

Abstract
Chlorogenic acid, a major compound among phenolic acids of Flaveria bidentis (L.) Kuntze and Flos lonicerae, was successfully separated by pH-zone-refining counter-current chromatography. Caffeic acid and chlorogenic acid were selected as test samples, which represent as phenolic acids for separation by the pH-zone-refining counter-current chromatography. The separation of these test samples was performed with two-phase solvent system composed of methyl-tert.-butyl-ether-acetonitrile-water at a volume ration of 4:1:5 (v/v) where trifluoroaceetic acid (8 mM) was added to the organic stationary phase as a retainer and NH4OH (10 mM) to the aqueous mobile phase as an eluter. For separation of crude extract from Flaveria bidentis (L.) Kuntze and Flos lonicerae. a slightly polar two-phase solvent system composed of methyl-tert.-butyl-ether-acetonitrile- n-butanol-water at a volume ratio of 4:1:1:5 (v/v) was selected where trifluoroacetic acid (3 mM) was added to the organic stationary phase as a retained and NH4OH (3 mM) to the aqueous mobile phase as an eluter. pH-zone-refining counter-current chromatography yielded 16.2 mg amount of chlorogenic acid with the purity of 91% from 1.4 g of Flaveria bidentis (L.) Kuntze, and 134 mg of chlorogenic acid at the purity of 99% from 1.3 g of crude extract of Flos lonicerae. These results suggest that pH-zone-refining counter-current chromatography is suitable for the isolation of the chlorogenic acid from the crude extracts of Flaveria bidentis (L.) Kuntze and Flos lonicerae.
Keywords: pH-zone-refining counter-current chromatography, Chlorogenic acid, Caffeic acid, Flaveria bidentis (L.) Kuntze, Flos lonicerae
1. Introduction
Flaveria bidentis (L.) Kuntze, native in South America mainly Brazil and Argentina, has spread to Heibei Provices in China in 2001 [1], severely destructing the agricultural ecosystem and also causing serious environmental problems [2]. Now, much attention has been taken to control it by manual, chemical and biological measures [3, 4]. Chlorogenic acid, which is found in F. bidentis (L.) Kuntze, has considerable antibacterial bioactivities. It is an ester formed between caffeic acid and quinic acid [5], and represents as one of major phenolic compounds in traditional Chinese medicinal herb [6]. Chlorogenic acid is also the main component in Flos lonicerae, which is a traditional Chinese medicinal herb widely used in pharmaceutical treatment. It has been used to clear heat, remove toxicity, relieve and treat many different human diseases [7, 8], especially in the prevalence of chronic and systemic diseases where the use of western medicines is limited by their side effects.
pH-zone-refining counter-current chromatography (CCC) was introduced by Dr. Ito. It is generally employed as a large-scale preparative technique for separating ionizable analytes according to their pKa and hydrophobility [9]. The method elutes highly concentrated rectangular peaks fused together with minimum overlapping while trace ionic compounds and impurities are concentrated and eluted between and the outside edges of the major peaks [10-12]. Many applications of this preparative technique for the separation of natural and synthetic mixtures had been widely reported including the separation of dye mixtures [13], alkaloids, organic acids [14-15], peptides [16] enantiomers [17] and so on. The present study aims to use pH-zone refining CCC to separate chlorogenic acid from Flaveria bidentis (L.) Kuntze (F. bidentis) and Flos lonicerae and to concentrate caffeic acid, a trace component in F. bidentis
2. Experimental
2.1. Apparatus
The analytical high-speed CCC instrument used in the present study was a Model GS20A multilayer coil planet centrifuge (Beijing Institute of New Technology Application, Beijing, China) equipped with a PTFE (polytetrafluoroethylene) multilayer coil of 70 m×0.85 mm I.D. with a total capacity of 40 ml. The β values of the coil values varied from 0.4 at the internal terminal to 0.7 at the external terminal. An optimum speed of 1600 rpm was used in the present studies.
The preparative CCC instrument used was a Model GS10AB multilayer coil planet centrifuge (Beijing Institute of New Technology Application, Beijing, China) equipped with a PTFE multilayer coil of 110 m×1.6 mm I.D. with a total capacity of 220 ml. The β values of the coil varied from 0.5 at the internal terminal to 0.75 at the external terminal. The revolution speed of the apparatus can be regulated with a speed controller in the range from 0 rpm to 1000 rpm where an optimum speed of 800 rpm was used in the present study.
The solvent was pumped into the column with a Model NS-1007 constant-flow pump (Beijing Institute of New Technology Application, Beijing, China). The continuous monitoring of the effluent was achieved with a Model 8823A-UV monitor (Beijing Institute of New Technology Application, Beijing, China) operating at 254 nm and a Model 330 pH meter (ATI Orion Research, Boston, MA, USA). A manual sample injection valve with 2 ml or 10 ml loop was used to introduce the sample into the column. Two portable recorders (Yokogama Model 3057, Sichuan Instrument Factory, Chongqin, China) were used to draw the chromatogram. A RE-52A rotary evaporator (Shanghai Yarong Biochemistry Instrument Factory, Shanghai, China) was used to concentrate fractions.
The analytical high-performance liquid chromatography (HPLC) equipment used was a Shimadzu LC-20AVP system equipped with two LC-20AT solvent delivery units, an SPD-M20AVP UV-VIS photodiode array detection (DAD) system, a Model 7725 injection valve with a 20 μl loop and an auto-sampler, an SCL-20AVP system controller, and a Class-VP-LC work station (Shimadzu, Tokyo, Japan).
2.2. Reagents and materials
All solvents used in pH-zone-refining CCC were of analytical grade (Tianjin Damao Chemical reagent Factory, China). Trifluoroacetic acid (TFA) (J&K Chemical Ltd. Beijing, China), ammonia and phosphoric acid (Beijing Chemical Factory, Beijing, China) were of reagent grade. Methanol used for HPLC analysis was of chromatographic grade.
F. bidentis was obtained from the Institute of Environment and Sustainable Development in Agriculture, Chinese Academy of Agricultural Sciences. Chlorogenic acid, caffeic acid test samples and crude sample of Flos lonicerae were purchased from Nanjing Zelang Medical Technology Company (Nanjing, China).
2.3. Preparation of the crude sample
The dried leafage of F. bidentis was powdered by disintegrator, and 80 g of the power was extracted by petroleum ether, ethyl acetate, and 80% ethanol in this order under 85°C. The extract of 80% ethanol was concentrated under reduced pressure in a rotary evaporator, yielding 12.5 g of the dried crude sample. The content of chlorogenic acid and caffeic acid in F. bidentis was 1.29% and 0.02%, respectively, while that of chlorogenic acid in Flos lonicerae was 15%.
2.4. Preparation of two-phase solvent and sample solution
For the separation of test samples of chlorogenic acid and caffeic acid, we selected a two-phase system composed of methyl tert.-butyl ether-acetonitrile-water (4:1:5 v/v). The solvent mixture was thoroughly equilibrated in a separatory funnel and the two phases separated shortly before use. Then, TFA (retainer) was added to the upper organic stationary phase and NH4OH (eluter) to the lower aqueous mobile phase.
For the separation of the crude extracts from F. bidentis and Flos lonicerae, we selected a two-phase solvent system composed of methyl tert.-butyl ether (MtBE)-acetonitrile (ACN)-n-butyl alcohol (BuOH)-water (4:1:1:5 v/v), where TFA (retainer) was added to the upper organic stationary phase at 3 mM and NH4OH (eluter) to the lower aqueous mobile phase at 3 mM.
The sample solution was prepared by dissolving the crude extract in about equal volumes of the stationary phase containing TFA and the mobile phase free of NH4OH.
2.5. Separation procedure
The column was first entirely filled with the organic stationary phase containing TFA, followed by injection of the sample solution. Then the mobile phase containing NH4OH was pumped into the inlet at a flow rate of 0.5 ml/min for analytical CCC and 2.0 ml/min for preparative CCC both in the head-to-tail elution mode, while the apparatus was run at a setting speed. The effluent from the outlet of the column was continuously monitored at 254 nm. Every peak fraction was collected according to the chromatogram. The retention of the stationary phase was computed from the volume of the stationary phase collected from the column after the separation was completed.
2.6. Analyses and identification of crude extract and pH-zone-refining CCC
The main peak fractions obtained by pH-zone-refining CCC were evaporated under reduced pressure at 60°C. Then each fraction was analyzed by HPLC under the optimum analytical conditions. The samples were analyzed by HPLC with Apollo C18 column (150×4.6 mm I.D.) at 324 nm and column temperature of 30°C. For test samples, the mobile phase was a solution of methanol (MeOH) and 0.05% phosphoric acid (27:73, v/v). For the crude extracts from Flos loniecrae and F. bidentis. gradient elution was performed using eluent A (MeOH) and eluent B (0.05% (v/v) H3PO4 in water) The gradient condition of Flos loniecrae was as follows: Methanol: 0-10 min, 22%; 12-28 min, 32-37%; 35-40 min, 42%。 The gradient condition of F. bidentis was as follows: Methanol: 0-20 min, 20-40%; 20-45 min, 40-58%; 45-60 min, 58-80%; 60-70 min, 80-100%. The flow-rate was 1.0 ml/min and the effluent was also monitored by absorbance at 324 nm.
3. Results and discussion
3.1. Solvent system selection of the pH-zone-refining CCC
The solvent system MtBE-ACN-water (4:1:5) used in the separation was selected in a typical way developed by Dr. Ito. Successful separation by pH-zone-refining CCC depends on the selection of a suitable two-phase solvent system that should have a suitable partition coefficient (K) values in both basic (Kbase≪1) and acidic (Kacid≫1) conditions. MtBE-ACN-water (4:1:5), MtBE-ACN-n-BuOH-water (4:1:1:5), and MtBE-ACN-n-BuOH-water (2:1:2:3) was tested during the solvent system selection process (Shown in Table 1). Among those, the volume ratio of MtBE-ACN-water (4:1:5) was selected for separation of the test sample and MtBE-ACN-n-BuOH-water (4:1:1:5) for separation of chlorogenic acid from the crude extracts in the present study. Using the MtBE-ACN-water (4:1:5) it is expected that chlorogenic acid will be eluted before the pH-zone and completely separated from caffeic acid. MtBE-ACN-n-BuOH-water (2:1:2:3) is not suitable for separation of caffeic acid and also it would be wasting the solvent because the volume of upper phase is 1.7 times of that of lower phase.
Table 1. Partition factor of solvent system.
| solvent system | Kbase (chlorogenic acid) | Kacid (chlorogenic acid) | Kbase (caffeic acid) | Kacid (caffeic acid) |
|---|---|---|---|---|
| MtBE-ACN-water (4:1:5) | ≪1 | 0.14 | ≪1 | ≫1 |
| MtBE-ACN-n-BuOH-water (4:1:1:5) | ≪1 | 1.2 | 0.52 | 5.6 |
| MtBE-ACN-n-BuOH-water (2:1:2:3) | 0.33 | 4.67 | 0.01 | 0.6 |
3.2. Selection of concentration of retainer and eluter
The molar concentration ratio between the retainer and the eluter determines the retention time of the pH-zone of analytes. Fig.1a to Fig.1c shows the pH-zone refining CCC of different concentration of retainer and eluter for test samples chlorogenic acid and caffeic acid separation. From Fig.1a to Fig.1c, the separations were performed with 1% (v/v) (132 mM) retainer and 1% (v/v) (161 mM) eluter; 10 mM retainer and 10 mM eluter; 8 mM retainer and 10 mM eluter, respectively. According to Fig.1a, when the concentration of retainer and eluter were 1% (v/v), two peaks were eluted earlier where the first peak was chlorogenic acid and the second peak was caffeic acid mixed with unknown impurity peak. By reducing the concentraton of retainer and eluter separation were much improved as seen from Fig.1b and Fig. 1c. Both Figs.1b and 1c show excellent separation of chlorogenic acid, while Fig. 1c gives shorter separation time. From the above results of pilot experiment with test samples, the optimum liquid-liquid system was methyl-tert.-butyl-ether-acetonitrile-water at at separation of the test sample, the optimum solvent system is considered to be the solvent volume ratio of 4:1:5 (v/v), with TFA (8 mM) in the organic stationary phase and NH4OH (10 mM) in the aqueous mobile phase.
Figure 1.
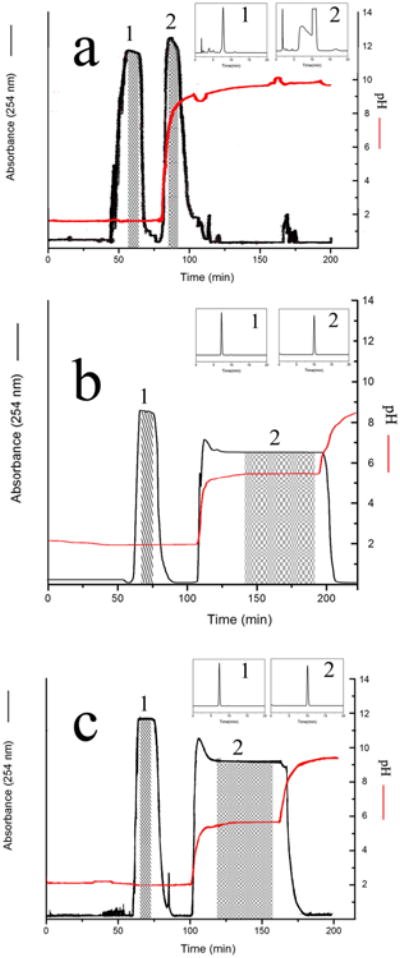
Separation of test samples by pH-zone-refining CCC Experimental conditions: Apparatus: the type-J coil planet centrifuge equipped with multilayer coil sparation column of 220 ml capacity; solvent system MtBE/acetonitrile/water (4:1:5), retainer/eluter: a (1% TFA/1% NH4OH), b (10 mM TFA/10 mM NH4OH) and c (8 mM TFA/10 m M NH4OH); flow rate: 2 ml/min; mobile phase: lower phase; elution mode: head to tail; revolution speed: 800 rpm; detection wavelength: 245 nm.
3.3. Separation of chlorogenic acid by pH-zone-refining CCC
Seen from Fig. 1c, the chlorogenic acid was eluted before the pH zone, so the solvent system should be changed to more hydrophilic solvent system to include the chlorogenic acid in the pH zone. Therefore, 1-butanol was added to increase hydrophilicity of solvent system. Fig. 2 is the analytical HPLC chromatogram of F. bidentis extracts. Since there are many compounds and relatively low concentration of chlorogenic acid in the crude sample of F. bidentis (Fig. 2), concentration of TFA should be reduced so that the width of the pH zone is increased; in this way, it would increase the yield of pure fractions. Therefore, the concentrations of both retainer (TFA) and eluter (NH4OH) were decreased to 3 mM. Fig. 3 shows the pH-zone refining CCC separation from 1.4 g of F. bidentis where 16.2 mg of chlorogenic acid was obtained with the purity of 91%. As seen from the chromatogram, chlorogenic acid formed a rectangular peak whereas impurities were highly concentrated at pH-zone front and near boundaries. The trace caffeic acid was concentrated in the mixing zone behind the rectangular peak of chlorogenic acid, which could be collected for further separation.
Figure 2.
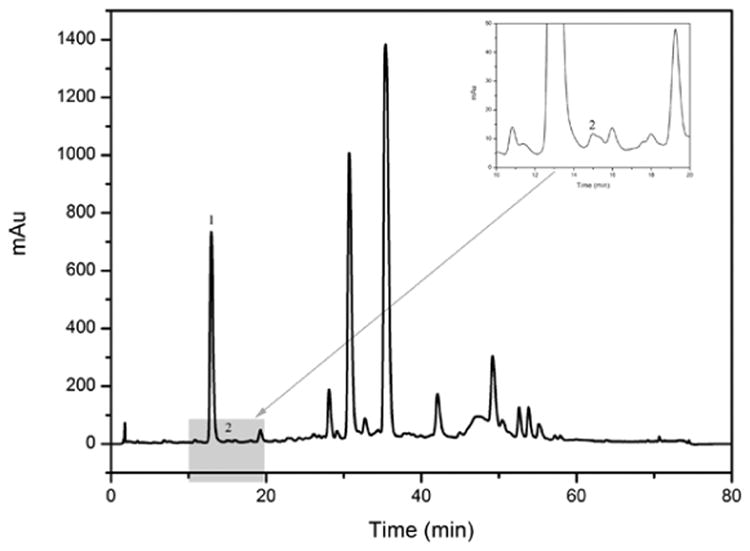
HPLC analysis of the crude extract of F. bidentis. Experimental conditions: Column: Apollo C18 column (150×4.6 mm I.D.); mobile phase: gradient condition: eluent A (MeOH) and eluent B (0.05% (v/v) H3PO4 in water); detection wavelength: 324 nm; column temperature: 30°C
Figure 3.
pH-zone-refining CCC separation of F. bidentis crude extract. Experimental conditions: solvent system: MtBE-acetonitrile-n- butanol-water (4:1:1:5) with 3 mM TFA in the upper phase and 3 mM NH4OH in the lower phase; sample size: 1.4 g; other conditions are described in Figure 1 caption.
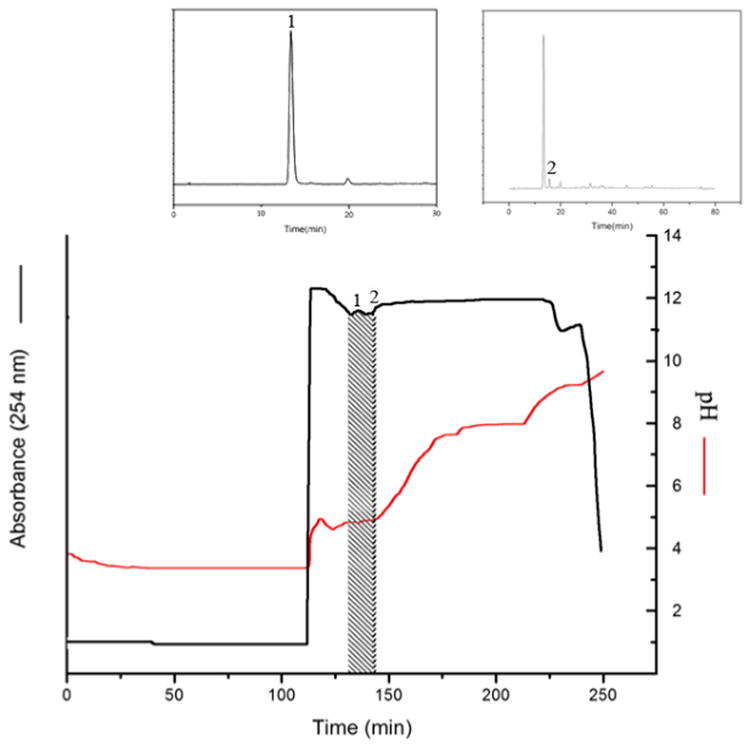
The same method was used to separate chlorogenic acid from the crude extract of Flos lonicerae, because the extract contains a large amount of chlorogenic acid. Fig. 4 is the analytical HPLC chromatogram of Flos lonicerae crude sample. Fig. 5 shows the pH-zone refining CCC of Flos lonicerae crude sample, where 134 mg of chlorogenic acid was separated with the purity of 99% from 1.3 g of the crude sample.
Figure 4.
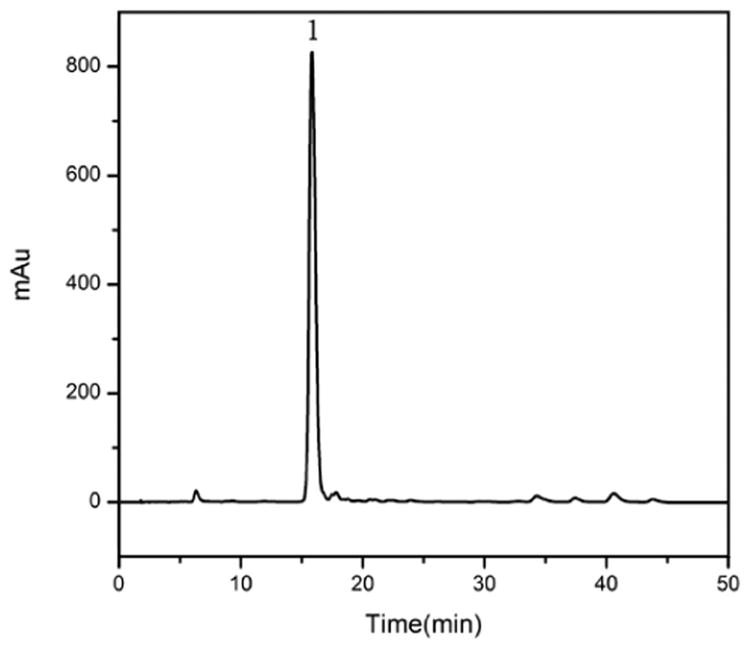
Analytical HPLC chromatogram of Flos lonicerae crude sample. Experimental conditions: See Figure 2 caption.
Figure 5.
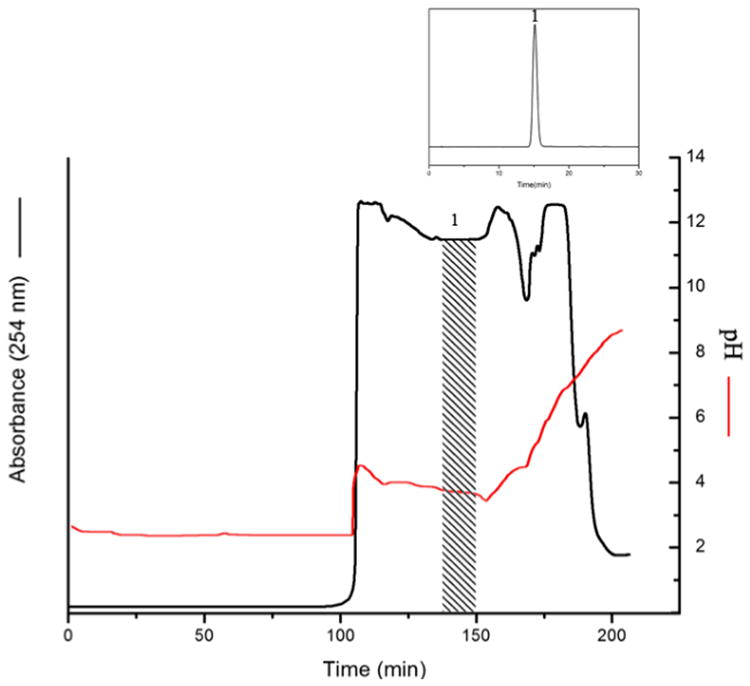
pH-zone refining CCC of Flos lonicerae crude extract. Experimental conditions: Sample size: 1.3 g; other conditions were described in Fig. 3 caption.
In the past chlorogenic acid from Flos lonicerae has been separated by the standard high-speed CCC method using a separation column of a 342 ml capacity where 16.8 mg of the pure target compounds was separated at 94.8% purity with two runs [18]. The present system utilizing pH-zone-refining CCC with a much smaller separation column (220 ml) could separate 8 fold of the target compound at much higher purity of 99% in a single run, indicating the powerful separation capability of pH-zone-refining CCC.
4. Conclusion
Using a pH-zone refining CCC composed of MtBE-ACN-n-BuOH-water (4:1:1:5) containing 3 mM TFA in the stationary phase as a retainer and 3 mM NH4OH in the mobile phase as an eluter, 16.2 mg of chlorogenic acid was obtained at the purity of 91% from 1.4 g extract of F. bidentis, while a trace compound of caffeic acid was also concentrated. The separation of the chlorogenic acid 134 mg at a high purity of 99% was achieved from 1.3 g of the crude sample of Flos lonicerae. The present study demonstrates a simple and efficient method to separate chlorogenic acid and concentrate a trace amount of compounds from natural products.
Acknowledgments
This work was supported by National Natural Science Foundation of China (NSFC, Grant No. 21075007), Program for New Century Excellent Talents in University (NCET-11-0563), Special Fund for Agro-scientific Research in the Public Interest (project 200803022 & 201103027) and Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT1205).
References
- 1.Xie QQ, Yin L, Zhang GL, Wei Y. J Sep Sci. 2012;35:159–165. doi: 10.1002/jssc.201100554. [DOI] [PubMed] [Google Scholar]
- 2.Zhang FJ, Guo JY, Chen FX, Guo AY, Wan FH. Archives of Agromomy and Soil Science. 2012;58:257–265. [Google Scholar]
- 3.Wei Y, Zhang K, Yin L, Du JL, Zhang GL. J Sep Sci. 2012;35:869–874. doi: 10.1002/jssc.201101027. [DOI] [PubMed] [Google Scholar]
- 4.Wei Y, Du JL, Lu YY. J Sep Sci. 2012;35:2608–2614. doi: 10.1002/jssc.201200266. [DOI] [PubMed] [Google Scholar]
- 5.Wei Y, Gao YL, Xie QQ, Zhang GL, Fu WD. Chromatographia. 2011;73:S97–S102. [Google Scholar]
- 6.Zhang JY, Zhang Q, Li N, Wang ZJ, Lu JQ, Qiao YJ. J Talanta. 2012;11:012. doi: 10.1016/j.talanta.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 7.Tong SQ, Yan JZ, Guan YX. J Chromatogr A. 2008;1212:48–53. doi: 10.1016/j.chroma.2008.09.114. [DOI] [PubMed] [Google Scholar]
- 8.Chen J, Song Y, Li P. J Chromatogr A. 2007;1157:217–226. doi: 10.1016/j.chroma.2007.05.063. [DOI] [PubMed] [Google Scholar]
- 9.Ito Y. J Chromatogr A. 2005;1065:145–168. doi: 10.1016/j.chroma.2004.12.044. [DOI] [PubMed] [Google Scholar]
- 10.Ito Y. J Chromatogr A. 2013;1271:71–85. doi: 10.1016/j.chroma.2012.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ito Y, Ma Y. J Chromatogr A. 1996;19:1451–1457. [Google Scholar]
- 12.Weisz A, Scher AL, Shinomiya K, Fales HM, Ito Y. J Am Chem Soc. 1994;116:74–78. [Google Scholar]
- 13.Weisz A, Mazzola PE, Matusik EJ, Ito Y. J Chromatogr A. 2001;923:87–96. doi: 10.1016/s0021-9673(01)00984-0. [DOI] [PubMed] [Google Scholar]
- 14.Zheng ZJ, Wang ML, Wang DJ, Duan WJ, Wang X, Zheng CC. J Chromatogr B. 2010;878:1647–1651. doi: 10.1016/j.jchromb.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Geng YL, Li FW, Gao QS, Shi XG. J Chromatogr A. 2006;1103:166–169. doi: 10.1016/j.chroma.2005.11.092. [DOI] [PubMed] [Google Scholar]
- 16.Ma Y, Ito Y. J Chromatogr A. 1997;771:81–88. doi: 10.1016/s0021-9673(97)00065-4. [DOI] [PubMed] [Google Scholar]
- 17.Ma Y, Ito Y, Foucault A. J Chromatogr A. 1995;704:75–81. doi: 10.1016/0021-9673(95)00148-g. [DOI] [PubMed] [Google Scholar]
- 18.Liu HT, Jiang Y, Chen F. J Chromatogr A. 2004;1026:185–190. doi: 10.1016/j.chroma.2003.11.004. [DOI] [PubMed] [Google Scholar]