

Abstract
We recently demonstrated whole genome sequencing of a human fetus using only parental DNA samples and plasma from the pregnant mother. This proof-of-concept study demonstrated how samples obtained noninvasively in the first or second trimester can be analyzed to yield a highly accurate and substantially complete genetic profile of the fetus, including both inherited and de novo variation. Here, we revisit our original study from a clinical standpoint, provide an overview of the scientific approach, and describe opportunities and challenges along the path towards clinical adoption of noninvasive fetal whole genome sequencing (NIFWGS).
The intent of this article is to review the methodological basis for NIFWGS as well as the challenges facing clinical translation. In the first section, we briefly review the history of noninvasive prenatal genetic testing. In the second section, we provide a primer on the technical basis for NIFWGS, including the prediction of both inherited and de novo variation, using samples obtained noninvasively. In the last section, we consider some of the key challenges to clinical adoption of NIFWGS as a diagnostic test.
Historical Perspective
Noninvasive prenatal prediction of fetal health has evolved rapidly over the past few decades. While fetal karyotyping following amniocentesis or chorionic villus sampling remains the gold standard for diagnostic testing, prenatal screening options offered to pregnant women have become increasingly predictive. Current screening now includes not only serum analytes and sonographic markers1,2 but also several techniques that have exploited the genetic material from fetal cells in the maternal circulation3–7. In 1997, the first report of fetal cell-free DNA (cfDNA) circulating in the plasma of pregnant women8 sparked a new phase of innovation (Table 1). Although the use of massively parallel sequencing9 of fetal cfDNA to screen for specific aneuploidies is increasingly available in clinics, confirmatory testing by karyotyping or diagnostic array remains essential10–12. Thus, while the refinement of fetal risk assessment with antenatal screening has greatly improved, all available methods continue to be non-diagnostic and, moreover, limited in genetic scope.
Table 1. Current approaches to interrogation of fetal cfDNA.
A variety of commercial and proof-of-concept methods use cfDNA in the maternal plasma to determine fetal genetic makeup in a targeted or holistic manner. Closed circles denote information explicitly obtained with each method. Open circles denote information that can be obtained with each method, but not explicitly addressed in the referenced publication. “Common SNPs” and “Rare SNVs” refer to genome-wide determination of common single-nucleotide polymorphisms and rare single-nucleotide variants. Relative cost estimates are denoted by dollar signs. “MPS”: massively parallel sequencing. “CNV”: copy-number variant.
| Study | Method overview | Trisomy / Monosomy | Sub-chrom. CNVs | Common SNPs | Rare SNVs | Single gene target | Gene panel / exome | De novo point mutations | Cost |
|---|---|---|---|---|---|---|---|---|---|
| Fan et al. (2008); Chiu et al. (2008) | MPS to determine relative over- or under-abundance of specific chromosomes using maternal plasma. Statistical models to infer gain or loss of entire chromosomes. | ● | $$ | ||||||
| Tynan et al. (2011) | Multiplex PCR to detect fetal sex and RHD status in RhD-negative pregnant women. | ● | $ | ||||||
| Lo et al. (2007) | Digital PCR to identify overrepresentation of specific allele in maternal plasma, and to assess chromosomal imbalance to infer trisomy 21. | ● | ○ | $ | |||||
| Srinivasan et al. (2013) | High-coverage MPS of DNA from plasma to identify over- or under- abundance of reads from 1 Mb genomic bins. Statistical model to infer CNVs. | ● | ● | $$ | |||||
| Fan et al. (2012) | DNA from maternal blood and plasma. Haplotype-based predictions of fetal inheritance using cfDNA at common genomic SNPs and exome. | ● | ● | ● | ○ | ● | $$$ | ||
| Kitzman et al. (2012) | DNA from blood (mom), saliva (dad), and plasma. Haplotype- based predictions of fetal inheritance using cfDNA and statistical modeling. | ● | ○ | ● | ● | ○ | ● | ● | $$$$ |
Scientific Approach
In 2010, Lo and colleagues reported that the entire fetal genome was represented in short cfDNA fragments in the maternal plasma, and suggested that the reconstruction of the inherited complement was technically attainable13. We pursued and recently reported a proof-of-concept study demonstrating, for the first time, the noninvasive determination of a fetal genome sequence14. We achieved substantial completeness and over 99% accuracy using only a sample of paternal saliva and a single tube of blood collected from the mother at 18.5 weeks gestation. Subsequently, another group achieved comparable accuracy using a similar maternal sample15. Although differing in key technical details, both studies inferred the fetal genotypes by first sequencing the maternal genome in order to identify alleles that could be transmitted from mother to fetus, and then analyzing the mother’s cfDNA to determine which alleles she actually transmitted.
A primary technical obstacle to sequencing fetal genomes from maternal plasma is that only a minority of total cfDNA fragments in maternal plasma are shed from the placenta16 and thus reflect the fetal inherited complement. For instance, the plasma specimens used in our study from two different pregnancies contained 8% and 13% fetoplacental content, which are representative examples given their collection at weeks 8.1 and 18.5, respectively. The remaining cfDNA is derived from maternal cells and is therefore uninformative in this context. Ideally, one might isolate the fetoplacental cfDNA, allowing a direct read-out of the inherited genome. However, despite attempts to separate these two fractions on the basis of size17 or methylation profile18, no technology has been developed to date that can do so with satisfactory yield and specificity.
Instead, efforts by our group and others demonstrate that by deeply sampling this mixture of fetal and maternal genetic material – along with statistical modeling – the fetal genotypes can be accurately inferred (Figure 1). This approach relies on the fact that the fetal genome is necessarily a composite of the parental chromosomes. By determining the parental genotypes, we can constrain the possible fetal genotypes on the basis of Mendelian inheritance – discounting, for the time being, the rare chance of a de novo mutation arising in the maternal or paternal germline. To determine the parental genotypes, we performed whole-genome shotgun sequencing (WGS) of the maternal and paternal genomes. This step could be performed at any time before or during pregnancy. In combination with individual and family medical histories, it would establish a set of recessive conditions for which each parent is a carrier.
Figure 1.
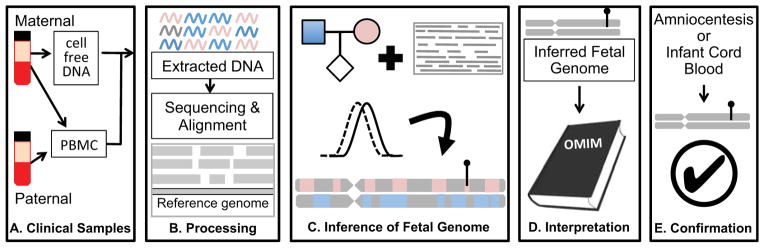
Overview of noninvasive fetal whole genome sequencing. (a). Sample collection. Parental blood samples are collected in the first or second trimester. After centrifugation, parental DNA is extracted from peripheral blood mononuclear cells (PBMC) or buffy coat, while cfDNA is isolated from the maternal plasma. (b). Sample processing. Extracted DNA is amplified for library preparation and sequenced to high depth. Reads are aligned to a reference genome to identify variant alleles carried by one or both parents. (c). Inference of fetal genome. A statistical model combines known parental genotypes and alleles observed in cfDNA reads to predict fetal inheritance. High-impact mutations, whether inherited or de novo, are identified (lollypop). (d). Interpretation. Identified variants are compared to catalogs of known disease-associated mutations. (e). Confirmation. A subset of clinically actionable predicted mutations is confirmed with conventional procedures such as amniocentesis. Accuracy of genome inference can be assessed post-hoc with DNA extracted from cord blood after delivery.
At the vast majority of sites in the genome (>99.9%), both parents are homozygous for the same allele, and the fetal genotype is therefore unambiguous: homozygous for that allele (Figure 2a). At a much smaller proportion of sites (typically fewer than 1x106, or 0.03% of sites, depending upon genetic ancestry), each parent will again be homozygous, but for different alleles; at these sites, the fetus is an obligate heterozygote. Uncertainty about fetal inheritance arises only at the remaining sites – those at which one or both parents are heterozygous.
Figure 2.

Inference of the fetal genome on a site-by-site basis. (a). Observed parental genotypes at a given site constrain the possible fetal genotypes. At the vast majority of sites, both parents are homozygotes and the fetal genotype is unambiguous. (b). Expected cfDNA makeup in the maternal plasma at maternal (top) and paternal (bottom) heterozygous sites. At these sites, either allele could be transmitted, yielding different expected allelic fractions. (c). Schematic of inference of fetal inheritance at maternal heterozygous sites. Numbers shown assume a constant sequencing depth of 100X at each site and do not represent real data. After sequencing the cfDNA, observed allele counts are compared to expected allele counts at each site, and in each case, the more likely scenario is chosen (purple boxes). (d). Schematic of inference of fetal inheritance at paternal heterozygous sites. Numbers are presented as in (c). At each site, presence of the B allele is unexpected in cases where the father transmits the “A” allele. At the rightmost site, the single observed “B” allele could be evidence of true “B” transmission or an error introduced during the sequencing process.
These uncertain cases can be further split into several possibilities. The most straightforward case is a site at which only the father is heterozygous. If the maternal cfDNA is sequenced sufficiently deeply, but the allele specific to the father is never observed, we infer that the father did not transmit that allele, but instead transmitted the shared allele (Figure 2b, 2d). This process is conceptually similar to determining the fetal sex by the presence of reads derived from the Y chromosome, which appear among the maternal cfDNA sequences only when the fetus is male, while their absence indicates the fetus is female. Noninvasively determining the fetal sex in this manner is straightforward, and only a small number of sequences must be sampled from the cfDNA in order to have a high degree of confidence in the presence or absence of an entire chromosome. By contrast, much deeper sampling is required to carry out the same task for each individual genomic site, and a key question is exactly how deep this sampling must be.
The answer to this question largely depends upon the proportion of fetal material among the maternal plasma cfDNA fragments. Accurately estimating this fraction is important not only for NIFWGS, but also key to current aneuploidy tests19. To estimate this, we can identify a set of informative genetic markers that would not be observed if the cfDNA were entirely maternal in origin. The homozygous alleles specific to the father (not carried by the mother) make an ideal set of markers. If the fetus is male, these may be supplemented by sequences specific to the Y chromosome. After deep sequencing of the plasma cfDNA, the frequency of these definitively fetal sequences is tallied, doubled to account for the equal inheritance from the mother, and used as a direct estimate of the percentage of fetal cfDNA in the maternal plasma.
Precisely estimating the fetal fraction of cfDNA is important for two reasons. First, as this fraction decreases, inaccuracies in the inferred fetal genotypes accumulate. If the fetal cfDNA level is too low – for example, less than 5% -- then the accuracy of the predicted fetal genome may drop below 95%14, potentially requiring a second plasma sample to be obtained later in pregnancy, when the fetal fraction may be higher. Second, the estimate of fetal concentration is a key parameter, along with the parental genotypes and the cfDNA sequencing reads, in the statistical model used to predict fetal inheritance.
This model is applied to infer the fetal genotypes at the remaining positions of uncertain inheritance: sites at which the mother is heterozygous and could transmit either allele. At these sites, the dosages of the two alleles among the plasma cfDNA sequences provide evidence for the maternal transmission of one or the other. For example, suppose maternal cfDNA is sequenced to a depth of 100X, with an estimated fetal fraction of 10%. At a given site, the homozygous father necessarily contributes the “A” allele, but the heterozygous mother could contribute either “A” or “B” (Figure 2c). On average, we will find 100 reads covering this particular site, of which 90% will be derived from the maternal genome and 10% from the fetal genome. The 90 maternal reads should have, again on average, an equal allele balance at this heterozygous site, meaning that 45 of the reads should contain the “A” allele and the other 45 should contain the “B” allele. The 10 fetal reads will consist of approximately five supporting the “A” allele contributed by the father, while the remainder represent the maternal contribution, which could be “A” or “B”. Thus, we expect that if the “A” allele is transmitted by the mother, we should observe this allele in 55 (45 + 5 + 5) of the reads, whereas if the “B” allele is transmitted, we should observe the A allele 50 (45 + 5 + 0) times. We can statistically test which of these two competing scenarios is more likely given the number of times we actually observe the A allele at this site. We can then repeat this process at all heterozygous sites to yield a set of site-by-site inheritance predictions.
Unfortunately, applying this straightforward model to the full genome yields unsatisfactory results. Suppose, from the previous example, we observe the “A” allele 59 times at this site. In this scenario, the hypothesis in which the mother transmits the “A” allele is almost four times as likely as the transmission of the “B” allele, strongly supporting the former possibility. Whole genome shotgun sequencing works by randomly sampling and sequencing fragments, and despite no change in the underlying inheritance or fetal fraction, the “A” allele at the next such site could be observed only 53 times by random fluctuation. In this event, the two hypotheses (“A” vs “B” transmitted) are nearly equally likely, suggesting that any prediction made in this scenario is roughly equivalent to a coin toss.
A simple means to overcome this limitation would be to sample the cfDNA more deeply to obtain clearer separation between the competing transmission hypotheses. For example, if we were able to sequence the cfDNA to 10,000X depth, and continued to observe the “A” allele in 53% of the reads, the transmission of the “A” allele would then be roughly 20,000 times more likely than the transmission of the “B” allele. Unfortunately, the expense of sequencing a human genome scales with the depth, such that sequencing to 10,000X would currently cost over $1 million. Even if expense were no object, this sampling depth is not achievable in many cases: a typical plasma specimen may not contain a sufficient number of distinct copies of the genome regardless of technical limitations of DNA isolation and sequencing library preparation steps.
Rather than sampling to an impractical depth at each genomic site in isolation, we employ an experimental technique to group together alleles from each parent, thereby realizing greater statistical power. This approach exploits the fact that the parental genomes are not inherited as a series of independent sites, but rather as haplotypes, or sets of variants jointly present on one of a given pair of homologous chromosomes. If we knew the haplotypes of the parental chromosomes, then we could search for evidence of joint transmission of large contiguous groups of genetic variants, allowing for a small number of crossover events during meiosis. However, long-range haplotypes that span all variants across the full length of a chromosome arm have to date remained largely recalcitrant to experimental methods, except in the context of multi-generation family studies where haplotypes can be inferred post-hoc by transmission patterns.
We recently developed a technique to ascertain smaller subsections of haplotypes, or “haplotype blocks,” each containing dozens or hundreds of heterozygous sites and covering tens to hundreds of kilobases20. At a given locus, we define two haplotype blocks, arbitrarily labeled “A” and “B”, representing the grouping, or “phase,” of genetic variants present on the two homologs (Figure 3a, 3b). Applying this technique to the parental genomes allows us to search for evidence of transmission of whole blocks “A” or “B”, instead of individual alleles “A” or “B”, by aggregating evidence of overrepresentation of each phased allele along the length of a haplotype block (Figure 3c, 3d). The signal generated by jointly considering large blocks of sites helps to mitigate the site-by-site noise described above. Moreover, sites at which both parents are heterozygous, where inheritance is particularly difficult to individually predict owing to the addition of a third possible fetal genotype, benefit from their inclusion in haplotype blocks with stronger evidence of inheritance.
Figure 3.

Inference of the fetal genome from haplotype blocks. (a). Phasing of maternal heterozygous sites into haplotype blocks (red bars). Haplotype blocks contain dozens or hundreds of such sites and cover over 300 kilobases on average. A single chromosome may have over 100 haplotype blocks; contiguity between blocks is not defined. Approximately 90% of all heterozygous sites are incorporated into haplotype blocks. Sites shown do not represent real data. (b). Phasing of paternal heterozygous sites into haplotype blocks (blue bars). Paternal and maternal blocks may overlap but are independently defined. (c). Schematic of inference of fetal inheritance of maternal haplotype blocks. Numbers shown assume a constant sequencing depth of 100X at each. After sequencing the cfDNA, evidence of deviations from expected allele counts is aggregated over each site in haplotype blocks “A” and “B”, and the more likely block is predicted. Block-level predictions in turn determine predictions at each contained site. The site in the center of the block would be incorrectly predicted if sites were considered independently; its inclusion in a haplotype block mitigates sampling noise and corrects the prediction. (d). Schematic of inference of fetal inheritance of paternal haplotype blocks. Numbers are presented as in (c). The observed “G” allele at the rightmost site, likely to cause an incorrect prediction if sites were considered independently, is now correctly identified as an error introduced during the sequencing process rather than evidence of transmission of the “G” allele. (e). The inferred fetal genome is a composite of the parental haplotype blocks.
The inferred fetal genome, then, consists of a set of predictions about inheritance of one or the other haplotype block from each of the parental genomes (Figure 3e). This composite picture of the fetal genome is substantially complete and highly accurate. However, several clear avenues for technical improvement remain. Intuitively, increasing the length of the haplotype blocks and ensuring they encompass every heterozygous site carried by each parent allows more evidence to be accumulated and yields more accurate predictions of inheritance. At the time of our study, we had determined haplotype blocks for only the maternal genome, and predicted paternal inheritance on a site-by-site basis. We subsequently phased the paternal genome in this same family, which increased the accuracy of prediction for paternal sites from 96.8% to 99.95%. Currently, the process of obtaining haplotypes blocks is laborious, although streamlined techniques21 promise to shorten the processing time required and improve the scalability of the method. Also, these approaches could be combined with other approaches that define longer but sparser blocks (e.g. phasing incomplete sets of heterozygous sites across entire chromosomes22–24). Leveraging even longer haplotype blocks while maintaining completeness in terms of the fraction of sites that are phased would improve prediction accuracy and additionally allow mapping of sites of meiotic recombination.
We now return to the question of de novo mutations, or mutations newly arising in the maternal or paternal germline. In principle, de novo mutations are easily identified as variants in the sequenced maternal cfDNA that are not found in either parent. In practice, despite ongoing improvement, WGS technology remains imperfect, and errors introduced during PCR or sequencing far outnumber the approximately 50 to 100 true de novo mutations that we would expect in any given fetus25. At a sequencing depth of 100X and fetal fraction of 10%, the two types of events yield signatures that are, on the whole, nearly indistinguishable: at a given site, a small handful of reads suggests the spontaneous emergence of a fetal genotype incompatible with Mendelian inheritance. Separating the true mutations from the spurious errors introduced during the sequencing process remains a challenge and a major area for improvement in both technology and analysis.
One way to address the large number of candidate de novo mutations is to apply an increasingly aggressive set of filters designed to improve the signal-to-noise ratio in the candidate set. For example, we might exclude any candidate with only one or two supporting reads. We might remove sites that are inside or adjacent to specific sequence motifs known to generate elevated error rates. We might discard any site also identified as a candidate in other samples within the same cohort. At each step, we may trade a small decrease in sensitivity for a suitably large gain in specificity. Even after extensive filtering, we are likely to be left with several thousand candidates – still too many for follow-up. However, only a very small percentage of these candidates are likely to fall within protein coding or regulatory regions, suggesting that manual review and/or validation of high-impact candidates may be plausible in a clinical setting.
Ideally, in order to systematically map de novo mutations, a sample must be collected from the father. Without knowledge of the paternal genotypes, any paternally transmitted alleles not shared with the mother are indistinguishable from de novo mutations in the maternal germline. However, even without a paternal sample, it may still be possible to identify likely de novo mutations by searching a predefined panel of genes known to be inherited in a dominant fashion with high penetrance; mutations in these genes could be ruled as unlikely given the father’s health status. Nevertheless, for all but the most stereotyped disorders, definitively separating deleterious mutations from benign ones remains an elusive goal, even for single-gene disorders.
Translation to Clinical Practice
Technical Aspects
Although NIFWGS can yield an accurate picture of the fetal genome, several challenges must be addressed and avenues for improvement explored before this technology can reach the bedside. One major hurdle facing care providers is establishing an informatics infrastructure to process and securely store large volumes of genomic data. Interpreting these data poses an even greater challenge: WGS provides measurements across over 20,000 protein-coding genes that are not readily summarized as a single result. The measurements themselves are complex: WGS reports an entire set of genotypes, far from providing a numerical read-out as analyte testing might, or a “normal/abnormal” status as trisomy screening would provide. While the report is comprehensive in breadth, most of the reported variants have little to no impact on patient health, placing the burden on the physician and genetic counselor to isolate the relevant information (if any) from that volume of data. Automated analyses have been applied in the context of neonatal sequencing to select only genetic variants in genes deemed relevant in order to streamline the process and to exclude incidental findings26, and a similar approach might be useful in this context. Additionally, the analytic method described here focuses primarily on single-nucleotide variants (which account for the majority of human genetic variation). In order to be truly complete, it is necessary to also consider other variants including insertions, deletions, and copy number variations. While necessary, these analyses will further complicate interpretation.
Beyond targeting the data analysis, the sequencing process itself could be targeted to a subset of genes where findings might be relevant. Technologies such as exome capture27 and molecular inversion probes28, originally developed for screening pre-defined panels of genes across large cohorts, could be adapted to cfDNA sequencing in order to interrogate only specific genes of interest. For instance, a panel could be established including the genes for all recessive conditions for which carrier testing is currently available. Targeted sequencing of this gene panel from cfDNA, along with targeted analysis of parental haplotypes at these loci, could establish parental carrier status and noninvasively test for transmission of risk alleles, while minimizing the burden of incidental findings. This approach would also likely decrease the cost and increase the turnaround time of the assay.
Translational Aspects
As discussed before, current approaches to prenatal diagnosis incorporate increasingly refined noninvasive screening techniques to identify pregnancies at high risk of fetal abnormalities, thus facilitating direction of invasive diagnostic approaches to a small number of pregnancies2. Ultimately, diagnostic approaches that are both noninvasive and comprehensive would replace screening altogether. Currently, though noninvasive approaches for detection of specific aneuploidies are commercially available, the test performance characteristics for these approaches drive consideration of these tests as sensitive screening tests with persistent reliance on invasive testing for diagnostic confirmation10. NIFWGS, with its extremely high sensitivity, has the potential to achieve the goal of noninvasive, broad diagnostic capability. However, in the context of prenatal diagnosis, in order to achieve this potential, we must keep in mind the need for absolute minimization of false positive results, matching or surpassing the accuracy of invasive testing (>99% in the case of amniocentesis29). Once technical aspects of the procedure are refined, scalability to larger validation studies carefully evaluating such test performance characteristics will be the essential next step.
One factor that must be considered in test performance evaluation and translation of NIFWGS to clinical practice is the placental origin of fetal cfDNA. As with CVS, which also samples placental material, confined placental mosaicism (CPM) must be considered in our interpretation of genetic results derived from fetal cfDNA30. Empiric evidence supporting the relevance of CPM to fetal cfDNA was recently described in a case report31. In practice, depending on the overall clinical picture, abnormal CVS results may require direct confirmatory testing of fetal cells through amniocentesis. Estimates of the incidence of CPM vary depending on preparation technique – whether performed directly or after culture -- but generally range between 1–2%32. These estimates derive primarily from first trimester samples evaluated for aneuploidy. Though CVS is typically performed in early pregnancy, some studies of CVS in the second and third trimesters have found an increased incidence of CPM with increasing gestational age33, a factor to consider in estimation of the effect in NIFWGS. CPM can result from a postyzygotic event generating genetic error in an initially normal pregnancy, or placental genetic rescue (e.g. trisomic rescue) in an initially abnormal pregnancy, which can result in fetal uniparental disomy (UPD) or segmental UPD34. It is important to consider that evaluation of CPM or UPD has typically focused on karyotypic analyses. Studies utilizing genome-wide or array-based approaches suggest greater detection of abnormalities through these techniques35,36. Indeed, CPM for subchromosomal changes across the genome would be expected to occur more frequently than CPM for aneuploidy.
Sorting out the effect of CPM on diagnostic performance of NIFWGS is complicated further by our increasing understanding of genetic diversity and even genetic flexibility within an individual. Throughout the field of genetics, technological advances are providing glimpses into the nonabsoluteness of genetic categorization34. In fact, “CPM” may not be confined to the placenta in a substantial proportion of cases, with true fetal mosaicism occurring more commonly than previously understood and with varying phenotypic manifestations37. While genetic flexibility in disease – for example, loss of heterozygosity at HLA loci to evade immune detection in cancer38 – has been known for some time, it is becoming increasingly clear that it is also present in health (e.g., somatic revertant mosaicism39) and development34. Genetic diversity within an individual, through mosaicism34 or microchimerism40 may in fact be the rule rather than the exception. Understanding the impacts of CPM and true fetal mosaicism at the level of WGS is an entirely new area and an essential component of bringing NIFWGS to clinical practice.
Clinical/Ethical Aspects
The complexity and ambiguity of NIFWGS results must be considered as the field of noninvasive prenatal diagnostics moves forward, and communication of the resultant ambiguity to our patients must be a priority. From a practical perspective, it is clear that as the field advances, there will be an increasing need for subspecialized genetic counseling, as it is unlikely that these discussions could reasonably be incorporated into busy obstetric or perinatal practices. Though a comprehensive discussion of the ethical implications of NIFWGS is precluded here, this is an essential area to consider41. While the questions raised by NIFWGS are similar to those facing the field of genetics more broadly, the intersection of these issues with prenatal decision making for families mandates careful consideration of how best to incorporate this technology into practice.
Initial targeting of NIFWGS to specific patient populations will likely include those with current or prior otherwise unexplainable fetal abnormalities or losses. Ultimately, after larger studies of test performance, couples at risk for genetic disorders on the basis of race/ethnicity or family history may benefit in the intermediate term. With increasing public awareness and accessibility of commercial genetic screening opportunities, genetic information obtained in other contexts may drive a particular patient population to seek NIFWGS. In the long term, after significant study, one can envision utilization of this approach for widespread screening, akin to or replacing neonatal screening, and thus enabling prenatal provision of information and also facilitating immediate neonatal intervention for specific conditions.
Overall, clinical translation of techniques to comprehensively assess fetal genetic health from a maternal blood sample has the potential to reshape the future of prenatal diagnosis. Scientific progress in this area has vastly evolved in the last 30 years and continues to accelerate rapidly. Thoughtful shepherding of this technology to the prenatal bedside must include technical refinement, careful evaluation of test performance, appropriate targeting of patient populations, and effective communication in the face of our increasing appreciation of genetic ambiguity.
What’s already known about this topic?
Noninvasive fetal genome sequencing was recently demonstrated to be technically achievable, yielding an accurate and substantially complete result. Genome-wide inherited and de novo variation can be determined during pregnancy without risk to the mother or fetus. However, technical, ethical, and translational challenges must be addressed before this technique can be introduced in the clinic.
What does this study add?
We present an overview of noninvasive fetal genome sequencing for a clinical audience. We discuss the methodology in an accessible format and consider the key challenges along the path to clinical adoption.
Acknowledgments
Funding sources
Our work was supported in part by grants from the NIH/National Human Genome Research Institute (J.S.), a gift from the Washington Research Foundation (J.S.), and an NSF Graduate Research Fellowship (J.O.K.).
Footnotes
Conflict of interest disclosures
J.S. is a member of the scientific advisory board or serves as a consultant for Ariosa Diagnostics, Stratos Genomics, Good Start Genetics, and Adaptive Biotechnologies. A provisional patent application has been deposited for aspects of these methods (M.W.S., J.O.K., and J.S.; “Non-invasive whole genome sequencing of a human fetus”; 61/651,356)
References
- 1.Merkatz IR, Nitowsky HM, Macri JN, Johnson WE. An association between low maternal serum alpha-fetoprotein and fetal chromosomal abnormalities. Am J Obstet Gynecol. 1984;148:886–894. doi: 10.1016/0002-9378(84)90530-1. [DOI] [PubMed] [Google Scholar]
- 2.ACOG Committee on Practice Bulletins ACOG Practice Bulletin No. 77: screening for fetal chromosomal abnormalities. Obstet Gynecol. 2007;109:217–227. doi: 10.1097/00006250-200701000-00054. [DOI] [PubMed] [Google Scholar]
- 3.Lapaire O, Holzgreve W, Oosterwijk JC, Brinkhaus R, Bianchi DW. Georg Schmorl on Trophoblasts in the Maternal Circulation. Placenta. 2007;28:1–5. doi: 10.1016/j.placenta.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Schröder J, De la Chapelle A. Fetal lymphocytes in the maternal blood. Blood. 1972;39:153–162. [PubMed] [Google Scholar]
- 5.Herzenberg LA, Bianchi DW, Schröder J, Cann HM, Iverson GM. Fetal cells in the blood of pregnant women: detection and enrichment by fluorescence-activated cell sorting. Proc Natl Acad Sci USA. 1979;76:1453–1455. doi: 10.1073/pnas.76.3.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lo YM. Noninvasive prenatal diagnosis using fetal cells in maternal blood. Methods Mol Med. 1996;5:237–244. doi: 10.1385/0-89603-346-5:237. [DOI] [PubMed] [Google Scholar]
- 7.Bianchi DW, et al. Fetal gender and aneuploidy detection using fetal cells in maternal blood: analysis of NIFTY I data. Prenat Diagn. 2002;22:609–615. doi: 10.1002/pd.347. [DOI] [PubMed] [Google Scholar]
- 8.Lo YM, et al. Presence of fetal DNA in maternal plasma and serum. Lancet. 1997;350:485–487. doi: 10.1016/S0140-6736(97)02174-0. [DOI] [PubMed] [Google Scholar]
- 9.Shendure J, Aiden EL. The expanding scope of DNA sequencing. Nature Biotechnology. 2012;30:1084–1094. doi: 10.1038/nbt.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.American College of Obstetricians and Gynecologists Committee on Genetics Committee Opinion No. 545: Noninvasive prenatal testing for fetal aneuploidy. Obstet Gynecol. 2012;120:1532–1534. doi: 10.1097/01.AOG.0000423819.85283.f4. [DOI] [PubMed] [Google Scholar]
- 11.Reddy UM, et al. Karyotype versus Microarray Testing for Genetic Abnormalities after Stillbirth. N Engl J Med. 2012;367:2185–2193. doi: 10.1056/NEJMoa1201569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wapner RJ, et al. Chromosomal Microarray versus Karyotyping for Prenatal Diagnosis. N Engl J Med. 2012;367:2175–2184. doi: 10.1056/NEJMoa1203382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lo YMD, et al. Maternal Plasma DNA Sequencing Reveals the Genome-Wide Genetic and Mutational Profile of the Fetus. Sci Transl Med. 2010;2:61ra91–61ra91. doi: 10.1126/scitranslmed.3001720. [DOI] [PubMed] [Google Scholar]
- 14.Kitzman JO, et al. Noninvasive whole-genome sequencing of a human fetus. Sci Transl Med. 2012;4:137ra76. doi: 10.1126/scitranslmed.3004323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan HC, et al. Non-invasive prenatal measurement of the fetal genome. 2012:1–7. doi: 10.1038/nature11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tjoa ML, Cindrova-Davies T, Spasic-Boskovic O, Bianchi DW, Burton GJ. Trophoblastic oxidative stress and the release of cell-free feto-placental DNA. Am J Pathol. 2006;169:400–404. doi: 10.2353/ajpath.2006.060161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan HC, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR. Analysis of the Size Distributions of Fetal and Maternal Cell-Free DNA by Paired-End Sequencing. Clinical Chemistry. 2010;56:1279–1286. doi: 10.1373/clinchem.2010.144188. [DOI] [PubMed] [Google Scholar]
- 18.Poon LLM, Leung TN, Lau TK, Chow KCK, Lo YMD. Differential DNA methylation between fetus and mother as a strategy for detecting fetal DNA in maternal plasma. Clinical Chemistry. 2002;48:35–41. [PubMed] [Google Scholar]
- 19.Jiang P, et al. FetalQuant: deducing fractional fetal DNA concentration from massively parallel sequencing of DNA in maternal plasma. Bioinformatics. 2012;28:2883–2890. doi: 10.1093/bioinformatics/bts549. [DOI] [PubMed] [Google Scholar]
- 20.Kitzman JO, et al. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nature Biotechnology. 2011;29:59–63. doi: 10.1038/nbt.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peters BA, et al. Accurate whole-genome sequencing and haplotyping from 10 to 20 human cells. Nature. 2012;487:190–195. doi: 10.1038/nature11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang H, Chen X, Wong WH. Completely phased genome sequencing through chromosome sorting. Proc Natl Acad Sci USA. 2011;108:12–17. doi: 10.1073/pnas.1016725108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma L, et al. Direct determination of molecular haplotypes by chromosome microdissection. Nat Meth. 2010;7:299–301. doi: 10.1038/nmeth.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan HC, Wang J, Potanina A, Quake SR. Whole-genome molecular haplotyping of single cells. Nature Biotechnology. 2010;29:51–57. doi: 10.1038/nbt.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kong A, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bell CJ, et al. Carrier Testing for Severe Childhood Recessive Diseases by Next-Generation Sequencing. Sci Transl Med. 2011;3:65ra4–65ra4. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ng SB, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Roak BJ, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.American College of Obstetricians Gynecologists. ACOG Practice Bulletin No. 88 December 2007. Invasive prenatal testing for aneuploidy. Obstet Gynecol. 2007;110:1459–1467. doi: 10.1097/01.AOG.0000291570.63450.44. [DOI] [PubMed] [Google Scholar]
- 30.Kalousek DK, Dill FJ. Chromosomal mosaicism confined to the placenta in human conceptions. Science. 1983;221:665–667. doi: 10.1126/science.6867735. [DOI] [PubMed] [Google Scholar]
- 31.Choi H, et al. Fetal aneuploidy screening by maternal plasma DNA sequencing: ‘False positive’ due to confined placental mosaicism. Prenat Diagn. 2012 doi: 10.1002/pd.4024. [DOI] [PubMed] [Google Scholar]
- 32.Hahnemann JM, Vejerslev LO. Accuracy of cytogenetic findings on chorionic villus sampling (CVS)--diagnostic consequences of CVS mosaicism and non-mosaic discrepancy in centres contributing to EUCROMIC 1986–1992. Prenat Diagn. 1997;17:801–820. doi: 10.1002/(sici)1097-0223(199709)17:9<801::aid-pd153>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 33.Carroll SG, Davies T, Kyle PM, Abdel-Fattah S, Soothill PW. Fetal karyotyping by chorionic villus sampling after the first trimester. Br J Obstet Gynaecol. 1999;106:1035–1040. doi: 10.1111/j.1471-0528.1999.tb08110.x. [DOI] [PubMed] [Google Scholar]
- 34.Engel E. A fascination with chromosome rescue in uniparental disomy: Mendelian recessive outlaws and imprinting copyrights infringements. Eur J Hum Genet. 2006;14:1158–1169. doi: 10.1038/sj.ejhg.5201619. [DOI] [PubMed] [Google Scholar]
- 35.Inbar-Feigenberg M, et al. Mosaicism for genome-wide paternal uniparental disomy with features of multiple imprinting disorders: Diagnostic and management issues. Am J Med Genet. 2012;161:13–20. doi: 10.1002/ajmg.a.35651. [DOI] [PubMed] [Google Scholar]
- 36.Filges I, et al. aCGH on chorionic villi mirrors the complexity of fetoplacental mosaicism in prenatal diagnosis. Prenat Diagn. 2011;31:473–478. doi: 10.1002/pd.2721. [DOI] [PubMed] [Google Scholar]
- 37.Stetten G, et al. Reevaluating confined placental mosaicism. Am J Med Genet. 2004;131A:232–239. doi: 10.1002/ajmg.a.30363. [DOI] [PubMed] [Google Scholar]
- 38.Vago L, et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. N Engl J Med. 2009;361:478–488. doi: 10.1056/NEJMoa0811036. [DOI] [PubMed] [Google Scholar]
- 39.Choate KA, et al. Mitotic Recombination in Patients with Ichthyosis Causes Reversion of Dominant Mutations in KRT10. Science. 2010;330:94–97. doi: 10.1126/science.1192280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nelson JL. The otherness of self: microchimerism in health and disease. Trends Immunol. 2012;33:421–427. doi: 10.1016/j.it.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tabor HK, et al. Non-invasive fetal genome sequencing: Opportunities and challenges. Am J Med Genet. 2012 doi: 10.1002/ajmg.a.35545. [DOI] [PMC free article] [PubMed] [Google Scholar]