

Genetic alterations in pulmonary arterial hypertension (PAH) have become increasingly recognized in both known familial or heritable as well as sporadic or idiopathic PAH. Unrecognized genetic alterations have now been found in up to 40% of the idiopathic PAH (IPAH), in which no familial predisposition is apparent.1 Bone morphogenetic protein receptor II (BMPR-II) is the most common gene implicated in this hereditary form of PAH; furthermore, it is implicated in the pathogenesis of non-hereditary forms of PAH with significant reduction in expression of BMPR-II in both IPAH and experimental animal models of pulmonary hypertension (PH).2 Bone morphogenetic proteins represent the largest group of cytokines in the TGF-β superfamily and regulate growth, differentiation and apoptosis in multiple cell types, while BMPR-II has been shown to have unique roles in differing cells.
BMPR-II is constitutively active at the cell membrane and ligand stimulation initiates cross- linking with BMPR-I to form a receptor complex that is necessary to activate intracellular signaling. BMPR-II is most highly expressed in endothelial cells in the pulmonary vasculature and BMPR-II activation leads to increased proliferation and decreased apoptosis through Smad signaling.3, 4 This is in contrast to pulmonary arterial smooth muscle cells (PASMCs) where BMP activation leads to inhibition of proliferation and increased apoptosis through Smad signaling in large vessels; though, in small pulmonary arteries a proliferative effect is seen through activation of ERK and MAPK which inhibits Smad signaling.5, 6 It is these unique, yet complementary functions that make BMPR-II mutations particularly damaging in the pulmonary circulation leading to development of PAH. A dysfunctional mutation of BMPR-II, as in heritable PAH (HPAH), or downregulation of protein expression, as in IPAH and animal models, can lead to endothelial dysfunction hallmarked by abnormal barrier function through increased apoptosis7, while also leading to vascular medial hypertrophy through increased proliferation and decreased apoptosis of distal arteriole PASMCs.
Autophagy represents a homeostatic mechanism essential for cell survival. Cell stress, including hypoxia, nutrient deprivation or reduction in growth factor stimulation, can all lead to autophagy responses in which cytoplasmic contents are collected and recycled to produce amino acids and fatty acids necessary for cellular response and ATP production.8 Another function is to clear unnecessary or toxic components of the cell cytoplasm, either in order to salvage the cell or as a mechanism to trigger cell death in a non-apoptotic fashion. After collecting cellular debris, mature autophagosomes fuse with lysosomes which results in degradation and recycling of this collected material. Each step of this process is highly regulated and dysfunction of this system has been implicated in multiple disease processes including malignancy, neurodegeneration, liver and heart disease.8 Recently, dysfunctional autophagy has been implicated in pulmonary diseases, with particularly strong evidence in COPD where cigarette smoke-induced emphysema is associated with increased numbers of autophagosomes, which is thought to be a result of imbalance in autophagosome production versus clearance.9 Many of the disease related autophagy studies have implicated this imbalance as the mechanism by which autophagy influences disease development and progression.
In IPAH, autophagy has been shown to be upregulated, with the marker for mature autophagosomes, LC3B-II, having increased expression compared to healthy controls.10 The role of autophagy in PH remains inconclusive but appears to play an important role in vascular remodeling.11 Pulmonary artery endothelial cells exposed to hypoxia have increased autophagy which is thought to be a protective mechanism, as LC3B-II knockout mice have exaggerated PH in response to chronic hypoxia.10 While persistent PH in fetal lambs, an experiment model of persistent PH of the newborn, is associated with increased autophagy thought to be detrimental to fetal angiogenesis and inhibiting autophagy can lead to restoration of adequate angiogenesis.12 Yet, as with any highly regulated cellular mechanism, proper cellular balance appears to be essential for normal function and this early data has confirmed the complicated nature of autophagy in disease processes. This appears to be the case with regard to BMPR-II and autophagy. Previous work by Morrell and his colleagues has shown that the lower levels of BMPR-II seen in experimental models of PH appear to be at least in part due to BMPR-II being targeted for ubiquitination and degradation via the lysosome.13
In this issue of Circulation Research, Long and colleagues discuss the role of autophagy and lysosomal BMPR-II degradation in the pathogenesis of experimental PH.14 They provide new evidence linking increased autophagy with downregulation of BMPR-II/Smad pathway in the development of PAH. In a disease in which new therapeutic targets are desperately needed, this may well be a novel target. The authors were able to show both prevention and partial reversal of monocrotaline (MCT)-induced PH in rats using chloroquine, an inhibitor of autophagy. They further were able to show that chloroquine's effect was through inhibition of autophagy both in vivo and in vitro, with downstream effects resulting in reduced proliferation and increased apoptosis of PASMCs. The diminished protein expression of BMPR-II and Smad signaling in this experimental rat model of PH was restored with the use of chloroquine. This inhibition of autophagy and lysosomal degradation is established in vitro by using knockdown of autophagy specific ATG5 as well as concanamycin A treatment, a specific inhibitor of lysosomal degradation, which were both associated with increased expression of BMPR-II and decreased LC3B-II. They have concluded that chloroquine ameliorates MCT-induced PH in the rat and this effect is mediated through downregulated autophagy inhibiting lysosomal degradation of BMPR-II.
The results from this study represent very interesting insight into the potential mechanism of attenuated BMPR-II expression in human PAH. Enhanced autophagy and lysosomal degradation may be a novel pathogenic mechanism and potential therapeutic target for this disease. A compound that has made a resurgence as of late, chloroquine and its less toxic cousin hydroxychloroquine, have not only remained mainstays in malaria treatment and chemoprophylaxis, but steroid-sparing anti-inflammatory effects of these medications have proved useful in systemic lupus erythematosus, rheumatoid arthritis, and sarcoidosis. Repurposing of a medication that is both well tolerated and inexpensive makes it an attractive agent to clinicians, especially to physicians in the developing countries where the commonly used drugs for PAH are not available. The current results provide a plausible mechanism by which the experimental PAH was both prevented and reversed, yet both chloroquine and hydroxycholoroquine have multiple known and unknown cellular and molecular targets in the human body and it is unlikely that inhibition of autophagy is the sole effect on the pulmonary vasculature.
There are growing data on the role of inflammation in PAH. Cytokines, particularly IL-6 have largely been implicated in the disease process, both affecting vascular remodeling as well as vasoconstriction, which should not be unexpected given the similarities between IPAH and associated PAH as a result of chronic inflammatory disorders such as systemic lupus erythematosus, rheumatoid arthritis, and systemic sclerosis. The fact that hydroxychloroquine has in more recent years been found to be a significant immunomodulator with steroid-sparing effects that are beneficial in many of these diseases would potentially implicate its anti-inflammatory properties in the treatment of PAH. Indeed, the fact that hydroxychloroquine treatment is associated with reduced levels of IL-6 in SLE patients, it would be reasonable to assume similar effects in the pulmonary vasculature which could have potential benefits in a disease characterized by inflammation and elevated IL-6 levels, though studies are lacking at this time.15 This immunomodulatory effect may also be contributing to PAH prevention and reversal in this animal model, though this effect would still support potential clinical efficacy and safety trials in human patients.
Animal models have always presented problems in research, and the complexity of PAH makes this particularly challenging and experimental models are invariably flawed. MCT, a plant derived pyrrolizidine alkaloid, leads to endothelial cell dysfunction and vascular remodeling through unclear mechanisms, though many believe that inflammatory mechanisms play an important role in the pathogenesis of MCT-induce experimental PH. Given the propensity for an exaggerated inflammatory response, it may not represent an ideal model of PAH and would presumably give preference to therapeutic agents with immune-related effects; though other experimental models such as hypoxia-induced PH or Sugen/hypoxia mediated PH are not without their limitations. It is more likely that these models each emphasize a small part of a large and complicated disease process, and using them in a complementary manner may likely provide better insight into a disease process than simply using a single model alone.
The complexity of human disease is becoming ever present. New and exciting molecular pathways and pathogenic mechanisms have met with both success and failure in translation to real world disease. The complexity that surrounds PAH and BMPR-II, in particular, makes it reasonable to assume that the answer will not come in a single pill. Still, with the undeniable knowledge that this single receptor plays such an important role in the pathogenesis of PAH, each new discovery is a piece of the puzzle leading toward a potential cure. This article provides a new and important piece to that puzzle, and a better understanding of the abnormal degradation of BMPR-II will ultimately lead to a better understanding of the mechanisms behind PAH. The current article by Long and colleagues provides encouraging data about chloroquine and hydroxychloroquine and further studies will help enhance understanding of the effects on the pulmonary circulation and the potential role in the treatment of patients with PAH.
Figure 1. Proposed role of autophagy in BMPR-II downreglation in PAH.
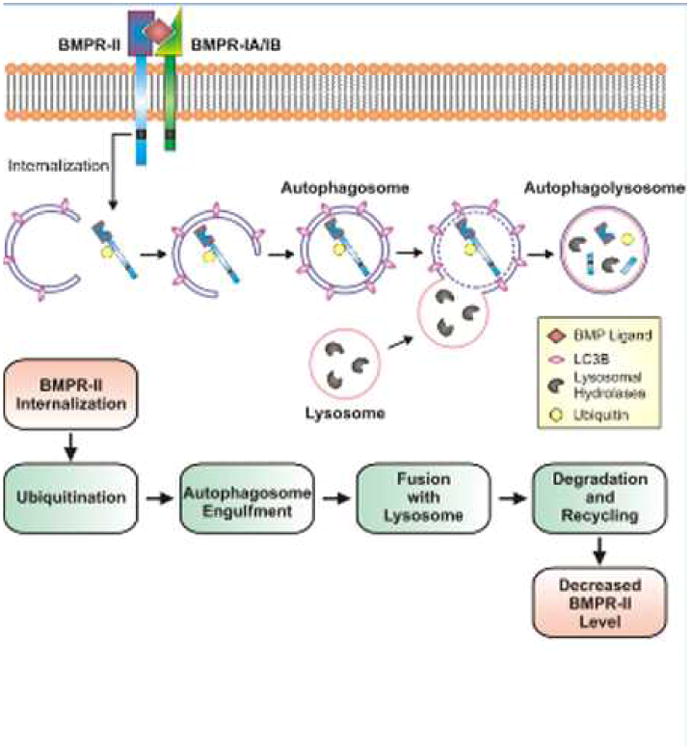
Decreased BMPR-II protein levels in pulmonary vascular smooth muscle and endothelial cells are observed in patients with IPAH and animals with experimental PH. Long and colleagues propose a mechanism in which BMPR-II is internalized from the plasma membrane and ubiquitinated. The ubiquitinated BMPR-II protein is then engulfed by the autophagosome and degraded through a lysosomal-mediated pathway. BMP, bone morphogenetic protein; BMPR, bone morphogenetic protein receptor; LC3B, microtubule-associated protein light chain 3.
Acknowledgments
Sources of Funding: N/A
Footnotes
Disclosures: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Humbert M, Elliott GC, Ward K, Yacoub M, Mikhail G, Rogers P, Newman J, Wheeler L, Higenbottam T, Gibbs JS, Egan J, Crozier A, Peacock A, Allcock R, Corris P, Loyd JE, Trembath RC, Nichols WC. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-b family. J Med Genet. 2000;37:741–745. doi: 10.1136/jmg.37.10.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morrell NW. Role of bone morphogenetic protein receptors in the development of pulmonary arterial hypertension. Adv Exp Med Biol. 2010;661:251–264. doi: 10.1007/978-1-60761-500-2_16. [DOI] [PubMed] [Google Scholar]
- 3.Cai J, Pardali E, Sanchez-Duffhues G, ten Dijke P. BMP signaling in vascular diseases. FEBS Lett. 2012;586:1993–2002. doi: 10.1016/j.febslet.2012.04.030. [DOI] [PubMed] [Google Scholar]
- 4.Yang X, Long L, Reynolds PN, Morrell NW. Expression of mutant BMPR-II in pulmonary endothelial cells promotes apoptosis and a release of factors that stimulate proliferation of pulmonary arterial smooth muscle cells. Pulm Circ. 2011;1:103–110. doi: 10.4103/2045-8932.78100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang X, Long L, Southwood M, Rudarakanchana N, Upton PD, Jeffery TK, Atkinson C, Chen H, Trembath RC, Morrell NW. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ Res. 2005;96:1053–1063. doi: 10.1161/01.RES.0000166926.54293.68. [DOI] [PubMed] [Google Scholar]
- 6.Zhang S, Fantozzi I, Tigno DD, Yi ES, Platoshyn O, Thistlethwaite PA, Kriett JM, Yung G, Rubin LJ, Yuan JX. Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2003;285:L740–754. doi: 10.1152/ajplung.00284.2002. [DOI] [PubMed] [Google Scholar]
- 7.Teichert-Kuliszewska K, Kutryk MJ, Kuliszewski MA, Karoubi G, Courtman DW, Zucco L, Granton J, Stewart DJ. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res. 2006;98:209–217. doi: 10.1161/01.RES.0000200180.01710.e6. [DOI] [PubMed] [Google Scholar]
- 8.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 9.Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee SJ, Ifedigbo E, Parameswaran H, Ryter SW, Choi AM. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A. 2010;107:18880–18885. doi: 10.1073/pnas.1005574107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee SJ, Smith A, Guo L, Alastalo TP, Li M, Sawada H, Liu X, Chen ZH, Ifedigbo E, Jin Y, Feghali-Bostwick C, Ryter SW, Kim HP, Rabinovitch M, Choi AM. Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med. 2011;183:649–658. doi: 10.1164/rccm.201005-0746OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin Y, Choi AM. Cross talk between autophagy and apoptosis in pulmonary hypertension. Pulm Circ. 2012;2:407–414. doi: 10.4103/2045-8932.105029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teng RJ, Du J, Welak S, Guan T, Eis A, Shi Y, Konduri GG. Cross talk between NADPH oxidase and autophagy in pulmonary artery endothelial cells with intrauterine persistent pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2012;302:L651–663. doi: 10.1152/ajplung.00177.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Durrington HJ, Upton PD, Hoer S, Boname J, Dunmore BJ, Yang J, Crilley TK, Butler LM, Blackbourn DJ, Nash GB, Lehner PJ, Morrell NW. Identification of a lysosomal pathway regulating degradation of the bone morphogenetic protein receptor type II. J Biol Chem. 2010;285:37641–37649. doi: 10.1074/jbc.M110.132415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Long L, Yang X, Southwood M, Lu J, Marciniak SJ, Dunmore BJ, Morrell NW. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal BMPR-II degradation. Circ Res. 2013 doi: 10.1161/CIRCRESAHA.111.300483. [DOI] [PubMed] [Google Scholar]
- 15.Wozniacka A, Lesiak A, Narbutt J, McCauliffe DP, Sysa-Jedrzejowska A. Chloroquine treatment influences proinflammatory cytokine levels in systemic lupus erythematosus patients. Lupus. 2006;15:268–275. doi: 10.1191/0961203306lu2299oa. [DOI] [PubMed] [Google Scholar]