.25 mM Na2HPO4, 0.44 mM KH2PO4, 1.3 mM CaCl2, 1 mM MgSO4, 4.2 mM NaHCO3, 1% glucose). To check cell quality, cell viability was determined using the trypan blue dye exclusion assay (viability <70% implies exclusion of the sample; Collins et al., 1995). For each sample, a positive control with cells treated with H2O2 (100 μM for 30 min at 4°C) was included. All steps described were performed in the dark, to prevent DNA damage by ultraviolet irradiation.
The samples were diluted in 0.5% low–melting point agarose and pipetted onto slides covered with 1.5% normal–melting point agarose (Sigma-Aldrich). The samples were gently spread by placing a coverslip on top and allowed to solidify at 4°C for 10 min. After removal of the coverslip, the slides were immersed in freshly prepared lysis solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, pH 10, with 1% Triton X-100 and 10% dimethyl sulfoxide) for 24 h at 4°C. After that, the slides were left in the electrophoresis solution (1 mM EDTA and 300 mM NaOH, pH 13, at 4°C) for 30 min to allow DNA unwinding and expression of alkali-labile damage, followed by electrophoresis (25 V, 300 mA for 20 min at 4°C). The slides were then washed three times in Tris buffer (0.4 M Tris, pH 7.5) and stained with ethidium bromide (20 μg/ml; Sigma-Aldrich). Nucleoids were evaluated visually in a blind test (Collins et al., 1995; Jaloszynski et al., 1997), using a Zeiss Axiophot 2 microscope equipped for fluorescence microscopy (excitation filter, 450 nm; barrier filter, 490 nm). One hundred comets were classified and assigned to five categories (0–4) according to the extent of DNA migration, as previously described (Jaloszynski et al., 1997; Ghiraldini and Mello, 2010). The extent of DNA damage (D) was determined as D = (L1 + 2L2 + 3L3 + 4L4)/(Σ/100), where L1—L4 are the numbers of nucleoids in classes 1–4, respectively, and Σ is the sum of all nucleoids counted, including category 0 (no tail).
AgNOR staining assay
AgNOR is a cytochemical method based on the specific argyrophilic affinity for some nucleolar proteins, such as B23, nucleolin, UBF, and some RNA polymerase I subunits (Rueschoff et al., 1990). Imprints freshly prepared from liver slices on glass slides were fixed in a mixture of absolute ethanol and glacial acetic acid (3:1, vol/vol) for 1 min, rinsed in 70% ethanol for 5 min, and air dried. AgNOR staining was done as described (Rueschoff et al., 1990; Derenzini and Ploton, 1991), preceded by treatment with a 1% Triton X-100 solution in the presence of 4 M glycerol for 15 min at 37°C (Vidal and Mello, 1995; Mello et al., 2008). Briefly, the cells were treated with a solution containing two volumes of 50% aqueous silver nitrate (Merck, Readington, PA) and one volume of 2% gelatin in 1% aqueous formic acid (vol/vol). The aqueous solutions were prepared with deionized water. The optimal staining time was 10 min at 37°C.
Native chromatin immunoprecipitation
Micrococcal nuclease digestion (30 U/mg DNA) of native chromatin from isolated hepatocyte nuclei was performed for 10 min at 25°C and stopped with EDTA (15 mM). After brief centrifugation, supernatant 1 (SN1) was collected and the digested chromatin was resuspended, released in TEEP buffer (10 mM Tris, pH 7.5, 0.5 mM EDTA; 0.5 mM ethylene glycol tetraacetic acid, 250 μM PMSF, and 0.05% Triton-X) with 5 mM NaCl, and left overnight at 4°C. Samples were centrifuged for 30 s at 5000 rpm, and supernatant 2 (SN2) was collected. Each ChIP reaction and input was performed with 5 μg of chromatin (2.5 μg from SN1 and 2.5 μg from SN2) using specific 5-methyl-cytidine (Abcam) and IgG anti-mouse (Santa Cruz Biotechnology) monoclonal antibodies for mock control. The hybridization of the complex antibody and magnetic beads (Invitrogen, Carlsbad, CA) with the chromatin was done overnight with constant rotation at 4°C. The samples were washed twice in TEEP buffer with 140 and 200 mM NaCl, respectively, and eluted in TEEP buffer with 50 mM NaCl and 1% SDS.
Nucleic acid extraction and quantitative PCR
DNA was extracted from the immunoprecipitated samples using phenol/chloroform. Primers were designed to amplify a 102-pb product from the 45S gene that corresponds to the 18S rDNA region. To analyze gene expression and corroborate the Western blotting results for nuclear sirtuins, RNA was extracted with the RNeasy Mini Kit (Qiagen, Hilden, Germany) from mouse livers stored in RNAlater (Ambion, Austin, TX). All the primers used to quantify the expression of nuclear Sirts, targets of PGC-1α, and 18S gene are described in Table 2. For the quantitative PCR assay, the FastStart Universal Syber Green Master (ROX) mix (Roche, Penzberg, Germany) was used in an ABI 7500 thermocycler (Applied Biosystems, Foster City, CA).
TABLE 2:
Primers used for gene expression and native-ChIP assay.
| Assay | Gene | Primer forward | Primer reverse |
|---|---|---|---|
| Gene expression | Sirt1 | CTCCTGTTGACCGATGGACT | ATCGGTGCAATCATGAGAT |
| Sirt6 | CCTGTAGAGGGGAGCTGAGA | GAGGTACCCAGGGTGACAGA | |
| Sirt7 | GAGAGCGAGGATCTGGTGAC | GCCCGTGTAGACAACCAAGT | |
| Foxo1 | GCTGGGTGTCAGGCTAAGAG | TTGCCAAGTCTGAGGAAAGG | |
| Pck1 | CCTGGAAGAACAAGGAGTGG | CTACGGCCACCAAAGATGAT | |
| Native-ChIP | 18S | CGAAAGCATTTGCCAAGAAT | AGTCGGCATCGTTTATGGTC |
Statistics
All statistical analyses were performed with Minitab 12 software (Minitab, State College, PA). Data comparison between the diabetic animals and their controls from five independent sets of experiments was performed by means of the analysis of variance (ANOVA) test, and for pairwise comparison Student's t test was used. p < 0.05 was considered the critical level for rejection of the null hypothesis.
Supplementary Material
Acknowledgments
We thank Maria C. G. Marcondes for the use of the microplate fluorimeter and the Laboratory of Molecular Genetics (FCM-Unicamp) for the use of the thermocycler. This work was supported by the São Paulo State Research Foundation (FAPESP; Grants 2010/50015-6 and 2008/58067-5) and the Brazilian National Research and Development Council (CNPq; Grants 471303/2009-7 and 301943/2009-5).
Abbreviations used:
- AgNOR
silver-stained nucleolar organizer region
- NOD
nonobese diabetic mice
- T1DM
type 1 diabetes mellitus
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-04-0186) on June 12, 2013.
REFERENCES
- Ahmed N. Advanced glycation endproducts—role in pathology of diabetic complications. Diabetes Res Clin Pract. 2005;67:3–21. doi: 10.1016/j.diabres.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Berg JM, Tymoczko JL, Stryer L. The integration of metabolism. In: Berg JM, Tymoczko JL, Stryer L, editors. Biochemistry. New York: WH Freeman; 2002. pp. 867–890. [Google Scholar]
- Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- Blazer S, Khankin E, Segev Y, Ofir R, Yalon-Hacohen M, Kra-Oz Z, Gottfried Y, Larisch S, Skorecki KL. High glucose-induced replicative senescence: point of no return and effect of telomerase. Biochem Biophys Res Commun. 2005;296:93–101. doi: 10.1016/s0006-291x(02)00818-5. [DOI] [PubMed] [Google Scholar]
- Blobel P, Potter VR. Nuclei from rat liver: isolation method that combines purity with high yield. Science. 1966;154:1662–1665. doi: 10.1126/science.154.3757.1662. [DOI] [PubMed] [Google Scholar]
- Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD +metabolism oxidative stress and sirt1 activity in Wistar rats. PLoS One. 2011;6:e19194. doi: 10.1371/journal.pone.0019194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao SX, Dhahbi JM, Mote PL, Spindler SR. Genomic profiling of short- and long-term caloric restriction effects in the liver of aging mice. PNAS. 2001;98:10630–10635. doi: 10.1073/pnas.191313598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Bruno J, Easlon E, Lin S, Cheng H, Alt FW, Guarente L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008;22:1753–1757. doi: 10.1101/gad.1650608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AR, Ma AG, Duthie SJ. The kinetics of repair of oxidative DNA-damage (strand breaks and oxidized pyrimidines) in human cells. Mutat Res. 1995;336:69–77. doi: 10.1016/0921-8777(94)00043-6. [DOI] [PubMed] [Google Scholar]
- Derenzini M, Ploton D. Interphase nuclear regions in cancer cells. Int Rev Exp Pathol. 1991;32:149–192. doi: 10.1016/b978-0-12-364932-4.50008-3. [DOI] [PubMed] [Google Scholar]
- Ford E, Voit R, Liszt G, Magin C, Grummt I, Guarente L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 2006;20:1075–1080. doi: 10.1101/gad.1399706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiraldini FG, Mello MLS. Micronucleus formation, proliferative status, cell death and DNA damage in ethosuximide-treated human lymphocytes. Cell Biol Int Rep. 2010;17:e00006. [Google Scholar]
- Ghiraldini FG, Silva IS, Mello MLS. Polyploidy and chromatin remodeling in hepatocytes from insulin-dependent diabetic and normoglycemic aged mice. Cytometry A. 2012;81:755–764. doi: 10.1002/cyto.a.22102. [DOI] [PubMed] [Google Scholar]
- Gonzalo S. Epigenetic alteration in aging. J Appl Physiol. 2010;109:586–597. doi: 10.1152/japplphysiol.00238.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grob A, Roussel P, Wright JE, McStay B, Hernandez-Verdun D, Sirri V. Involvement of SIRT7 in resumption of rDNA transcription at the exit from mitosis. J Cell Sci. 2009;122:489–498. doi: 10.1242/jcs.042382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis MC, Guarente LP. Mammalian sirtuins—emerging role in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- Herrman CE, Sanders RA, Klaunig JE, Schwarz LR, Watkins JB. Decreased apoptosis as a mechanism for hepatomegaly in streptozotocin-induced diabetic rats. Toxicol Sci. 1999;50:146–151. doi: 10.1093/toxsci/50.1.146. [DOI] [PubMed] [Google Scholar]
- Herzog B, Cardenas J, Hall RK, Villena JA, Budge PJ, Giguere V, Granner DK, Kralli A. Estrogen-related receptor α is a repressor of phosphoenolpyruvate carboxykinase gene transcription. J Biol Chem. 2005;281:99–106. doi: 10.1074/jbc.M509276200. [DOI] [PubMed] [Google Scholar]
- Imai S. The NAD world: a new systemic regulatory network for metabolism and aging—Sirt1, systemic NAD biosynthesis, and their importance. Cell Biochem Biophys. 2009;53:65–74. doi: 10.1007/s12013-008-9041-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaloszynski P, Kujawaski M, Czub-Swierczek M, Markowska J, Szyfter K. Bleomycin-induced DNA damage and its removal in lymphocytes of breast cancer patients studied by comet assay. Mutat Res. 1997;385:223–233. doi: 10.1016/s0921-8777(97)00046-3. [DOI] [PubMed] [Google Scholar]
- Kim H, et al. Hepatic-specific disruption of Sirt6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010;12:224–236. doi: 10.1016/j.cmet.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibiger IB, Berggren P. Sirt1: a metabolic master switch that modulates lifespan. Nature. 2006;12:34–36. doi: 10.1038/nm0106-34. [DOI] [PubMed] [Google Scholar]
- Lombard DB, Schwer B, Alt FW, Mostoslavsky R. SIRT6 in DNA repair, metabolism and aging. J Intern Med. 2008;263:128–141. doi: 10.1111/j.1365-2796.2007.01902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machwe A, Orren DK, Bohr VA. Accelerated methylation of ribosomal RNA genes during the cellular senescence of Werner syndrome fibroblasts. FASEB J. 2000;14:1715–1724. doi: 10.1096/fj.99-0926com. [DOI] [PubMed] [Google Scholar]
- Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of mice. Exp Anim. 1980;29:1–13. doi: 10.1538/expanim1978.29.1_1. [DOI] [PubMed] [Google Scholar]
- Matunis MJ. Isolation and fractionation of rat liver nuclear envelopes and nuclear pore complexes. Methods. 2006;39:277–283. doi: 10.1016/j.ymeth.2006.06.003. [DOI] [PubMed] [Google Scholar]
- McCord R, et al. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break point. Aging. 2009;1:109–121. doi: 10.18632/aging.100011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown PC, Shaw PJ. Chromatin: linking structure and function in the nucleolus. Chromosoma. 2009;118:11–23. doi: 10.1007/s00412-008-0184-2. [DOI] [PubMed] [Google Scholar]
- Mehta IS, Figgitt M, Clements CS, Kill IR, Bridger JM. Alterations to nuclear architecture and genomic behavior in senescent cells. Ann NY Acad Sci. 2007;1100:250–263. doi: 10.1196/annals.1395.027. [DOI] [PubMed] [Google Scholar]
- Mello MLS, Aldrovani M, Moraes AS, Guaraldo AMA, Vidal BC. DNA content, chromatin supraorganization, nuclear glycoproteins and RNA amounts in hepatocytes of mice expressing insulin-dependent diabetes. Micron. 2009;40:577–585. doi: 10.1016/j.micron.2009.02.010. [DOI] [PubMed] [Google Scholar]
- Mello MLS, Vidal BC, Russo J, Planding W, Schenck U. Image analysis of the AgNOR response in ras-transformed human breast epithelial cells. Acta Histochem. 2008;110:210–216. doi: 10.1016/j.acthis.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E, McCord RA, Boxer LD, Barber MF, Hong T, Gozani O, Chua KF. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human Sirt6. Cell Cycle. 2009;8:2664–2666. doi: 10.4161/cc.8.16.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes AS, Guaraldo AMA, Mello MLS. Chromatin supraorganization and extensibility in mouse hepatocytes with development and aging. Cytometry A. 2007;71:28–37. doi: 10.1002/cyto.a.20356. [DOI] [PubMed] [Google Scholar]
- Moraes AS, Mondin M, Beletti ME, Aguiar-Perecin MLR, Guaraldo AMA, Mello MLS. Age-related association of rDNA and telomeres with the nuclear matrix in mouse hepatocytes. Cell Biol Int. 2010;34:925–931. doi: 10.1042/CBI20090457. [DOI] [PubMed] [Google Scholar]
- Mostoslavsky R, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Murayama A, et al. Epigenetic control of rDNA loci in response to intracellular energy status. Cell. 2008;133:627–639. doi: 10.1016/j.cell.2008.03.030. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Kawakami K, Kametani F, Nakamoto H, Goto S. Biological significance of protein modifications in aging and calorie restriction. Ann NY Acad Sci. 2010;1197:33–39. doi: 10.1111/j.1749-6632.2009.05374.x. [DOI] [PubMed] [Google Scholar]
- Nelson DL, Cox MM. Fatty acid catabolism. In: Nelson DL, Cox MM, editors. Principles of Biochemistry. New York: WH Freeman; 2004. pp. 631–655. [Google Scholar]
- Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci USA. 2008;105:9793–9798. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic adaptations through the PGC-1α and Sirt pathways. FESB Lett. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueschoff J, Plate KH, Contractor H, Stern S, Timmermann R, Thomas C. Evaluation of nucleolus organizer regions (NORs) by automatic image analysis: a contribution to standardization. J Pathol. 1990;161:113–118. doi: 10.1002/path.1711610205. [DOI] [PubMed] [Google Scholar]
- Sarg B, Koutzumani E, Helliger W, Rundquist IN, Lindner HH. Postsynthetic trimethylation of histone H4 at lysine 20 in mammalian tissues is associated with aging. J Biol Chem. 2002;277:39195–39201. doi: 10.1074/jbc.M205166200. [DOI] [PubMed] [Google Scholar]
- Schwer B, Verdin E. Conserved metabolic regulatory function of sirtuins. Cell Metab. 2008;7:104–112. doi: 10.1016/j.cmet.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Sedivy JM, Banumathy G, Adams PD. Aging by epigenetics—a consequence of chromatin damage. Exp Cell Res. 2007;10:1909–1917. doi: 10.1016/j.yexcr.2008.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DM, Kucia M, Ratajczak MZ. Nuclear and chromatin reorganization during cell senescence and aging—a mini-review. Gerontology. 2011;57:76–84. doi: 10.1159/000281882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175:184–191. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- Spindler SR, Dhahbi JM, Mote PL. Protein turnover, energy metabolism, aging, and caloric restriction. Adv Cell Aging Gerontol. 2003;14:69–85. [Google Scholar]
- Swisshelm K, Disteche CM, Thorvaldsen J, Nelson A, Salk D. Age-related increase in methylation of ribosomal genes and inactivation of chromosome-specific rRNA gene clusters in mouse. Mutat Res. 1990;237:131–146. doi: 10.1016/0921-8734(90)90019-n. [DOI] [PubMed] [Google Scholar]
- Tennen RI, Chua KF. Chromatin regulation and genome maintenance by mammalian SIRT6. Trends Biochem Sci. 2011;36:39–46. doi: 10.1016/j.tibs.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai YC, Grecot TM, Boonmee A, Miteva Y, Cristea IM. Functional proteomics establishes the interaction of Sirt7 with chromatin remodeling complexes and expands its role in regulation of RNA polymerase I transcription. Mol Cell Proteomics. 2012;11:60–76. doi: 10.1074/mcp.A111.015156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gool F, Galli M, Gueyda C, Kruys V, Prevot PP, Bedalov A, Mostoslavsky R, Alt FW, Smedt T, Leo O. Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin-dependent manner. Nat Med. 2009;15:206–210. doi: 10.1038/nm.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal BC, Mello MLS. Re-evaluating the AgNOR staining response in Triton X-100-treated liver cells by image analysis. Anal Cell Pathol. 1995;9:39–43. [PubMed] [Google Scholar]
- Xiao C, Kim HS, Lahusen T, Wang RH, Xu X, Gavrilova O, Jou W, Gius D, Deng CX. SIRT6 deficiency results in severe hypoglycemia by enhancing both basal and insulin-stimulated glucose uptake in mice. J Biol Chem. 2010;285:36776–36784. doi: 10.1074/jbc.M110.168039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Zwaans BM, Eckersdorff M, Lombard DB. The sirtuin Sirt6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cyle. 2009;8:2662–2663. doi: 10.4161/cc.8.16.9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1α. Cell. 2010;140:280–293. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.