

Abstract
Middle-ear mucosa maintains middle-ear pressure. However, the majority of surgical cases exhibit inadequate middle-ear mucosal regeneration, and mucosal transplantation is necessary in such cases. The aim of the present study was to assess the feasibility of transplantation of isolated mucosal cells encapsulated within synthetic self-assembling peptide nanofiber scaffolds using PuraMatrix, which has been successfully used as scaffolding in tissue engineering, for the repair of damaged middle-ear. Middle-ear bullae with mucosa were removed from Sprague Dawley (SD) transgenic rats, transfected with enhanced green fluorescent protein (EGFP) transgene and excised into small pieces, then cultured up to the third passage. After surgical elimination of middle-ear mucosa in SD recipient rats, donor cells were encapsulated within PuraMatrix and transplanted into these immunosuppressed rats. Primary cultured cells were positive for pancytokeratin but not for vimentin, and retained the character of middle-ear epithelial cells. A high proportion of EGFP-expressing cells were found in the recipient middle-ear after transplantation with PuraMatrix, but not without PuraMatrix. These cells retained normal morphology and function, as confirmed by histological examination, immunohistochemistry, and electron microscopy, and multiplied to form new epithelial and subepithelial layers together with basement membrane. The present study demonstrated the feasibility of transplantation of cultured middle-ear mucosal epithelial cells encapsulated within PuraMatrix for regeneration of surgically eliminated mucosa of the middle-ear in SD rats.
Keywords: nanofiber, synthetic self-assembling peptide scaffolds, regeneration, middle-ear mucosa, in situ tissue engineering
Introduction
Despite advances in public health and medical care, otitis media remains a worldwide clinical problem associated in some patients with hearing loss. Middle-ear surgery is one of the most effective strategies for noncholesteatomatous and cholesteatomatous chronic otitis media. The main goals of middle-ear surgery are hearing improvement and the complete removal of active disease.1 In any middle-ear surgery, it is necessary to preserve middle-ear mucosa, since it plays an important role in the maintenance of middle-ear pressure.2 However, it is often difficult to preserve middle-ear mucosa, due to the complete removal of the active focus of otitis media. Impairment of gas exchange in the mastoid mucosa, which follows middle-ear surgery, can lead to retraction of the tympanic membrane, causing recurrence of otitis media or hearing loss.2 Regeneration of the mucosa from the tympanic cavity to the mastoid after surgery could potentially prevent adhesion within the mastoid caused by scar contraction and restoration of mastoid aeration. We reported previously the usefulness of a large silicone sheet for recovery of mastoid aeration after staged ear surgery with soft-wall reconstruction and mastoidectomy.3 In some of these cases, regeneration of the mucosal epithelium in the mastoid cavity was noted, though it was incomplete.3
Clinically, there is a need for therapeutic trials based on regenerative medicine. Previous experimental studies described the use of a sheet of mucosal cells grown on a collagen gel to promote mucosal regeneration, which was successfully implanted in the middle-ear after surgery.4,5 However, with regard to the clinical application of this technology, cell-sheet engineering has certain limitations related to the size, location, and irregularity of middle-ear mucosal defect caused by middle-ear surgery. In comparison, in situ tissue engineering is a new field aimed at promotion and acceleration of regeneration and repair of defective and damaged tissues based on the natural healing potential of patients themselves.6 Thus, in situ tissue engineering can be considered another suitable method for regeneration of middle-ear mucosa.
The basic concept of biomaterial-based tissue engineering was originally introduced by Langer and Vacanti.7 To achieve the therapeutic efficacy of cell transplantation, it is necessary to provide an environment suitable for cell function and survival.6–9 Tissue engineering requires scaffolds that serve as substrates for seeding cells, provide physical support that guides new tissue formation, promote the regeneration of natural tissues, or enhance the creation of biological substitutes for defective or lost organs.10,11 Many types of scaffolds, such as biological extracellular matrix (ECM) and biological or synthetic hydrogels, have been developed for cell transplantation over the last several decades. Biological ECM, eg, Matrigel (BD Biosciences, San Jose, CA, USA), which consists of type IV collagen, laminin, and heparan sulfate proteoglycan, seems to help create a suitable microenvironment.12,13 Moreover, due to their innate structural and compositional similarities to ECM, hydrogels have been used as the material of choice in many applications in regenerative medicine and have been used as drug and cell carriers in the field of tissue engineering.14,15 Hydrogels are three-dimensional networks formed from hydrophilic homopolymers, copolymers, or macromers. Biological hydrogels prepared from agarose, alginate, chitosan, hyaluronan, fibrin, collagen, and many other materials have been used in tissue engineering.14–16 However, with regard to their clinical application, biological ECM and biological hydrogels carry the risk of contamination.8,13 On the other hand, synthetic hydrogels, consisting of neutrally charged synthetic monomers, such as poly(2-hydroxyethyl methacrylate) (PHEMA), poly(vinyl acetate), and poly(ethylene glycol) (PEG) hydrogels do not have the drawback of contamination with infectious pathogens and might be ideal for tissue engineering. Much of the success with synthetic hydrogels in tissue engineering is due to the use of PHEMA hydrogels.15 One of the earliest uses of PHEMA was artificial cornea and keratoprosthesis, and the success of PHEMA hydrogels resulted in the important development of synthetic scaffolds.15,17 On the other hand, poly(vinyl acetate) hydrogels were mainly developed for avascular tissues based on their elasticity, low coefficient of friction, and structural properties similar to natural cartilage.18 However, these hydrogels are polymerized chemically (for example, by photopolymerization, including free radical polymerization, by disulfide-bond formation, or by reaction between thiols and acylate), and have the potential of toxic environments, which may denature incorporated proteins, embedded cells, and surrounding tissues.15,19,20 PEG is hydrophilic and biocompatible, with properties that limit immunogenicity and antigenicity.21,22 As with many other synthetic hydrogels, PEG hydrogels have been used successfully in tissue-engineering applications that do not require the scaffold to be vascularized, such as skin and cartilage.15 PEG hydrogels are also polymerized chemically, but copolymers of PEG with biodegradable biocompatible aliphatic polyesters, eg, polylactide, polyglycolide, poly(ɛ-caprolactam), or poly([R]-3-hydroxy-butyrate), can be polymerized physically (eg, by temperature, pH, or both).20 Thus, synthetic polymer hydrogels consisting of PEG or block copolymers have been the most successful tissue-engineering applications to date.15,20 On the other hand, another important class of synthetic hydrogels is self-assembled peptide hydrogels that assemble physically to form fibers or other kinds of nanoscale structures. Self-assembled peptides provide a number of unique advantages, such as the ability to form gels and relatively easy gel functionalization,15 compared to the aforementioned synthetic polymer hydrogels. Some of the important properties as matrices for tissue engineering are degradability, injectability, ease of handling, and water content.14 Furthermore, most self-assembled peptide hydrogels conform to these properties.10,15 One of the attractive self-assembling peptide hydrogels used as three-dimensional scaffolds for tissue engineering comprises ionic self-complementary peptides, which form stable β-sheet structures that self-assemble to form nanofibers.11 These nanofibers form interwoven matrices that further form a high-water-content scaffold hydrogel.11
In the present study, we evaluated the synthetic self-assembling peptide nanofiber PuraMatrix (BD Biosciences) scaffolds for cell transplantation and in situ tissue engineering for regeneration of middle-ear mucosa in an animal model. PuraMatrix is a 16-amino acid synthetic peptide (Ac-[RADA]4-CONH2), characterized by stable B-sheet structure with ionic side-chain interactions and known to undergo self-assembly into hydrogels containing ~99.5% w/v water.8,9,23 Upon the introduction of millimolar amounts of monovalent cations (physiological condition), PuraMatrix undergoes self-assembly into nanofibers on a scale (average pore size 5–200 nm) similar to the ECM and provides a suitable biological scaffold allowing encapsulated cells to migrate within that microenvironment and create their own microenvironments rapidly, including production of their own ECM, although other synthetic polymer hydrogels are often made of microfibers after gelation with diameters of 10–100 μm, which are much larger than native ECM.8 Moreover, these peptides have the motif arginine-alanine-aspartic acid (RAD), which is similar to the ubiquitous integrin receptor-binding site arginine-glycine-aspartic acid (RGD), which has been studied in the context of cell attachment.8,11,24 At present, several clinical trials of cell therapies using PuraMatrix have been reported for cartilage,25,26 myocardial,27 neural,28–31 and hepatic32,33 regeneration, indicating the potential usefulness of PuraMatrix.
For analysis of cell transplantation, it is important to clarify the origin of the regenerating tissue. In the model used in this study, donor cells were prepared from Sprague Dawley (SD) transgenic (Tg) rats carrying the enhanced green fluorescence protein (EGFP) transgene,34,35 and EGFP expression was used as a tracer of donor cells after transplantation. Thus, in the present study, we evaluated the usefulness of isolated middle-ear mucosal epithelial cells encapsulated within PuraMatrix, which formed nanofiber scaffolds for in situ tissue engineering of middle-ear mucosal tissue, and identified the origin of regenerating tissue by EGFP expression of donor cells.
Materials and methods
Experimental animals
Ten male SD-Tg rats (4–5 weeks old weight 90–140 g) were used for primary culture of middle-ear epithelial cells, for calculation of the doubling time (DT), and as donor cells. Fourteen male SD rats (6–7 weeks old weight 150–160 g) were used as recipients. All animal-care and experimental procedures were conducted in accordance with the Guidelines for Animal Experimentation of Nagasaki University and were approved by the Institute of Animal Care and Use Committee.
Primary culture of middle-ear epithelial cells from SD-Tg rats
Donor cells were prepared by primary explant culture, as described originally by Harrison36 and Carrel.37 The primary explant culture was prepared by allowing cells to migrate out of fragments of tissue adhering to a suitable substrate. The technique for cultured epithelium can be applied to middle-ear epithelium, which has been successfully cultured from rats, guinea pigs, gerbils, chinchillas, and humans.38 SD-Tg rats (n = 4) were killed by intraperitoneal injection of 200 mg/kg pentobarbital. The middle-ear bullae with attached mucosa were removed and minced into small pieces, which were then seeded onto a collagen I-coated dish containing serum-free culture medium (1:1 mixture of Dulbecco’s Modified Eagle’s Medium [Life Technologies, Carlsbad CA, USA] and bronchial epithelial cell growth medium [Lonza, Basel, Switzerland]) with supplemental kit (Lonza) containing hydrocortisone (0.5 μg/mL), insulin (5.0 μg/mL), transferrin (10 μg/mL), triiodothyronine (6.5 ng/mL), epinephrine (0.5 μg/mL), bovine pituitary extract (10 μg/mL), epidermal growth factor (0.5 ng/mL), retinoic acid (0.1 ng/mL), gentamicin (50 μg/mL), and amphotericin B (50 ng/mL).39 The culture dish was maintained under a humidified atmosphere of 5% CO2 in air at 37°C, and the outgrowth cells were subcultured up to the third passage.
Cell-viability test
The number of viable cells present in a cell suspension was determined by the trypan blue dye-exclusion test (trypan blue stain 0.4% [Life Technologies]).
Calculation of cell doubling time
Cells were prepared from six SD-Tg rats by explant culture, as previously described. The cells were cultured up to the second passage and collected after treatment with 0.05% trypsin/phosphate-buffered saline (PBS) and then detached from the culture dish for calculation of the DT. A total of 7.5 × 104 cells were seeded per 35 mm dish. Using a hemocytometer, the cell number in three dishes was determined after trypsinization, and the average number was calculated every day. On day 9, the growth curve was constructed and the DT was calculated using the following formula: DT = t log (2)/(log n – log n0), where t = total time elapsed n = final number of cells, and n0 = initial number of cells.
Preparation of donor cells before transplantation
Donor cells were prepared from four SD-Tg rats by explant culture, as previously described. The cells were cultured up to the third passage and collected after treatment with 0.05% trypsin/PBS and then detached from the culture dish for transplantation. PuraMatrix consists of standard amino acids (1% w/v) and 99% water, and sol–gel transition occurs upon interaction of the peptide monomers in water with the electrolyte solution. The cells were washed in 10% sucrose solution and encapsulated within PuraMatrix. An equal volume of the culture medium was added to induce gelation (0.25% w/v) and prepared donor cells at a final density of 0.5 × 106 (low density) or 1.0 × 106 cells/mL (high density) (Figure 1A). Because PuraMatrix exhibits a low pH, this process was performed rapidly to minimize the contact time between cells and material prior to the addition of culture media. The cell densities selected in this study have been described previously in a protocol for cell seeding with PuraMatrix and used for myocardial progenitor cell transplantation with PuraMatrix.24 The cells encapsulated within PuraMatrix were incubated under a humidified atmosphere and 5% CO2 in air at 37°C overnight after gelation with culture medium.
Figure 1.

(A and B) Schematic diagram of in situ tissue engineering model of rat middle-ear epithelium. (A) Donor cells (gray dots) were prepared from Sprague Dawley transgenic (SD-Tg) rats by explant culture and encapsulated within PuraMatrix before transplantation. (B) Donor cells were transplanted into the middle-ear bullae of recipient rats that had undergone surgical elimination of the middle-ear mucosa before transplantation. Creation of a small hole by surgical drill (arrow) (a); mucosa elimination (arrows) (b); before transplantation (c); transplantation of epithelial cells encapsulated within PuraMatrix (d); filling of the hole with bone wax (black circle) (e).
Transplantation of cultured cells with peptide hydrogel
Recipient SD rats (n = 14) were anesthetized by intraperitoneal injection of 30 mg/kg pentobarbital. A small incision was made behind the ear, and a small hole with a 3 mm diameter was created in the posterior-superior surface of the middleear bulla using a surgical drill (Figure 1B [a]). Using this approach, the internal mucosa of the middle-ear bulla was eliminated as thoroughly as possible under a surgical microscope (Figure 1B [b]) (the middle-ear mucosa-eliminated model). The donor cells encapsulated within PuraMatrix were transplanted into the bullae of recipients through the hole at 100 μL of cell mixture per ear (Figure 1B [c and d]). The hole was subsequently filled with bone wax (TMI, Tokyo, Japan) (Figure 1B [e]). As a control, donor cells with culture medium were also transplanted by injection. FK506 (Astellas Pharma, Tokyo, Japan) was administered at a dose of 0.32 mg/kg/day for 5 consecutive days per week,40 together with penicillin G at a dose of 22 U/g/day every day by intramuscular injection after transplantation. At days 0, 7, 14, and 28, recipient rats were killed by intraperitoneal injection of pentobarbital, and middle-ear bullae were collected for analysis.
Antibodies
The antibodies used in the present study are listed in Table 1. A mouse monoclonal antibody against cytokeratins 5, 6, 8, and 18 (pancytokeratin [5/6/8/18]) (dilution 1:200 or 1:20) (Novocastra, Newcastle, UK) was used as an epithelial cellular marker,41–43 and a mouse monoclonal antibody against vimentin (Dako, Glostrup, Denmark) (dilution 1:800) was used as a mesenchymal cell marker.41,44,45 A rabbit polyclonal antibody against collagen type III (dilution 1:400) (Abcam, Cambridge, UK) was used as a marker of elaboration of ECM component,46,47 and a rabbit polyclonal antibody against collagen type IV (dilution, 1:50) (LSL, Tokyo, Japan) was used as a marker of basement membrane.46,48 A monoclonal antibody against E-cadherin (dilution 1:1000) (BD Biosciences) was used as a marker of the adhesive core of adherens junctions.48,49 Normal goat immunoglobulin (Ig)-G, normal mouse IgG, and normal rabbit IgG were purchased from Sigma-Aldrich (St Louis, MO, USA). Horseradish peroxidase (HRP) goat anti-mouse IgG was purchased from Chemicon International (Temecula, CA, USA) and HRP goat anti-rabbit IgG was purchased from Medical and Biological Laboratories (Nagoya, Japan). Cy3 goat anti-mouse IgG was purchased from Amersham Biosciences (Piscataway, NJ, USA).
Table 1.
List of primary antibodies used for immunohistochemistry
| Antigen | Antibody | Working dilution |
|---|---|---|
| Cytokeratin 5, 6, 8 and 18 | Monoclonal; NCL-L-PAN-CK | 1:200 or 1:20 (3.2 or 32 μg/mL) |
| Vimentin | Monoclonal; anti-vimentin clone V9 | 1:800 (0.025 μg/mL) |
| Collagen type III | Polyclonal; anti-collagen type III antibody | 1:400 (0.25 μg/mL) |
| Collagen type IV | Polyclonal; anti-collagen type IV antibody | 1:50 |
| E-Cadherin | Monoclonal; anti-E-cadherin antibody | 1:1000 (0.025 μg/mL) |
Note: NCL-L-PAN-CK = Pancytokeratin (Novocastra Laboratories).
Cell and tissue preparation
Cells prepared from donor middle-ear bullae and cultured up to the third passage were pelleted and resuspended at 8.0 × 104 cells/mL in PBS. For cytospin preparation, the cells were spun down to 3-aminopropytriethoxysilane-coated slides at 3.2 × 104 cells/slide at 800 rpm for 2 minutes. They were then air-dried before storage at −80°C.
At days 0, 7, 14, and 28, recipients were killed by intraperitoneal injection of pentobarbital, and middle-ear bullae were collected for analysis. Each sample was divided horizontally into two halves. Half of the bullae collected at day 0 were fixed with 4% paraformaldehyde/PBS at 4°C overnight, decalcified by 10% ethylenediaminetetraacetic acid at 4°C for 7 days,50 and embedded in paraffin. Serial sections (5 μm thick) were prepared and then mounted on 3-aminopropytriethoxysilane-coated slides. These were used for immunohistochemistry for pancytokeratin and vimentin. The other half of the bullae collected at days 7, 14, and 28 were frozen immediately on dry ice and embedded in optimal cutting temperature compound (Sakura Finetek, Tokyo, Japan). Serial sections (5 μm thick) using the film method51 were prepared and used for the detection of EGFP, periodic acid-Schiff (PAS) staining, and immunohistochemistry. Bullae of the other half were fixed in 2.5% glutaraldehyde in 0.01 M PBS overnight at 4°C for electron microscopy (EM). For histological examination, hematoxylin and eosin staining was performed using a standard procedure.
Immunohistochemistry
Immunohistochemistry was performed with the indirect enzyme-labeled antibody method as described previously.52,53 The cytospin slides and frozen sections were warmed up in the sealed black case at room temperature (RT) for 2 hours and fixed with 4% paraformaldehyde/PBS at RT for 30 minutes for enzyme immunohistochemistry. The sections were deparaffinized with toluene and rehydrated with serially graded ethanol solutions. The sections were treated with 1% trypsin 250 and 1% calcium chloride in 50 mM tris-buffered saline (pH 7.8) to detect pancytokeratin, collagen type III, collagen type IV, and E-cadherin. The cytospin slides, frozen sections, and paraffin sections were microwaved in 10 mM citrate buffer (pH 7.4) for 3 minutes to detect vimentin. After inactivation of endogenous peroxidase activity with 0.3% H2O2 in methanol (RT, 15 minutes) the slides were preincubated with 500 μg/mL normal goat IgG in 1% bovine serum albumin (Sigma-Aldrich)/PBS for 1 hour to block nonspecific reaction. Then, the slides were incubated overnight with anti-pancyto keratin (cytospin slides and paraffin sections, 3.2 μg/mL; frozen sections, 32 μg/mL), anti-vimentin (0.025 μg/mL), anti- collagen type III (2.5 μg/mL), 50-fold dilution for anti- collagen type IV, and anti-E-cadherin (0.025 μg/mL) antibody (Table 1). The slides were washed four times with 0.075% Brij 35 (Sigma-Aldrich) in PBS, and reacted with HRP goat antimouse IgG (dilution 1:00) for anti-pancytokeratin and antivimentin antibodies or HRP goat anti-rabbit IgG (dilution, 1:00) for anti-collagen type III and anti-collagen type IV antibodies for 1 hour as the secondary antibody. After washing the slides with 0.075% Brij 35 in PBS, the HRP sites were visualized with 3,3′- diaminobenzidine-4HCl (Dojin Laboratories, Kumamoto, Japan) in H2O2 solution (pH 7.6) with 2% methyl green counterstaining. Cy3 goat anti-mouse IgG (dilution 1:100) was used as the secondary antibody for anti-E-cadherin antibody and visualized by fluorescence conjugate. For negative control, normal mouse IgG or normal rabbit IgG was used instead of the first antibodies, respectively, in every experiment.
Fluorescent images were obtained using a Zeiss fluorescent microscope or a Zeiss LSM 510 confocal laser scanning microscope connected to a charge-coupled-device camera (AxioCam; Carl Zeiss Meditec, Jena, Germany).
PAS staining
The frozen sections were warmed up in a sealed black case at RT for 2 hours, and fixed with 100% ethanol at RT overnight for PAS staining. PAS staining was performed according to McManus’s method54 using Schiffs reagent (Muto, Tokyo, Japan).
Quantitative analysis
For quantitative analysis, more than 1,000 cells were counted in random fields at 400× magnification, and the numbers of pancytokeratin- and vimentin-positive cells were expressed as percentage of positive cells per total number of counted cells (mean ± standard deviation, %, n = 3). The staining intensity of pancytokeratin and vimentin was graded as positive or negative compared to the background staining with normal mouse IgG.
Results
Analysis of donor cells
Mucosal epithelial cells from SD-Tg rats showed a cobblestone appearance and expressed EGFP up to the third passage (Figure 2A and B). Immunohistochemical analysis identified staining of mucosal epithelial cells for pancytokeratin but not vimentin, (96.0% ± 1.5%, n = 3) (Figure 2C and D). The growth curve of the donor cells from SD-Tg rats indicated logarithmic growth during the first 9 days at the third passage, with a calculated DT of 22.1 hours (Figure 2E). The viability of trypsinized cells was >90%. These results indicate that donor cells are almost epithelial cells at the third passage.
Figure 2.
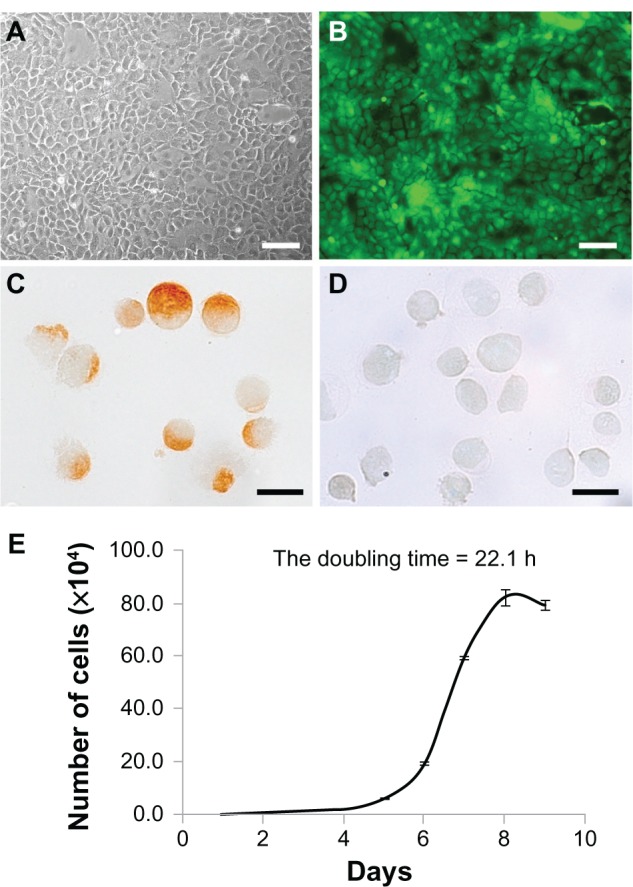
(A–E) Characterization of donor cells cultured by the primary explant-culture method. (A and B) Bright-field images and fluorescence images of donor cells by phase-contrast microscopy. Donor cells of Sprague Dawley transgenic rats showed a cobblestone appearance (A), and enhanced green fluorescent protein expression was stable up to the third passage (B). Scale bars, 100 μm. (C and D) Immunohistochemical analysis of donor cells collected by cytospin. The majority (98.6%) of donor cells were positive for pancytokeratin (C), with only a few positive for vimentin (D) at the third passage collected by cytospin. Scale bars, 20 μm. (E) In vitro cell-growth curve. The calculated doubling time was 22.1 hours.
Analysis of middle-ear mucosa-elimination model
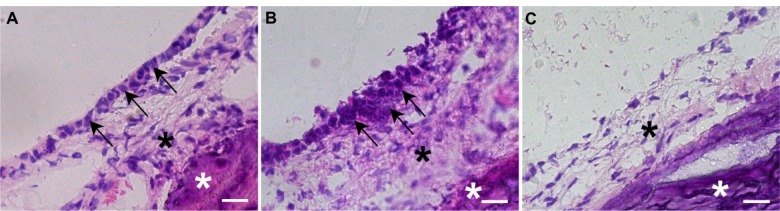
To confirm middle-ear mucosal elimination in the recipient rats, we performed immunohistochemical analysis using anti-pancytokeratin and anti-vimentin antibodies. The middle-ear regions of these rats contained only a few pancytokeratin-positive cells (Figure 3A and D), compared with abundant vimentin-positive cells (Figure 3B and F). On the other hand, examination of the outer-ear regions demonstrated the presence of pancytokeratin-positive cells in the epithelium (Figure 3A and E) and vimentin-positive cells in the subepithelial layer (Figure 3B and G). No staining was found in regions of the middle and external ears in the control sections stained with normal mouse IgG instead of the first antibody (Figure 3C, H, and I). These results confirmed the complete elimination of the epithelial mucosal layer and partial elimination of the subepithelial layer during the surgical preparation of the middle-ear of recipient rats.
Figure 3.

(A–I) Immunohistochemical analysis of the middle-ear of the recipients after mucosal tissue elimination before transplantation. (A, D and E) Anti-pancytokeratin; (B, F and G) anti-vimentin; (C, H and I) normal mouse immunoglobulin G. In the middle-ear lesion, vimentin-positive cells were detected (arrows in B and F), and only a few pancytokeratin-positive cells were present (A and D). In the external ear, pancytokeratin-positive cells were detected in the epithelial lesion (arrows in A and E), while vimentin-positive cells were detected in the subepithelial lesion (arrows in B and G). No staining was detected in sections reacted with normal mouse immunoglobulin G instead of the primary antibody (C–I).
Note: Arrowheads, tympanic membrane; arrows, positive cells; scale bars 100 μm (A–C), 50 μm (D–I).
Analysis of mucosal regeneration after transplantation
The middle-ear bullae with transplanted cells were analyzed at 7, 14, and 28 days after transplantation. In all cases, no apparent inflammatory changes were observed on the otomicroscope before killing. At 7 days after transplantation, similar to normal middle-ear bullae (Figure 4A), an epithelioid layer could be detected on the surface of the subepithelial layer of the recipient middle-ear bullae in which the cells were transplanted with PuraMatrix (Figure 4B), but not without PuraMatrix (Figure 4C). At the same time point, EGFP-expressing donor cells were found on the surface of the subepithelial layer of the recipient middle-ear bullae in all three cases in which the cells were transplanted with PuraMatrix at a density of 0.5 × 106 cells/mL (Figure 5A) and in two of the three cases in which they were transplanted with PuraMatrix at a density of 1.0 × 106 cells/mL (Table 2). In contrast, EGFP-expressing cells were not found on the subepithelial surface in middle-ear bullae of recipients in which the cells were transplanted without PuraMatrix (n = 3) (Table 2). The EGFP-expressing cells found on the recipient middle-ear bullae were positive for pancytokeratin but not for vimentin (Figure 5C and D).
Figure 4.
(A–C) Hematoxylin and eosin staining of middle-ear bullae after cell transplantation at postoperative day 7. (A) Normal middle-ear bulla; (B and C) a middle-ear bulla in which the cells were transplanted with PuraMatrix at a density of 5.0 × 105 cells/mL (B) or without PuraMatrix (C). (A) A monolayer structure of middle-ear mucosal epithelium was confirmed on the inner surface of the normal middle-ear bulla (arrows). (B) Epithelioid layer formation was identified on the surface of the subepithelial layer of the recipient middle-ear bulla in which the cells were transplanted with PuraMatrix (arrowheads). (C) No epithelioid layer formation was observed on the surface of the subepithelial layer of the recipient middle-ear bulla in which the cells were transplanted without PuraMatrix.
Note: Black asterisks, subepithelial layer; white asterisks, inner bone surface of the middle-ear bulla; scale bars, 20 μm.
Figure 5.

(A–K) Analysis of transplanted mucosal epithelial cells in the recipient tissues at postoperative day 7. (A–H) Serial frozen sections of middle-ear bullae after transplantation at a concentration of 0.5 × 106 cells/mL Enhanced green fluorescent protein-expressing cells were found on the internal surface of the recipient middle-ear bullae under laser scanning microscope (green, enhanced green fluorescent protein; blue, 4′,6-diamidino-2-phenylindole) (A) and were localized on the surface of the subepithelial layer and compared with hematoxylin and eosin staining (B). These cells were positive for pancytokeratin (C, arrows) but not for vimentin (D). Collagen III-positive cells were detected mainly in the subepithelial layer (E) while collagen IV-positive cells were detected under the donor cells, resembling basement membrane (F) (arrowheads). Some epithelial cells were positive for periodic acid–Schiff staining (G, arrows). E-cadherin-positive cells were detected between adjacent epithelial cells (red, E-cadherin) (H). Scale bars, 20 μm. (I–K) Ultrastructural analysis. Scanning electron microscopy confirmed the morphology of the regenerating epithelial surface, which consisted of flat polygonal squamous cells (I), scale bar 50 μm. Transmission electron microscopy identified the formation of adhesion-junction-like structures between adjacent epithelial cells (J and K, white circle), scale bar 1 μm.
Table 2.
Localization of EGFP-expressing cells after transplantation
| Samples | Hydrogel | POD7 | POD14 | POD28 | Engraftment rate |
|---|---|---|---|---|---|
| 0.5 × 106 cells/mL (n = 3) | + | 3/3 | 3/3 | 2/3 | 88.9% |
| 1.0 × 106 cells/mL (n = 3) | + | 2/3 | 3/3 | 0/3 | 44.4% |
| 1.0 × 106 cells/mL (n = 3) | – | 0/3 | – | – | 0.0% |
Abbreviations: POD, postoperative day; EGFP, enhanced green fluorescent protein.
In these samples, the donor cells were still stratified and lacked a monolayer structure (Figure 5A and B). Collagen III-positive regions were detected mainly in the subepithelium after transplantation (Figure 5E), indicating active remodeling of the middle-ear after mucosal tissue elimination.46–48 Collagen IV-positive regions were detected under the donor cells (Figure 5F), indicating attachment of donor cells to the ECM and basement membrane conformation.46,49 Moreover, some of the cells found in the subepithelial layer were positive for PA S staining, indicating they were functional, producing some mucus protein (Figure 5G). To confirm these results, we also performed ultrastructural analysis. Scanning electron microscopy indicated that the regenerating epithelial surface consisted of flat polygonal squamous cells bordered by microvilli (Figure 5I), which resembled the normal middle-ear mucosa.55,56 Furthermore, transmission electron microscopy identified the formation of adherens junction-like structures between adjacent epithelial cells (Figure 5J and K). Immunohistochemical analysis also identified E-cadherin-positive regions between adjacent cells (Figure 5H). These findings indicate the migration of transplanted donor cells into the recipient tissue and their reorganization to almost normal ultrastructure of the middle-ear epithelial cells at 7 days after transplantation.
At 14 days after transplantation, EGFP-expressing donor cells were detected in all recipients that received donor cells transplanted with PuraMatrix at density of 0.5 × 106 cells/mL (n = 3) (Figure 6A and B) and 1.0 × 106 cells/mL (n = 3) (Table 2), together with the formation of a monolayer structure of mucosal epithelial cells (Figure 6A). At 28 days after transplantation, EGFP-expressing donor cells were detected in two of three recipients that received donor cells transplanted with PuraMatrix at a density of 0.5 × 106 cells/mL (n = 3) (Figure 6G and H), but not in recipients that received donor cells transplanted with PuraMatrix at a density of 1.0 × 106 cells/mL (n = 3) (Table 2). At 14 and 28 days after transplantation, EGFP-expressing cells found in the recipient middle-ear bullae were positive for pancytokeratin (Figure 6C and I), but not for vimentin (Figure 6D and J), similar to the results at 7 days after transplantation. Collagen III-positive regions were also detected mainly in the subepithelium (Figure 6E and K), and the regions were reduced in size at 28 days after transplantation. Collagen IV-positive regions were also detected under the monolayer structure of donor cells at 14 (Figure 6F) and 28 days (Figure 6L) after transplantation.
Figure 6.

(A–L) Analysis of transplanted mucosal epithelial cells in recipient tissues at postoperative days 14 and 28. (A–F) Serial frozen sections of middle-ear bullae after transplantation (0.5 × 106 cells/mL) at postoperative day 14, and (G–L) postoperative day 28. (A, B, G and H) Fluorescence images at several time points. Enhanced green fluorescent protein (EGFP)-expressing cells were detected on the internal surface of recipient middle-ear bullae (green, EGFP; blue, 4′,6-diamidino-2-phenylindole) (A and G). results of immunostaining with (C and I) anti-pancytokeratin, (D and J) anti-vimentin, (E and K) anti-collagen III, and (F and L) anti-collagen IV antibody. EGFP-expressing cells were positive for pancytokeratin (C and I, arrows), but not for vimentin (D and J). Collagen III-positive regions were detected mainly in the subepithelium (E and K). Collagen IV-positive regions were detected under the monolayer structure of donor cells at 14 and 28 days after transplantation (F and L, arrowheads). Scale bars, 50 μm.
Discussion
In this study, we evaluated the usefulness of PuraMatrix as scaffolding for in situ tissue engineering of middle-ear mucosa. We generated a rat model of surgically damaged middle-ear transplanted with cultured middle-ear epithelial cells encapsulated within PuraMatrix. Donor cells were middle-ear epithelial cells from SD-Tg rats, and their EGFP expression was sufficient up to the third passage. The transplanted cells were confirmed to have normal epithelial cell features, both morphologically and functionally. After the transplantation of EGFP-expressing cells within PuraMatrix, these cells were found on the surface of the subepithelial lesion in the recipient middle-ear, had retained their normal morphology and function, and formed new epithelial and subepithelial layers, separated by a basement membrane. In contrast, donor cells transplanted without PuraMatrix were not settled in our model. These results suggest that proper scaffolds are indispensable for cell transplantation and that PuraMatrix forms effective nanofiber scaffolds in in situ tissue engineering of middle-ear mucosal tissue in this animal model.
Synthetic scaffolds with several biopolymers have been developed for biomedical and pharmaceutical applications, such as drug delivery57,58 and tissue engineering.8,20,59–61 Injectable biodegradable polymer hydrogels are ideal for in situ tissue engineering, because cells can be easily incorporated into polymer solutions and rapidly convert into a gel under physiological conditions, comprising three-dimensional hydrophilic polymeric networks.8,20 However, these biomaterials are often made of microfibers after gelation, which are much larger than native ECM.8 In our study, PuraMatrix (synthetic peptide hydrogel) could provide a suitable biological environment for cell-to-cell interaction based on such structural features as nanofiber formation on a scale similar to the ECM, and also its ability to retain water (water content up to 99.5% w/v).8,24,25
Examination of the regenerating mucosa in the damaged middle-ear demonstrated increased synthesis of collagen III around the migrated donor cells at day 7 after transplantation, indicating active tissue remodeling.46–48 Collagen type IV-positive regions were also detected under the epithelial cells during the same period, indicating the formation of the basement membrane.46,49 Moreover, the migrated epithelial cells were PAS-positive, indicating that the cells produced mucus, which is a functional characteristic of middle-ear epithelial cells. Furthermore, the scanning electron microscopy findings indicated that the regenerating cells resembled middle-ear epithelial cells morphologically, while transmission electron microscopy demonstrated the presence of adherens junction-like structures. In epithelial tissues, intercellular junctions consist of tight junctions, adherens junctions, and desmosomes. Adherens junctions are first assembled in epithelial cells, and are one of the most important components of cell-to-cell adhesion in epithelial tissues.48 The adhesive elements, notably E-cadherin, stably connect epithelial cells together and play essential roles in overall tissue organization and proper physiological function of tissue.48,49 The results of immunohistochemical studies also confirmed the presence of E-cadherin-positive regions among donor cells. Considered together, these results indicate that cell adhesion had already begun and that transplanted donor cells with PuraMatrix migrated into the appropriate parts of recipient tissue, reorganizing the normal structure of the middle-ear epithelial layer soon after transplantation.
Recently, Kopesky et al25 reported chondrogenic differentiation of bone marrow stromal cells after rapid cell-to-cell contact during the first week after encapsulation in PuraMatrix under conditioned medium. In another study, the formation of neovascular networks was observed within 7 days of injection of human endothelial progenitor cells and mesenchymal progenitor cells with PuraMatrix into the dorsal flanks of immune-deficient mice, and the effect was similar to the biological ECM Matrigel.62 Our results provide support to these studies and indicate that transplanted donor cells with PuraMatrix migrated into the appropriate parts of recipient tissue following early cell-to-cell contact at 7 days after transplantation. Specifically, nanoscale surface topography could promote cell-to-cell contact and an RAD motif of PuraMatrix, which is similar to the sequence of RGD identified as a cell attachment sequence in various adhesive proteins present in the ECM, thus providing support for cell adhesion, migration, and proliferation.8,10,11 Furthermore, the findings using our model suggest that the remnant subepithelial layer of the recipient middle-ear seems to facilitate homing of donor cells to their destination.
At 14 days after transplantation, the engraftment rate was 100% in both the high- and low-cell-density groups with PuraMatrix. Interestingly, at 28 days after transplantation, the engraftment rate was lower in the high-cell-density group than the low-cell-density group (Table 2). Generally, the proliferation rate is seeding density-dependent, but it is possible that a high seeding density could induce cell-to-cell contact inhibition in a three-dimensional environment.63 The exact reason for this finding was not investigated in this experiment, but a previous study using bovine chondrocytes embedded in agarose hydrogels suggested that high seeding density may be associated with limitation of nutrient supply.64,65 In fact, it was difficult to maintain primary cultured cells obtained from SD-Tg rats in PuraMatrix at high density in vitro (data not shown). Employing cartilage tissue-engineering technology, Park et al65 demonstrated higher ECM protein expression following inoculation of rabbit bone marrow mesenchymal stem cells embedded in oligo(poly[ethylene glycol] fumarate) at low cell density compared with high cell density.65 On the other hand, peptide hydrogels are mechanically weak, and this feature is disadvantageous for tissue engineering,15 because adherent tissue cells must, in general, attach to a solid substrate for their survival, and mechanical stiffness of scaffold materials for tissue engineering is important for cell proliferation.63,66,67 However, one previous study indicated that adhesion and proliferation of human umbilical vein endothelial cells for 5 days was higher in PuraMatrix (0.275% w/v) than PEG (7.84% w/v), although the elastic and viscous moduli of PuraMatrix were lower than those of PEG.68 Taken together, the results indicate a better engraftment rate with the use of low cell density compared with high cell density. These phenomena need to be investigated in more detail, including comparison with other scaffolds.
The limitation of cell source should be solved in extensive tissue damage to ensure adequate tissue regeneration. To address this issue, cell sheets engineered from autologous oral mucosal epithelial cells and used for reconstruction of the ocular surface resulted in restoration of vision in patients with severe bilateral ocular diseases.69 When the cell source is not exhausted, it is ideal to transplant cells obtained from the same origin into the damaged lesion for proper tissue regeneration. In our study, the required cells for regeneration of a damaged middle-ear mucosa were less than one-twentieth of the requirement for a middle-ear cell sheet.4 In situ tissue engineering using cell transplantation with PuraMatrix could potentially result in successful regeneration of mucosal tissue.
Conclusion
The present study demonstrated the feasibility of transplantation of engineered middle-ear mucosal epithelial cells encapsulated within PuraMatrix for regeneration of surgically damaged mucosa of the middle-ear in SD rats.
Acknowledgments
FK506 was provided by Astellas Pharma Inc (Tokyo, Japan). This study was supported by a Grant-in-Aid for Young Scientists (B) from the Japanese Ministry of Education, Culture, Sports, Science, and Technology (7870005 to N Akiyama).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Tos M. Manual of Middle Ear Surgery. Vol. 1. Stuttgart: Thieme; 1993. Indications for surgery and preoperative management; pp. 2–6. [Google Scholar]
- 2.Takahashi H. Ars B. Chronic Otitis Media. Pathogenesis-Oriented Therapeutic Management. Amsterdam: Kugler; 2008. Middle ear cleft mucosal conditions and gas-exchange function; pp. 197–212. [Google Scholar]
- 3.Kazama K, Takahashi H, Kaieda S, et al. Effect of a large-sized silicone sheet upon recovery of mastoid aeration after mastoidectomy. Otolaryngol Head Neck Surg. 2008;138:738–742. doi: 10.1016/j.otohns.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 4.Wada K, Tanaka Y, Kojima H, Inamatsu M, Yoshizato K, Moriyama H. In vitro reconstruction of a three-dimensional middle ear mucosal organ and its in vivo transplantation. Acta Otolaryngol. 2006;126:801–810. doi: 10.1080/00016480500507385. [DOI] [PubMed] [Google Scholar]
- 5.Yaguchi Y, Wada K, Uchimizu H, Tanaka Y, Kojima H, Moriyama H. Middle ear mucosa regeneration by grafting of artificial mucosa. Acta Otolaryngol. 2007;127:1038–1044. doi: 10.1080/00016480701200285. [DOI] [PubMed] [Google Scholar]
- 6.Tabata Y. Biomaterial technology for tissue engineering applications. J R Soc Interface. 2009;6(Suppl 3):S311–S324. doi: 10.1098/rsif.2008.0448.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Langer R, Vacanti JP. Tissue engineering. Science. 1993;260:920–926. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- 8.Zhang S, Ellis-Behnke R, Zhao X, Spirio L. Self-assembling peptide nanofiber scaffolds. In: Elisseeff PX, Ma J, editors. Scaffolding in Tissue Engineering. Boca Raton (FL): CRC; 2005. pp. 217–238. [Google Scholar]
- 9.Zhang S, Holmes TC, DiPersio CM, Hynes RO, Su X, Rich A. Self-complementary oligopeptide matrices support mammalian cell attachment. Biomaterials. 1995;16:1385–1393. doi: 10.1016/0142-9612(95)96874-y. [DOI] [PubMed] [Google Scholar]
- 10.Hosseinkhani H. 3D in vitro technology for drug discovery. Curr Drug Saf. 2012;7:37–43. doi: 10.2174/157488612800492753. [DOI] [PubMed] [Google Scholar]
- 11.Hosseinkhani H, Hong PD, Yu DS. Self-assembled proteins and peptides for regenerative medicine. Chem Rev. 2013 Apr 2; doi: 10.1021/cr300131h. Epub. [DOI] [PubMed] [Google Scholar]
- 12.Kleinman HK, McGarvey ML, Liotta LA, Robey PG, Tryggvason K, Martin GR. Isolation and characterization of type IV procollagen, laminin, and heparan sulfate proteoglycan from the EHS sarcoma. Biochemistry. 1982;21:6188–6193. doi: 10.1021/bi00267a025. [DOI] [PubMed] [Google Scholar]
- 13.Irwin EF, Gupta R, Dashti DC, Healy KE. Engineered polymer-media interfaces for the long-term self-renewal of human embryonic stem cells. Biomaterials. 2011;32:6912–6919. doi: 10.1016/j.biomaterials.2011.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffman AS. Hydrogels for biomedical applications. Adv Drug Deliv Rev. 2002;54:3–12. doi: 10.1016/s0169-409x(01)00239-3. [DOI] [PubMed] [Google Scholar]
- 15.Slaughter BV, Khurshid SS, Fisher OZ, Khademhosseini A, Peppas NA. Hydrogels in regenerative medicine. Adv Mater. 2009;21:3307–3329. doi: 10.1002/adma.200802106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malafaya PB, Silva GA, Reis RL. Natural-origin polymers as carriers and scaffolds for biomolecules and cell delivery in tissue engineering applications. Adv Drug Deliv Rev. 2007;59:207–233. doi: 10.1016/j.addr.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 17.Wichterle O, Lim D. Hydrophilic gels for biological use. Nature. 1960;185:117–118. [Google Scholar]
- 18.Pan Y, Xiong D. Study on compressive mechanical properties of nanohydroxyapatite reinforced poly(vinyl alcohol) gel composites as biomaterial. J Mater Sci Mater Med. 2009;20:1291–1297. doi: 10.1007/s10856-008-3679-8. [DOI] [PubMed] [Google Scholar]
- 19.Schmedlen RH, Masters KS, West JL. Photocrosslinkable polyvinyl alcohol hydrogels that can be modified with cell adhesion peptides for use in tissue engineering. Biomaterials. 2002;23:4325–4332. doi: 10.1016/s0142-9612(02)00177-1. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen MK, Lee DS. Injectable biodegradable hydrogels. Macromol Biosci. 2010;10:563–579. doi: 10.1002/mabi.200900402. [DOI] [PubMed] [Google Scholar]
- 21.Elisseeff J, McIntosh W, Anseth K, Riley S, Ragan P, Langer R. Photoencapsulation of chondrocytes in poly(ethylene oxide)-based semi-interpenetrating networks. J Biomed Mater Res. 2000;51:164–171. doi: 10.1002/(sici)1097-4636(200008)51:2<164::aid-jbm4>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 22.Alcantar NA, Aydil ES, Israelachvili JN. Polyethylene glycol-coated biocompatible surfaces. J Biomed Mater Res. 2000;51:343–351. doi: 10.1002/1097-4636(20000905)51:3<343::aid-jbm7>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 23.Nagai Y, Unsworth LD, Koutsopoulos S, Zhang S. Slow release of molecules in self-assembling peptide nanofiber scaffold. J Control Release. 2006;115:18–25. doi: 10.1016/j.jconrel.2006.06.031. [DOI] [PubMed] [Google Scholar]
- 24.Hosseinkhani H, Hosseinkhani M, Tian F, Kobayashi H, Tabata Y. Osteogenic differentiation of mesenchymal stem cells in self-assembled peptide-amphiphile nanofibers. Biomaterials. 2006:4079–4086. doi: 10.1016/j.biomaterials.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 25.Kopesky PW, Vanderploeg EJ, Sandy JS, Kurz B, Grodzinsky AJ. Self-assembling peptide hydrogels modulate in vitro chondrogenesis of bovine bone marrow stromal cells. Tissue Eng Part A. 2010;16:465–477. doi: 10.1089/ten.tea.2009.0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang QG, Hughes N, Cartmell SH, Kuiper NJ. The composition of hydrogels for cartilage tissue engineering can influence glycosaminoglycan profile. Eur Cell Mater. 2010;19:86–95. doi: 10.22203/ecm.v019a09. [DOI] [PubMed] [Google Scholar]
- 27.Tokunaga M, Liu ML, Nagai T, et al. Implantation of cardiac progenitor cells using self-assembling peptide improves cardiac function after myocardial infarction. J Mol Cell Cardiol. 2010;49:972–983. doi: 10.1016/j.yjmcc.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 28.Holmes TC, de Lacalle S, Su X, Liu G, Rich A, Zhang S. Extensive neurite outgrowth and active synapse formation on self-assembling peptide scaffolds. Proc Natl Acad Sci USA. 2000;97:6728–6733. doi: 10.1073/pnas.97.12.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Semino CE, Kasahara J, Hayashi Y, Zhang S. Entrapment of migrating hippocampal neural cells in three-dimensional peptide nanofiber scaffold. Tissue Eng. 2004;10:643–655. doi: 10.1089/107632704323061997. [DOI] [PubMed] [Google Scholar]
- 30.Thonhoff JR, Lou DI, Jordan PM, Zhao X, Wu P. Compatibility of human fetal neural stem cells with hydrogel biomaterials in vitro. Brain Res. 2008;1187:42–51. doi: 10.1016/j.brainres.2007.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koutsopoulos S, Zhang S. Long-term three-dimensional neural tissue cultures in functionalized self-assembling peptide hydrogels, matrigel and collagen I. Acta Biomater. 2013;9:5162–5169. doi: 10.1016/j.actbio.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 32.Semino CE, Merok JR, Crane GG, Panagiotakos G, Zhang S. Functional differentiation of hepatocyte-like spheroid structures from putative liver progenitor cells in three-dimensional peptide scaffolds. Differentiation. 2003;71:262–270. doi: 10.1046/j.1432-0436.2003.7104503.x. [DOI] [PubMed] [Google Scholar]
- 33.Wang S, Nagrath D, Chen PC, Berthiaume F, Yarmush ML. Three-dimensional primary hepatocyte culture in synthetic self-assembling peptide hydrogel. Tissue Eng Part A. 2008;14:227–236. doi: 10.1089/tea.2007.0143. [DOI] [PubMed] [Google Scholar]
- 34.Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- 35.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–200. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 36.Harrison RG. Observations on the living developing nerve fiber. Exp Biol Med (Maywood) 1907;4:140–143. [Google Scholar]
- 37.Carrel A. On the permanent life of tissues outside of the organism. J Exp Med. 1912;15:516–528. doi: 10.1084/jem.15.5.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakamura A, Kumazawa T, Lim DJ, Demaria TF, van Blitterswijk CA. Culture of middle ear epithelium: a review. Acta Otolaryngol Suppl. 1993;500:75–79. doi: 10.3109/00016489309126185. [DOI] [PubMed] [Google Scholar]
- 39.Moon SK, Lim DJ, Lee HK, Kim HN, Yoon JH. Mucin gene expression in cultured human middle ear epithelial cells. Acta Otolaryngol. 2000;120:933–939. doi: 10.1080/00016480050218654. [DOI] [PubMed] [Google Scholar]
- 40.Inamura N, Nakahara K, Kino T, et al. Prolongation of skin allograft survival in rats by a novel immunosuppressive agent, FK506. Transplantation. 1988;45:206–209. doi: 10.1097/00007890-198801000-00042. [DOI] [PubMed] [Google Scholar]
- 41.Yamamoto-Fukuda T, Aoki D, Hishikawa Y, Kobayashi T, Takahashi H, Koji T. Possible involvement of keratinocyte growth factor and its receptor in enhanced epithelial-cell proliferation and acquired recurrence of middle-ear cholesteatoma. Lab Invest. 2003;83:123–136. doi: 10.1097/01.lab.0000050763.64145.cb. [DOI] [PubMed] [Google Scholar]
- 42.Moll R, Franke WW, Schiller DL, Geiger B, Krepler R. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell. 1982;31:11–24. doi: 10.1016/0092-8674(82)90400-7. [DOI] [PubMed] [Google Scholar]
- 43.Gatter KC, Abdulaziz Z, Beverley P, et al. Use of monoclonal antibodies for the histopathological diagnosis of human malignancy. J Clin Pathol. 1982;35:1253–1267. doi: 10.1136/jcp.35.11.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Azumi N, Battifora H. The distribution of vimentin and keratin in epithelial and nonepithelial neoplasms. A comprehensive immunohistochemical study on formalin- and alcohol-fixed tumors. Am J Clin Pathol. 1987;88:286–296. doi: 10.1093/ajcp/88.3.286. [DOI] [PubMed] [Google Scholar]
- 45.Osborn M, Debus E, Weber K. Monoclonal antibodies specific for vimentin. Eur J Cell Biol. 1984;34:137–143. [PubMed] [Google Scholar]
- 46.Kleinman HK, Klebe RJ, Martin GR. Role of collagenous matrices in the adhesion and growth of cells. J Cell Biol. 1981;88:473–485. doi: 10.1083/jcb.88.3.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hashizume R, Fujimoto KL, Hong Y, et al. Morphological and mechanical characteristics of the reconstructed rat abdominal wall following use of a wet electrospun biodegradable polyurethane elastomer scaffold. Biomaterials. 2010;31:3253–3265. doi: 10.1016/j.biomaterials.2010.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gumbiner BM. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- 49.Simpson CL, Patel DM, Green KJ. Deconstructing the skin: cytoarchitectural determinants of epidermal morphogenesis. Nat Rev Mol Cell Biol. 2011;12:565–580. doi: 10.1038/nrm3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamamoto-Fukud T, Shibata Y, Hishikawa Y, et al. Effects of various decalcification protocols on detection of DNA strand breaks by terminal dUTP nick end labelling. Histochem J. 2000;32:697–702. doi: 10.1023/a:1004171517639. [DOI] [PubMed] [Google Scholar]
- 51.Kawamoto T. Use of a new adhesive film for the preparation of multipurpose fresh-frozen sections from hard tissues, whole-animals, insects and plants. Arch Histol Cytol. 2003;66:123–143. doi: 10.1679/aohc.66.123. [DOI] [PubMed] [Google Scholar]
- 52.Yamamoto-Fukuda T, Hishikawa Y, Shibata Y, Kobayashi T, Takahashi H, Koji T. Pathogenesis of middle ear cholesteatoma: a new model of experimentally induced cholesteatoma in Mongolian gerbils. Am J Pathol. 2010;176:2602–2606. doi: 10.2353/ajpath.2010.091182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Song N, Liu J, An S, Nishino T, Hishikawa Y, Koji T. Immunohistochemical analysis of histone H3 modifications in germ cells during mouse spermatogenesis. Acta Histochem Cytochem. 2011;44:183–190. doi: 10.1267/ahc.11027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McManus JF. Histological and histochemical uses of periodic acid. Stain Technol. 1948;23:99–108. doi: 10.3109/10520294809106232. [DOI] [PubMed] [Google Scholar]
- 55.Chole RA, Chiu M. Ultrastructure of middle ear mucosa in the Mongolian gerbil, Meriones unguiculatus. Acta Otolaryngol. 1985;100:273–288. doi: 10.3109/00016488509104790. [DOI] [PubMed] [Google Scholar]
- 56.Albiin N, Hellström S, Stenfors LE, Cerne A. Middle ear mucosa in rats and humans. Ann Otol Rhinol Laryngol Suppl. 1986;126:2–15. doi: 10.1177/00034894860950s501. [DOI] [PubMed] [Google Scholar]
- 57.Siepmann J, Peppas NA. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC) Adv Drug Deliv Rev. 2001;48:139–157. doi: 10.1016/s0169-409x(01)00112-0. [DOI] [PubMed] [Google Scholar]
- 58.Deshmukh M, Kutscher HL, Gao D, et al. Biodistribution and renal clearance of biocompatible lung targeted poly(ethylene glycol) (PEG) nanogel aggregates. J Control Release. 2012;164:65–73. doi: 10.1016/j.jconrel.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu W, Chen B, Deng D, Xu F, Cui L, Cao Y. Repair of tendon defect with dermal fibroblast engineered tendon in a porcine model. Tissue Eng. 2006;12:775–788. doi: 10.1089/ten.2006.12.775. [DOI] [PubMed] [Google Scholar]
- 60.Weng Y, Wang M, Liu W, et al. Repair of experimental alveolar bone defects by tissue-engineered bone. Tissue Eng. 2006;12:1503–1513. doi: 10.1089/ten.2006.12.1503. [DOI] [PubMed] [Google Scholar]
- 61.Zhou G, Liu W, Cui L, Wang X, Liu T, Cao Y. Repair of porcine articular osteochondral defects in non-weightbearing areas with autologous bone marrow stromal cells. Tissue Eng. 2006;12:3209–3221. doi: 10.1089/ten.2006.12.3209. [DOI] [PubMed] [Google Scholar]
- 62.Allen P, Melero-Martin J, Bischoff J. Type I collagen, fibrin and PuraMatrix matrices provide permissive environments for human endothelial and mesenchymal progenitor cells to form neovascular networks. J Tissue Eng Regen Med. 2011;5:e74–e86. doi: 10.1002/term.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hadjipanayi E, Mudera V, Brown RA. Close dependence of fibroblast proliferation on collagen scaffold matrix stiffness. J Tissue Eng Regen Med. 2009;3:77–84. doi: 10.1002/term.136. [DOI] [PubMed] [Google Scholar]
- 64.Mauck RL, Seyhan SL, Ateshian GA, Hung CT. Influence of seeding density and dynamic deformational loading on the developing structure/function relationships of chondrocyte-seeded agarose hydrogels. Ann Biomed Eng. 2002;30:1046–1056. doi: 10.1114/1.1512676. [DOI] [PubMed] [Google Scholar]
- 65.Park H, Temenoff JS, Tabata Y, Caplan AI, Mikos AG. Injectable biodegradable hydrogel composites for rabbit marrow mesenchymal stem cell and growth factor delivery for cartilage tissue engineering. Biomaterials. 2007;28:3217–3227. doi: 10.1016/j.biomaterials.2007.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kshitiz, Park J, Kim P, et al. Control of stem cell fate and function by engineering physical microenvironments. Integr Biol (Camb) 2012;4:1008–1018. doi: 10.1039/c2ib20080e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kyle S, Felton SH, McPherson MJ, Aggeli A, Ingham E. Rational molecular design of complementary self-assembling peptide hydrogels. Adv Health Mater. 2012;1:640–645. doi: 10.1002/adhm.201200047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shroff K, Rexeisen EL, Arunagirinathan MA, Kokkoli E. Fibronectin-mimetic peptide-amphiphile nanofiber gels support increased cell adhesion and promote ECM production. Soft Matter. 2010;6:5064–5072. [Google Scholar]
- 69.Nishida K, Yamato M, Hayashida Y, et al. Corneal reconstruction with tissue-engineered cell sheets composed of autologous oral mucosal epithelium. N Engl J Med. 2004;351:1187–1196. doi: 10.1056/NEJMoa040455. [DOI] [PubMed] [Google Scholar]