

Abstract
Hydrogen sulfide (H2S) plays important roles in the enteric system in the wall of the gastrointestinal tract. There have been no studies on whether H2S is endogenously generated in peripheral sympathetic ganglia and, if so, its effect on synaptic transmission. In this study, we examined the effect of H2S on cholinergic excitatory fast synaptic transmission in the mouse superior mesenteric ganglion (SMG). Our study revealed that NaHS and endogenously generated H2S selectively potentiated cholinergic fast EPSPs (F-EPSPs) evoked by splanchnic nerve stimulation but not F-EPSPs evoked by colonic nerve stimulation. The H2S-producing enzyme cystathionine-γ-lyase (CSE) was expressed in both neurons and glial cells. The CSE blocker PAG (dl-propargylglycine) significantly reduced the amplitude of F-EPSPs evoked by splanchnic nerve stimulation but not F-EPSPs evoked by colonic nerve stimulation. Inhibiting the breakdown of endogenously generated H2S with stigmatellin potentiated the amplitude of F-EPSPs evoked by splanchnic nerve stimulation but not F-EPSPs evoked by colonic nerve stimulation. Splanchnic F-EPSPs but not colonic F-EPSPs were reduced in CSE knock-out (KO) mice. Functional studies showed that NaHS enhanced the inhibitory effect of splanchnic nerve stimulation on colonic motility. Colonic motility in CSE-KO mice was significantly higher than colonic motility in wild-type mice. We conclude that endogenously generated H2S acted selectively on presynaptic terminals of splanchnic nerves to modulate fast cholinergic synaptic input and that this effect of H2S modulates CNS control of gastrointestinal motility. Our results show for the first time that the facilitatory effect of endogenous H2S in the mouse SMG is pathway specific.
Introduction
Sympathetic innervation to the gastrointestinal and urogenital organs is supplied mainly by noradrenergic neurons, the cell bodies of which lie in the celiac ganglion, superior mesenteric ganglion, and inferior mesenteric ganglion, collectively referred to as the abdominal prevertebral ganglia (PVGs). PVG neurons are essential for the control of transit through the gastrointestinal tract, secretion, absorption, and gastrointestinal blood flow. PVG neurons receive preganglionic fast excitatory nicotinic cholinergic synaptic input via splanchnic nerves from the intermediolateral column of the spinal cord and fast excitatory nicotinic cholinergic synaptic input from a unique subset of enteric ganglion neurons referred to as the intestinofugal neurons (Szurszewski and Linden, 2012). Axons of PVG neurons, in turn, make synaptic contact with neuronal structures in the enteric nervous system to modify programmed enteric circuits (Wood, 1999). The intestinofugal pathway together with the PVG can be considered as an extended neural network that coordinates enteric neuronal activity within and between different regions of the gastrointestinal tract and its accessory organs.
Hydrogen sulfide (H2S) is considered a signaling molecule in the mammalian CNS and peripheral nervous system (Wang, 2012). In the CNS, H2S enhances NMDA receptor-mediated responses during tetanic nerve stimulation in the hippocampus (Abe and Kimura, 1996; Tan et al., 2010; Austgen et al., 2011), reversibly inhibits synaptic responses in dorsal raphe serotonergic neurons (Kombian et al., 1993), and augments synaptic neurotransmission in the rat nucleus tractus solitarius (Austgen et al., 2011). In the peripheral nervous system, H2S induces contraction of the bladder wall (Patacchini et al., 2004) and acts on enteric neurons in the human and guinea pig colon to increase chloride secretion (Schicho et al., 2006; Krueger et al., 2010).
There have been no studies on the effect of H2S in mammalian abdominal sympathetic PVG. Because of the central role that the PVGs have in modulating reflex activity between different regions of the gastrointestinal tract, and because of the emerging importance of H2S in the CNS and enteric nervous system, we designed experiments to test the hypothesis that enzymes that generate endogenous H2S are expressed in the mouse superior mesenteric ganglion (SMG) and that H2S modulates synaptic transmission in the SMG. Our results show that NaHS and endogenously generated H2S selectively potentiated fast excitatory cholinergic nicotinic synaptic input from splanchnic nerves without having any effect on fast excitatory synaptic input from mechanosensory intestinofugal neurons. We also showed that the H2S-mediated modulation of splanchnic nerve input modulates the inhibitory effect of splanchnic nerve activation on colonic motility.
Materials and Methods
Adult (5- to 8-week-old) SJL/J mice (The Jackson Laboratory), cystathionine-γ-lyase (CSE) knock-out (KO) mice and wild-type (WT) control mice, and adult guinea pigs (Hartley, either sex, ∼500 g of body weight) were used in this study. CSE-KO mice were obtained from Dr. Rui Wang (Lakehead University, Thunder Bay, ON, Canada) (Yang et al., 2008) and backcrossed at the Mayo Clinic for 10 generations until they were congenic with C57BL/6 mice (The Jackson Laboratory). CSE-KO and WT control animals used in this study were littermates obtained via heterozygous breeding. Genotype was determined by a single PCR with one primer (5′-TGATCTTGGCCAGAGTAGACACGC-3′) corresponding to a sequence 365 bp 5′ of the transcriptional start site, one primer (5′-AATGCTGGAAACTAGGCAGGAAGC-3′) complementary to a sequence in exon 1 downstream of the ATG start codon, and one primer (5′-TGAATGGAAGGATTGGAGCTACGG-3′) that is complementary to a sequence within the neor cassette. This reaction yields a 437 bp product from a wild-type allele and a 547 bp product from a transgenic allele. The specificity of both products was confirmed by sequencing. The use of animal tissue was approved by the Institutional Animal Care and Use Committee.
Electrophysiology.
Immediately after the animals were killed with CO2, the abdomen was opened, and the SMG and attached splanchnic and colonic nerves were dissected and placed in a recording chamber. Bipolar stimulating electrodes were placed on each nerve trunk, and two duel-channel stimulators (Grass S88, Grass Medical Instruments) were used for electrical stimulation. Single stimuli were applied at rate of 1 per 20 s. The minimum stimulation interval among different nerve trunks was at least a 2 s. When paired-pulse stimulations were applied, each pulse was of equal strength at an interval of 80–100 ms. When paired pulses were delivered to different nerve trunks in the same preparation, they were separated in time by at least 1 min. In some experiments, the SMG was attached to a segment of colon. The SMG with an attached segment of distal colon (∼4 cm) was dissected and placed in a recording chamber that had two compartments (Miller and Szurszewski, 1997). The SMG was pinned down into one compartment, and the attached colon was placed in the adjacent compartment. The orad end of the colon was tied closed, and the caudad end was cannulated and attached to a pressure transducer to monitor intraluminal pressure. Basal intraluminal pressure was set at 5 cm H2O. The two compartments were superfused separately with preoxygenated normal Krebs solution (NKS) at 37°C. The preparations were incubated in the recording chamber for 1–2 h before recording.
The composition of the Krebs solution was as follows (in mm): 137.4 Na+, 5.9 K+, 2.5 Ca2+, 1.2 Mg2+, 124 Cl−, 15.5 HCO3−, 1.2 H2PO4−, and 11.5 glucose. It was continuously bubbled with 97% O2, 3% CO2, and maintained at pH 7.4. Sharp glass microelectrodes filled with 3 M KCl (with input resistances of ∼70 MΩ) were used to record intracellular values. Only cells that had a stable resting membrane potential equal to or more negative than −40 mV and that responded to a depolarizing current injection with action potentials (with overshoot) were used. Recorded signals were amplified through an amplifier (Intra 767, WPI), digitized (Digidata 1322A, Molecular Devices), analyzed, and stored in a computer. Acetylcholine (ACh) was delivered through a fine glass micropipette by pressure ejection (Picospritzer, General Valve Corporation).
Immunohistochemistry.
The mouse SMG was dissected immediately after the animal was killed with CO2 gas. The SMG was fixed with 4% paraformaldehyde at 4°C for 2 h. After fixation, the SMG was stained for CSE and vesicular ACh transporter (VAChT), and for cystathionine-β-synthase (CBS) and VAChT using mouse anti-CSE (1:1000 dilution; monoclonal, Abnova), mouse anti-CBS (1:1000 dilution; monoclonal, Abnova), and goat anti-VAChT (1:10,000 dilution; Phoenix Pharmaceuticals) using previously published methods (Sha et al., 2010). Tissue without the primary antibody was used as a negative control. The CSE and CBS antibodies we used were previously tested and provided good immunostaining in a previous publication (Linden et al., 2008). The preparations were mounted onto slides with Slowfade antifade reagent (Life Technologies) and examined under a fluorescence microscope.
H2S release.
The method used for measuring endogenous production and release of H2S was previously described (Linden et al., 2008). Guinea pig celiac ganglion, SMG and inferior mesenteric ganglion were dissected immediately after the animal was killed with CO2 gas. The tissue was homogenized in ice-cold 50 mm potassium phosphate, pH 6.8, at 40 mg of tissue/ml. The homogenized tissue was put into an incubation chamber at 37°C. The incubation solution (1 ml in each incubation chamber) contained 10 mm l-cysteine, 2 mm pyridoxal 5′-phosphate, 100 mm potassium phosphate buffer, pH 7.4, and 12 mg of tissue homogenate. Air was bubbled from the bottom of the incubation chamber through the incubating solution at a rate of 2–3 ml/min. The incubating chamber was connected by SILASTIC tubing and hypodermic tubing connectors to a 2 ml glass vial containing 1 ml of 10 mm zinc acetate. The air from the incubating chamber was then bubbled though the zinc acetate. The reaction was started by adding l-cysteine to the solution, then stopped at 30 min by applying 50 μl of 500% (w/v) trichloroacetic acid into the incubation solution. Air flow was allowed to continue for an additional 30 min to ensure complete trapping of H2S in the zinc acetate solution. The zinc acetate solutions were then sealed in vials until measured using gas chromatography (Levitt et al., 1999).
Measurement of colonic pressure.
To record colonic pressure in vitro, the SMG with an attached segment of distal colon (∼4 cm) was dissected from adult SJL/J mice and placed in a recording chamber that had two compartments. The SMGs with attached splanchnic nerve trunks were placed in one compartment, and the colon attached to the SMG via the colonic nerve was placed in the adjacent compartment. The orad end of the colon was closed, while the caudad end was cannulated and attached to a pressure transducer to monitor intraluminal pressure. Both compartments were superfused separately with NKS at 37°C. Basal intraluminal pressure was set at 5 cm H2O. A bipolar stimulating electrode was placed on the splanchnic nerves to stimulate splanchnic nerves. The pressure signal was digitized (Digidata 1322A, Molecular Devices), analyzed, and stored in a computer.
To record colonic pressure in vivo in conscious mice, mice were briefly anesthetized with isoflurane by inhalation, and a miniaturized pressure transducer catheter (Miller Instruments) lubricated with medical grade lubricant was introduced into the colon such that the middle of the pressure sensor was 2.5 cm proximal to the anus. The catheter was secured to the tail with tape. Conscious mice were kept in a restraint tube for the duration (90 min) of the experiments. The pressure signal was digitized (PowerLab/4SP, ADinstruments), analyzed, and stored in a computer.
NaHS, hexamethonium, dl-propargylglycine (PAG), aminooxyacetic acid (AOAA), bicuculline, and stigmatellin were dissolved in NKS and applied to the recording chamber through superfusion. ACh was dissolved in distilled water and applied by pressure ejection through a glass micropipette brought into close proximity to the cell that was impaled by the microelectrode. NaHS was purchased from Cayman Chemical; isoflurane was from Piramal Healthcare; and hexamethonium, nifedipine, mibefradil, PAG, AOAA, ACh, bicuculline, and stigmatellin were from Sigma.
Values are given as the mean ± SE. Paired Student's t test was used in statistical comparisons between the value in the test group and the value in the control group. A p value of ≤0.05 was considered to be statistically significant.
Results
NaHS selectively augmented cholinergic fast EPSPs evoked by splanchnic nerve stimulation
We recorded from 86 neurons in 52 mouse SMG preparations. The neurons had a membrane potential of −51.7 ± 1.2 mV and membrane input resistance of 78.4 ± 9.7 MΩ.
In 35 neurons tested, 25 responded to splanchnic nerve stimulation with fast EPSPs (F-EPSPs) and 31 neurons responded to colonic nerve stimulation with F-EPSPs. Antidromic action potentials were recorded in 12 neurons during colonic nerve stimulation. No antidromic action potentials were observed following splanchnic nerve stimulation.
In three neurons tested from three preparations in NKS, F-EPSPs evoked by splanchnic nerve stimulation had an amplitude of 12.3 ± 1.1 mV, and those evoked by colonic nerve stimulation had an amplitude of 15.7 ± 1.6 mV. Hexamethonium (50 μm) completely abolished nerve stimulation-evoked F-EPSPs, indicating that they were cholinergic and mediated by nicotinic receptors.
To determine the effect of NaHS on cholinergic F-EPSPs, we measured the amplitude, area, duration at half-amplitude, and the area above half-amplitude of F-EPSPs evoked by electrical stimulation of the splanchnic and colonic nerves in NKS and 10–15 min after application of 200 μm NaHS (Fig. 1A). NaHS significantly (p < 0.05) increased the amplitude of F-EPSPs evoked by splanchnic nerve stimulation (12.8 ± 2.0 mV compared with 7.8 ± 1.2 mV in NKS, n = 9; Fig. 1B; Table 1). The area of F-EPSPs and the area above half-amplitude were also increased significantly (Table 1). The effect of NaHS was reversible 20 min after washout of NaHS (Fig. 1B). NaHS (200 μm) had no effect on F-EPSPs evoked by colonic nerve stimulation (Fig. 1C; Table 1) as there were no significant changes in the amplitude, area, duration at half-amplitude, or area above half-amplitude (Table 1).
Figure 1.

NaHS-potentiated F-EPSPs evoked by splanchnic nerve stimulation, but not F-EPSPs, evoked by colonic nerve stimulation. NaHS had no effect on the postsynaptic membrane. A, Measurements made of evoked F-EPSPs. B, C, Effect of NaHS (200 μm) on F-EPSPs evoked by splanchnic nerve stimulation (B) and colonic nerve stimulation (C) in the same neuron. Control recordings are in black; recordings made with NaHS are in red; washout of NaHS is in blue. D, NaHS (200 μm) potentiated paired F-EPSPs evoked by splanchnic nerve stimulation without significantly altering the paired-pulse ratio. E, NaHS had no effect on paired F-EPSPs evoked by paired colonic nerve stimulation. Recordings in D and E were made from the same neuron. F, NaHS had no significant effect on the depolarizing response to ACh applied by pressure ejection. Both traces were recorded from the same neuron. G, NaHS had no effect on the current–voltage relationship of the same neuron. Current (−1 to 1 nA, 200–500 ms duration) was injected into the recorded neuron via the recording electrode. The membrane potential changes were plotted against the injected currents.
Table 1.
Effect of NaHS, PAG plus AOAA, PAG alone, and AOAA alone on F-EPSPs
| Nerve trunk | Condition | Amplitude (mV) | Area (mV·ms) | Duration at 0.5 A (ms) | Area above 0.5 A (mV·ms) | |
|---|---|---|---|---|---|---|
| Effect of NaHS | Splanchnic (n = 9) | Control | 7.8 ± 1.2 | 128 ± 26 | 14.6 ± 1.8 | 38 ± 9 |
| NaHS | 12.8 ± 2.0** | 336 ± 81* | 17.9 ± 2.2 | 69 ± 15** | ||
| Colonic (n = 13) | Control | 7.3 ± 1.2 | 186 ± 44 | 18.4 ± 2.0 | 45 ± 10 | |
| NaHS | 7.5 ± 1.5 | 202 ± 56 | 18.3 ± 2.1 | 48 ± 12 | ||
| Effect of PAG plus AOAA | Splanchnic (n = 5) | Control | 13.0 ± 3.2 | 322 ± 152 | 15.2 ± 2.7 | 72 ± 35 |
| PAG + AOAA | 9.7 ± 2.9** | 197 ± 68 | 16.1 ± 2.6 | 43 ± 17 | ||
| Colonic (n = 6) | Control | 14.0 ± 2.3 | 385 ± 116 | 16.2 ± 2.7 | 68 ± 16 | |
| PAG + AOAA | 12.4 ± 2.9 | 368 ± 103 | 21.3 ± 4.7 | 76 ± 20 | ||
| Effect of PAG | Splanchnic (n = 6) | Control | 27.0 ± 4.1 | 1230 ± 414 | 25.1 ± 3.0 | 169 ± 36 |
| PAG | 23.7 ± 3.9* | 825 ± 288* | 19.5 ± 3.1** | 130 ± 33* | ||
| Colonic (n = 5) | Control | 26.9 ± 7.6 | 855 ± 241 | 26.8 ± 5.2 | 164 ± 73 | |
| PAG | 30.2 ± 6.1 | 808 ± 235 | 23.3 ± 5.8 | 197 ± 84 | ||
| Effect of AOAA | Splanchnic (n = 4) | Control | 20.2 ± 10.9 | 438 ± 318 | 11.3 ± 3.2 | 84 ± 62 |
| AOAA | 23.6 ± 12.1* | 651 ± 455 | 16.9 ± 3.6 | 130 ± 79* | ||
| Colonic (n = 6) | Control | 28.9 ± 8.1 | 1084 ± 402 | 25.2 ± 4.1 | 232 ± 89 | |
| AOAA | 36.2 ± 8.2 | 1609 ± 468* | 28.0 ± 4.6 | 307 ± 103 | ||
| Effect of AOAA with bicuculline | Splanchnic (n = 3) | Control | 11.9 ± 2.8 | 346 ± 153 | 16.4 ± 1.9 | 54 ± 15 |
| AOAA | 11.5 ± 4.2 | 303 ± 152 | 16.2 ± 1.4 | 51 ± 18 | ||
| Colonic (n = 5) | Control | 17.5 ± 3.8 | 630 ± 188 | 19.5 ± 2.4 | 88 ± 27 | |
| AOAA | 16.7 ± 3.8 | 540 ± 195 | 20.6 ± 3.6 | 101 ± 37 | ||
| Effect of stigmatellin | Splanchnic (n = 4) | Control | 18.4 ± 5.3 | 338 ± 108 | 14.6 ± 1.4 | 83 ± 28 |
| Stigmatellin | 21.0 ± 4.8* | 441 ± 107** | 15.3 ± 2.0 | 94 ± 31 | ||
| Colonic (n = 4) | Control | 24.7 ± 5.3 | 539 ± 153 | 14.2 ± 1.9 | 97 ± 24 | |
| Stigmatellin | 24.1 ± 4.1 | 570 ± 154 | 16.1 ± 2.6 | 104 ± 24 | ||
| WT control for CSE-KO | Splanchnic (n = 6) | 21.4 ± 3.7 | 1200 ± 647 | 27.4 ± 9.6 | 156 ± 63 | |
| Colonic (n = 6) | 15.9 ± 2.5 | 633 ± 146 | 30.3 ± 5.4 | 132 ± 29 | ||
| CSE-KO | Splanchnic (n = 5) | 8.7 ± 2.2*** | 198 ± 57 | 17.4 ± 5.7 | 46 ± 17 | |
| Colonic (n = 6) | 11.1 ± 2.2 | 549 ± 162 | 28.4 ± 3.7 | 99 ± 34 |
*p < 0.05 compared to its control, paired t test.
**p < 0.01 compared to its control, paired t test.
***p < 0.01 compared to WT control.
When the PVGs in the mouse and guinea pig remain attached to the colon, neurons in these ganglia receive ongoing cholinergic F-EPSPs from mechanosensitive intestinofugal neurons (Miller and Szurszewski, 2002; Ermilov et al., 2004). In the present study, we recorded from three neurons in three preparations when the SMG was attached to a segment of the distal colon. Ongoing F-EPSPs occurred at a frequency of 95.3 ± 34.6 per minute and had an amplitude of 3.6 ± 1.0 mV in NKS. When NaHS (200 μm) was applied only on the SMG side of the two-compartment organ bath, the frequency of ongoing F-EPSPs was 96.0 ± 35.7 per minute and the amplitude was 3.7 ± 0.7 mV. Neither were significantly different (p > 0.05, n = 3) when compared with the values obtained in NKS. These data indicated that NaHS did not potentiate ongoing cholinergic F-EPSPs from colonofugal neurons consistent with the observation that NaHS did not potentiate F-EPSPs that were evoked by colonic nerve stimulation.
NaHS had no effect on the postsynaptic membrane
To determine whether the potentiation of F-EPSPs that were evoked by splanchnic nerve stimulation was due to an action of NaHS on the postsynaptic membrane, we tested the effect of NaHS (200 μm, applied for 10–15 min) on the membrane potential and membrane input resistance. There were no significant changes (p > 0.05) in resting membrane potential or membrane input resistance (resting membrane potential: −48.2 ± 1.0 mV with NaHS compared with −46.7 ± 0.8 mV in NKS, n = 13; membrane input resistance: 85.3 ± 12.5 MΩ in NaHS compared with 87.1 ± 10.9 MΩ in NKS, n = 6). We also tested the effect of NaHS (200 μm) on the depolarizing response evoked by microejection of ACh. We used parameters of microejection of ACh that evoked a depolarizing response that matched F-EPSPs evoked by splanchnic nerve stimulation in the same neuron. In NKS, microejection of ACh (200 mm, 10 psi, 20–150 ms) evoked a depolarization that had an amplitude of 10.5 ± 1.2 mV and a duration of 6.7 ± 3.8 s. In the presence of NaHS (200 μm, 10 min), the amplitude was 9.3 ± 0.7 mV and the duration was 7.0 ± 4.0 s (n = 3). Neither were significantly different (p > 0.05) when compared with ACh-evoked responses observed in NKS (Fig. 1F). We also tested the effect of NaHS (200 μm, 10–15 min) on the I–V relationship. In four neurons from four preparations, there was no significant difference in the slope of the I–V relationship, which indicated that NaHS had no significant effect on the conductance of the postsynaptic membrane (Fig. 1G).
L-type and T-type calcium channel blockers had no effect on potentiation of F-EPSPs by NaHS
It has been reported that NaHS increases intracellular Ca2+ in synaptic terminals augmenting synaptic release of glutamate in the nucleus of the solitary tract (Austgen et al., 2011). H2S has been shown to activate L-type Ca2+ channels (García-Bereguiaín et al., 2008) and T-type Ca2+ channels (Kawabata et al., 2007). To test whether NaHS potentiated splanchnic F-EPSPs by activating L-type or T-type Ca2+ channels, we used nifedipine (1 μm) to block L-type Ca2+ channels and mibefradil (1 μm) to block T-type Ca2+ channels.
In four preparations, F-EPSPs evoked by splanchnic nerve stimulation in NKS had an amplitude of 8.9 ± 1.4 mV. When NaHS was applied (200 μm, 10 min), F-EPSPs evoked by splanchnic nerve stimulation had an amplitude of 13.7 ± 1.9 mV (p < 0.05, compared with the values in NKS). The failure of L-type and T-type Ca2+ channel blockers to block NaHS-induced F-EPSP potentiation suggests that potentiation of F-EPSPs by NaHS was not through activation of either L-type or T-type calcium channels.
NaHS and short-term synaptic plasticity of splanchnic and colonic nerve terminals
A paired-pulse stimulation protocol was used to evoke paired F-EPSPs. To avoid F-EPSPs from reaching the threshold to trigger action potentials, a negative current was applied through the recording microelectrode to hold the membrane potential to approximately −100 mV. F-EPSPs were evoked in 20 neurons (10 preparations) by paired-pulse stimulation of the splanchnic nerve. The amplitude of the first F-EPSP was 13.0 ± 1.4 mV, and the amplitude of the second F-EPSP was 17.6 ± 1.5 mV. The paired-pulse ratio was 1.46 ± 0.10, indicating synaptic facilitation in splanchnic nerve terminals (Fig. 1D). In the same 20 neurons, F-EPSPs were evoked from 19 neurons following paired-pulse stimulation of the colonic nerve. The amplitude of the first F-EPSP was 16.5 ± 2.0 mV, and the amplitude of the second F-EPSP was 14.8 ± 1.8 mV. The paired-pulse ratio was 0.90 ± 0.03, indicating synaptic depression in colonic nerve terminals (Fig. 1E). In seven neurons from seven preparations, we tested the effect of NaHS (200 μm) on the paired-pulse ratios. The amplitudes of the first and second F-EPSP evoked by paired-pulse stimulation of the splanchnic nerve were 12.7 ± 0.6 and 16.9 ± 1.6 mV, respectively, before NaHS, and 15.1 ± 0.8 and 19.7 ± 2.0 mV, respectively, in the presence of NaHS (p < 0.01). Although NaHS potentiated the amplitude of both F-EPSPs (Fig. 1D), the paired-pulse ratio was not significantly changed (1.37 ± 0.13 before NaHS; 1.29 ± 0.10 with NaHS; p > 0.05). NaHS had no significant effect on the F-EPSPs evoked by colonic nerve stimulation (13.8 ± 3.0 and 13.4 ± 3.2 mV before NaHS; 13.5 ± 3.3 and 12.7 ± 3.0 mV with NaHS; p > 0.05) and on the paired-pulse ratios of F-EPSPs (0.95 ± 0.04 before NaHS; 0.98 ± 0.07 with NaHS; p > 0.05; Fig. 1E).
H2S was produced in SMG by CSE and CBS
The results above show that exogenous NaHS selectively potentiated cholinergic F-EPSPs evoked by splanchnic nerve stimulation via a presynaptic mechanism. We next devised experiments to determine whether H2S-producing enzymes are expressed in the mouse SMGs, and whether there is ongoing endogenous production and release of H2S.
Immunostaining was performed with antibodies for CSE or CBS. CSE immunoreactivity (IR) was found in 50% of SMG neurons and in structures around neurons, presumably glial cells. VAChT, a marker of synaptic terminals, was also immunostained together with CSE or CBS. We found no colocalization of CSE-IR and VAChT-IR, suggesting that CSE was not expressed in synaptic terminals (Fig. 2, top left). Although CBS-IR was found in a few glial cell-like structures between neurons, no SMG neurons contained CBS-IR (Fig. 2, top right). There was no colocalization of CBS-IR and VAChT-IR.
Figure 2.
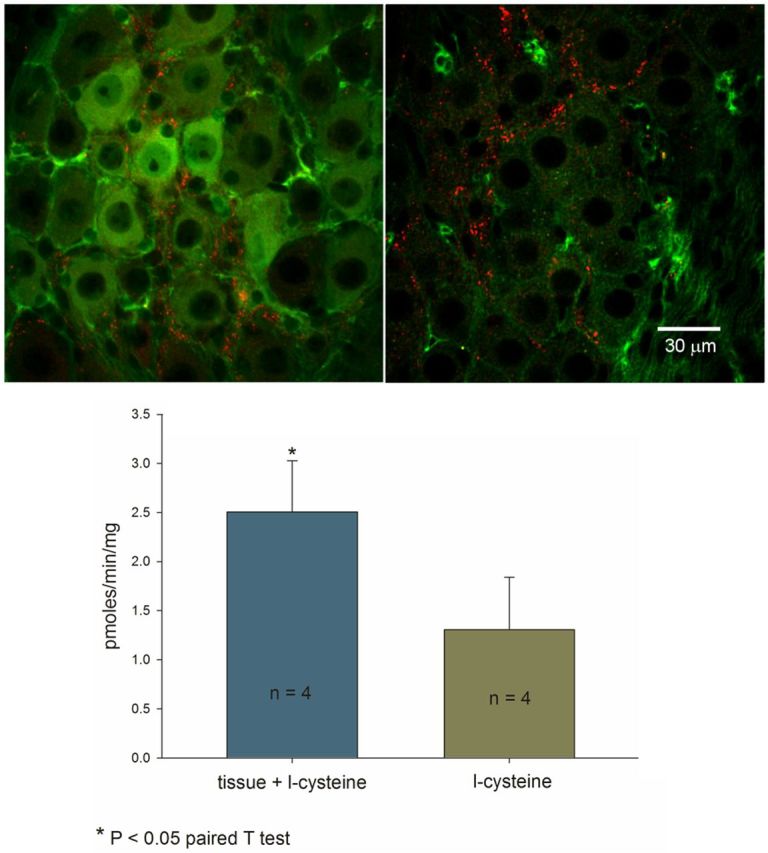
Top, Double staining for CSE (green, left) and VAChT (red, left), and double staining for CBS (green, right) and VAChT (red, right) in a mouse SMG. CSE immunoreactivity was expressed in both neurons and glia cells. CBS immunoreactivity was expressed only in glia cells. Bottom, Endogenous H2S was generated and released in guinea pig PVG.
It was not possible to determine whether H2S was endogenously generated and released in the mouse SMG as the amount of tissue, including the celiac ganglion, the SMGs, and the inferior mesenteric ganglia was <1 mg, far below the amount (30 mg) required to measure H2S production and release. Since the mouse and guinea pig PVGs share many of the same physiological properties (Szurszewski and Linden, 2012), we used PVGs from the guinea pig as a surrogate for mouse PVGs. Our results show that H2S was endogenously generated and released from homogenized guinea pig sympathetic PVGs at a rate of 2.52 ± 0.52 pmol/min/mg tissue weight (Fig. 2, bottom).
Endogenously H2S-modulated F-EPSPs evoked by splanchnic nerve stimulation
Based on the results obtained using guinea pig PVGs, we hypothesized that a mouse PVG also has the capacity to endogenously generate and release H2S. Therefore, we tested the effect of blocking the endogenous production of H2S and the effect of blocking the breakdown of endogenous H2S on evoked F-EPSPs in the mouse SMG.
First, we tested the effect of PAG, a blocker of CSE activity, and AOAA, a blocker of CBS activity. In five neurons from five preparations, PAG (1 mm) and AOAA (0.5 mm) applied together for 10–15 min significantly reduced the amplitude of F-EPSPs evoked by splanchnic nerve stimulation (Fig. 3A1; Table 1). In six neurons from six preparations, PAG and AOAA had no effect on any of the parameters of F-EPSPs evoked by colonic nerve stimulation (Fig. 3A2; Table 1).
Figure 3.

A1, A2, The CSE blocker PAG together with the CBS blocker AOAA inhibited F-EPSPs evoked by splanchnic nerve stimulation (A1), but not F-EPSPs evoked by colonic nerve stimulation (A2). The inhibition of F-EPSPs evoked by the splanchnic nerve stimulation was reversed when NaHS (200 μm) was added to the bath (see text for further details). B1, B2, PAG alone inhibited F-EPSPs evoked by splanchnic nerve stimulation (B1) but not F-EPSPs evoked by colonic nerve stimulation (B2). C1, C2, AOAA alone potentiated splanchnic (C1) and colonic F-EPSPs (C2). D1, D2, Stigmatellin potentiated F-EPSPs evoked by the central splanchnic nerve stimulation (D1) but not F-EPSPs evoked by the peripheral colonic nerve stimulation (D2). Traces in each group were from the same neuron.
The inhibition of F-EPSPs evoked by splanchnic nerve stimulation when H2S production was blocked by the CSE and CBS inhibitors was reversed with the addition of NaHS. In three neurons from the three preparations tested, F-EPSPs evoked by splanchnic nerve stimulation in NKS had an amplitude of 15.8 ± 3.9 mV, an area of 446 ± 234 mV·ms, and an area above half-amplitude of 103 ± 54 mV·ms. After 15 min with PAG and AOAA, the amplitude of the F-EPSPs was reduced significantly to 11.2 ± 3.9 mV (p < 0.05). The area and the area above half-amplitude were reduced to 251 ± 95 and 58 ± 24 mV·ms, respectively. When NaHS (200 μm; 10–15 min) was added when both blockers were present, the amplitude of F-EPSPs was restored to 15.8 ± 2.4 mV, as were the area of the F-EPSPs (416 ± 74 mV·ms) and the area above half-amplitude (109 ± 25 mV·ms).
We also tested the effect of each blocker separately. To avoid interference from action potentials, the membrane potential of the recorded neurons was hyperpolarized to −100 to −130 mV by constant current injection through the recording microelectrode. In NKS, F-EPSPs evoked by splanchnic nerve stimulation had an amplitude of 27.0 ± 4.1 mV (six neurons from six preparations). With the application of PAG (1 mm) for 10–15 min, the amplitude of F-EPSPs evoked by splanchnic nerve stimulation was significantly reduced to 23.7 ± 3.9 mV (p < 0.05, Fig. 3B1). The area of F-EPSPs, the duration at half-amplitude, and the area above half-amplitude were also significantly reduced (Table 1). F-EPSPs evoked by colonic nerve stimulation were also tested in four neurons from four preparations. In contrast to the effect of PAG on F-EPSPs evoked by splanchnic nerve stimulation, PAG (1 mm) had no significant effect on the amplitude, area, duration at half-amplitude, and area above half-amplitude of the F-EPSPs evoked by colonic nerve stimulation (Fig. 3B2; Table 1).
We also tested the effect of AOAA (0.5 mm, 10–15 min) applied alone. There was a significant increase or trend of an increase in F-EPSPs evoked by splanchnic nerve stimulation, and colonic nerve stimulation (Fig. 3C1,C2; Table 1). Since AOAA is also an inhibitor of GABA transaminase (Sawynok and LaBella, 1982; Pagliusi et al., 1983), it was possible that the potentiating effect of AOAA on F-EPSPs was due to the inhibition of GABA transaminase. To test for this possibility, we pretreated five preparations with the GABAA receptor antagonist bicuculline (20–50 μm). In the presence of bicuculline, AOAA failed to potentiate F-EPSPs evoked by splanchnic nerve stimulation (n = 3) and by colonic nerve stimulation (n = 5; Table 1).
Stigmatellin is a sulfide quinone reductase (SQR) inhibitor that inhibits the breakdown of endogenously produced H2S (Linden et al., 2012). We tested the effect of stigmatellin on F-EPSPs evoked by splanchnic nerve stimulation and colonic nerve stimulation. Stigmatellin increased the amplitude of F-EPSPs evoked by splanchnic nerve stimulation in NKS from 18.4 ± 5.3 to 21.0 ± 4.8 mV (n = 4, p < 0.05). The area of F-EPSPs also increased significantly when stigmatellin was present in the bathing solution (p < 0.01; Fig. 3D1;Table 1). In sharp contrast, stigmatellin had no significant effect on F-EPSPs evoked by colonic nerve stimulation (p > 0.05; Fig. 3D2; Table 1).
F-EPSPs evoked by splanchnic nerve stimulation were significantly reduced in CSE-KO mice
In this set of experiments, six CSE-KO mice and six WT control mice were used. The membrane potential of recorded neurons was hyperpolarized to −100 to −130 mV with constant negative current injection to allow maximal stimulation while avoiding the interference of action potentials. In WT control mice, the amplitude of F-EPSPs evoked by splanchnic nerve stimulation was 21.4 ± 3.7 mV, and the area was 1200 ± 647 mV·ms (n = 6 preparations; Table 1). F-EPSPs evoked by colonic nerve stimulation had an amplitude of 15.9 ± 2.5 mV and an area of 633 ± 146 mV·ms (n = 6; Table 1). In CSE-KO mice, the amplitude of F-EPSPs evoked by splanchnic nerve stimulation was significantly reduced to 8.7 ± 2.2 mV (n = 5, p < 0.01). The areas of F-EPSPs (198 ± 57 vs 1200 ± 647 mV·ms, p = 0.06) and area above half-amplitude (46 ± 17 vs 156 ± 63 mV·ms, p = 0.08) were not significantly different from WT controls (Table 1). There was no change in duration at half-amplitude (p = 0.2; Table 1). The amplitude, area, duration at half-amplitude, and area above half-amplitude of F-EPSPs evoked by colonic nerve stimulation in CSE-KO mice were unchanged (n = 6, p > 0.05; Table 1) compared with the WT control. These results show that F-EPSPs evoked by splanchnic nerve stimulation but not by colonic nerve stimulation were reduced in CSE-KO mice.
NaHS enhanced the inhibitory effect of splanchnic nerve activation on colonic motility
Ongoing splanchnic nerve activity modulates colonic motility in vivo (Szurszewski and Linden, 2012). In this set of experiments, we investigated the functional significance of endogenously generated H2S on colonic motility in preparations that consisted of the SMG attached to a segment of distal colon. Colonic motility was monitored by measuring the intraluminal pressure, and splanchnic nerves were activated by electric nerve stimulation. All colon preparations generated short-duration (2–9 s) and low-amplitude (1–12 mm water) phasic increases in intraluminal pressure, and low-frequency (0.3 ± 0.1/min), long-duration (24–104 s duration), high-amplitude (8–16 mm H2O) phasic increases in pressure. The average area under the pressure wave curve was 481 ± 116 mm H2O·ms/s (n = 4; Fig. 4A) above baseline in normal Krebs solution without splanchnic nerve stimulation. Splanchnic nerve stimulation at 3–5 Hz and 60 V completely abolished both low- and high-amplitude phasic pressure waves. To test the effect of NaHS on colonic motor activity during splanchnic nerve stimulation, we used parameters of stimulation that only partially inhibited the amplitude of pressure waves. In four preparations, splanchnic nerve stimulation at 3–5 Hz with decreased intensity (12–40 V) partially inhibited the amplitude of the low- and high-amplitude pressure waves (389 ± 124 mm H2O·ms/s; p > 0.05, n = 4; Fig. 4B). When only the SMG was superfused with 200 μm NaHS, the same splanchnic nerve stimulation significantly enhanced the inhibition of colonic pressure waves (189 ± 36 mm H2O·ms/s; p < 0.05, n = 4; Fig. 4C). Superfusion of the SMG with 200 μm NaHS alone without the splanchnic nerve stimulation had no effect on colonic pressure waves (532 ± 74 mm H2O·ms/s; p > 0.05 compared with without NaHS). These results show that NaHS added only to the SMG that potentiated the inhibitory effect of splanchnic nerve stimulation on colonic motility in vitro.
Figure 4.

NaHS enhanced the inhibitory effect of splanchnic nerve activation on colonic motility (left panels), and colonic motility was higher in CSE-KO mice than colonic motility in wild-type mice (right panels). A–C, left, Recordings in the same preparation. A, Colonic pressure changes in normal Krebs solution without splanchnic nerve stimulation. B, Colonic pressure changes in normal Krebs solution with splanchnic nerve stimulation. C, Colonic pressure changes with the splanchnic nerve stimulation and NaHS added only to the SMG. The top trace in the right panel is intraluminal pressure recorded in a conscious wild-type mouse. The bottom trace is intraluminal pressure recorded in a CSE-KO mouse.
Colonic motility in conscious WT and CSE-KO mice
To determine whether colonic motility is altered in mice with altered H2S production, we measured colonic motility in CSE-KO mice and compared it with colonic motility in littermate wild-type mice. Colonic motility was recorded in five CSE-KO mice and five littermate wild-type control mice. A representative trace of colonic intraluminal pressure in a wild-type mouse and a CSE-KO mouse is shown in the right panel of Figure 4. Phasic changes of intraluminal pressure had greater amplitude in CSE-KO mice compared with wild-type mice. The area under the colonic motility curve above baseline was measured for each mouse. The average area under the curve in wild-type mice was 125.7 ± 12.3 mm Hg·s/min. The average area under the curve in CSE-KO mice was 222.3 ± 31.2 mm Hg·s/min, which was significantly higher than the control (p < 0.05).
Discussion
Preganglionic splanchnic nerves innervate adrenergic neurons in the PVGs. Sympathetic PVG neurons provide inhibitory innervation to the colon suppressing colonic motility (De Groat and Krier, 1979; Sarna, 1991; Szurszewski and Linden, 2012). In this study, we found that NaHS and endogenously generated H2S selectively modulated preganglionic splanchnic nerve transmission in mouse SMG. Colonic motility was inhibited during NaHS-induced facilitation of fast splanchnic input to SMG neurons supporting the hypothesis that H2S-mediated modulation on synaptic transmission in splanchnic nerve terminals has physiological significance. The potentiation of splanchnic nerve stimulation to inhibit colonic motility by NaHS and the increase in ongoing colonic contractions in CSE-KO mice support the general hypothesis that endogenous production and release of H2S in the PVGs facilitate central splanchnic inhibition of ongoing colonic motor activity.
Our study shows that CSE was the predominant H2S-generating enzyme, as it was found in 50% of the neurons and in presumptive glial cells, and that NaHS and endogenously generated H2S potentiated fast excitatory cholinergic nicotinic synaptic transmission through an action on presynaptic cholinergic terminals. Importantly, this effect was pathway specific. NaHS and endogenously generated H2S potentiated F-EPSPs evoked by stimulation of central preganglionic nerves without any measurable effect on F-EPSPs evoked by electrical stimulation of peripheral colonic nerves. Furthermore, neither NaHS nor endogenously generated H2S had any effect on ongoing F-EPSPs arising from colonic mechanosensory cholinergic intestinofugal neurons. The inhibition of endogenous production of H2S during pharmacological blockade of CSE and the genetic deletion of CSE significantly reduced the amplitude of F-EPSPs evoked by stimulation of central preganglionic nerves. In contrast, the amplitude of F-EPSPs evoked by stimulation of central preganglionic nerves was potentiated when H2S breakdown was inhibited by stigmatellin, an inhibitor of SQR. When considered together, our results strongly suggest that constitutive release of H2S in the mouse SMG potentiates F-EPSPs arising from central preganglionic nerves. This argues for a specific action of H2S on subsets of synaptic terminals rather than a ubiquitous synaptic facilitator. Our functional study revealed that the H2S-mediated potentiation of splanchnic nerve input enhanced the inhibitory effect of splanchnic nerve activation on colonic motility.
The potentiating effect of NaHS on central preganglionic synaptic input was not due to a postsynaptic site of action. NaHS did not alter the membrane potential and membrane input resistance, suggesting that the postsynaptic membrane channels that contribute to membrane voltage and resting conductance were insensitive to NaHS. NaHS also did not change the depolarization evoked by acetylcholine applied exogenously by pressure ejection, suggesting that NaHS also had no effect on postsynaptic cholinergic nicotinic receptors. These results suggest that the potentiating effect of NaHS was due to a presynaptic effect. In the CNS, NaHS has been shown to act both presynaptically and postsynaptically (Kimura, 2000; Matsunami et al., 2009; Tan et al., 2010).
Both CSE and CBS were expressed in the mouse SMG. CBS was found in only a few glial cell-like structures and not in neurons. In contrast, CSE had a much wider distribution, as it was found in neurons and in glial-like cells as well as in neurons. We did not observe colocalization of either CSE or CBS in punctate structures with immunoreactivity for vesicular acetylcholine transporter, which would be suggestive of cholinergic synaptic terminals.
The results we obtained when using CSE and CBS enzyme inhibitors are consistent with the hypothesis that CSE was the predominant enzyme generating H2S in the mouse SMG. The CSE inhibitor PAG selectively inhibited F-EPSPs evoked by stimulation of central preganglionic nerves, whereas the CBS inhibitor AOAA slightly potentiated F-EPSPs evoked by stimulation of central preganglionic nerves and peripheral colonic nerves. Because AOAA lacks specificity (Hosoki et al., 1997; Kabil and Banerjee, 2010) and is an inhibitor of GABA transaminase (Sawynok and LaBella, 1982; Pagliusi et al., 1983), GABA could have increased acetylcholine release from central preganglionic nerves as it does in the rabbit celiac ganglion (Ercoli et al., 2007). When we tested the effect of bicuculline, a GABAA receptor antagonist (Parkman et al., 1993; Stapelfeldt et al., 1993), we found that bicuculline blocked the increase in F-EPSP amplitude evoked by splanchnic nerve stimulation when AOAA was present in the recording chamber. These results suggest that stimulation of central preganglionic nerves released not only acetylcholine but also GABA, as it does in the rabbit celiac ganglion (Ercoli et al., 2007). Our experiments suggest that the potentiation of F-EPSPs by AOAA was primarily due to the inhibition of GABA transaminase, which prevented the degradation of GABA, and not the inhibition of CBS.
F-EPSPs evoked by splanchnic nerve stimulation showed paired-pulse facilitation, while F-EPSPs evoked by colonic nerve stimulation showed paired-pulse depression. It has been previously reported that splanchnic nerve terminals responded to paired-pulse stimulations with facilitation in rabbit celiac ganglion (Ercoli et al., 2007). Our results extend this observation to include the mouse SMG. The facilitation observed in the rabbit celiac ganglion is mediated by the activation of presynaptic GABAA receptors (Ercoli et al., 2007). Our results when using AOAA suggest that GABA was involved in modulating splanchnic F-EPSPs. Thus, it is possible that the paired-pulse facilitation we observed when stimulating the splanchnic nerve had a similar mechanism. Although our results during GABAA receptor blockage with bicuculline indicated that GABAA receptors were present on colonic nerve terminals as well, no paired-pulse facilitation was observed. Rather, the colonic nerve terminals responded to paired-pulse stimulations with paired-pulse depression. Whether paired-pulse depression seen during colonic nerve stimulation was due to the depletion of vesicles in the readily release pool (Stevens and Wang, 1995) or was due to the vesicle positioning mechanism (Wu and Borst, 1999; Wadel et al., 2007) is not known.
The failure of NaHS and of endogenously generated H2S to potentiate F-EPSPs evoked by colonic nerve stimulation may in part be related to the known structural differences between splanchnic nerve terminals and peripheral intestinofugal cholinergic nerve terminals. Many synaptic boutons of intestinofugal neurons in the celiac ganglion fail to form recognizable contact at the ultrastructural level (Gibbins et al., 2003) and of those that do, some have no detectable SNAP-25 (Gibbins et al., 2003). Synapses that lack the target SNARE protein SNAP-25 have a low release probability. In contrast, SNAP-25 immunoreactivity as well as vesicular acetylcholine transporter and choline acetyltransferase immunoreactivity are much higher in synaptic boutons belonging to splanchnic nerves (Gibbins et al., 2003). This heterogeneity in synaptic structure between central preganglionic and peripheral colonic nerve inputs may in some way account for the differential effects of NaHS and H2S in the mouse SMG. Although H2S has been reported to act on L- and T-type calcium channels elevating intracellular calcium levels in astrocytes, glial cells, and nociceptive neurons (Nagai et al., 2004; Lee et al., 2006; Maeda et al., 2009; Matsunami et al., 2009; Yong et al., 2010), our data suggest that L- and T-type calcium channels were not involved in the H2S-mediated F-EPSP potentiation.
Finally, the effect of stigmatellin remains to be discussed. Previous studies have shown that stigmatellin is an inhibitor of SQR (Linden et al., 2012) and that SQR-like enzymatic activity, while present in the colonic external muscularis and liver, is absent in the brain (Lagoutte et al., 2010; Linden et al., 2010, 2012; Mimoun et al., 2012). The results of the present study suggest that SQR is expressed in the mouse SMG, which contributes to the removal of endogenously generated H2S, thus modulating the level of synaptic transmission from preganglionic spinal neurons.
In conclusion, our study demonstrates a novel, pathway-specific effect of NaHS and endogenously generated H2S on fast excitatory cholinergic nicotinic synaptic input in the mouse SMG. H2S increases cholinergic synaptic transmission only between central preganglionic terminals and SMG neurons. The current study supports the hypothesis that endogenously generated H2S persistently upregulates acetylcholine release from central preganglionic nerve terminals, and that H2S plays an important role in maintaining or modulating gastrointestinal function and coordinating peripheral and central neuronal input to enteric ganglia and to gastrointestinal smooth muscle cells.
Footnotes
This work is supported by National Institutes of Health Grants DK17238 and DK076665. We thank Dr. Rui Wang for providing us with CSE-KO mice. We also thank Julie K. Furne and Dr. Michael D. Levitt for H2S measurement; Dr. Purna C. Kashyap for providing the in vivo colonic pressure measurement technique; and Jan Applequist for preparing this manuscript.
The authors declare no competing financial interests.
References
- Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci. 1996;16:1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austgen JR, Hermann GE, Dantzler HA, Rogers RC, Kline DD. Hydrogen sulfide augments synaptic neurotransmission in the nucleus of the solitary tract. J Neurophysiol. 2011;106:1822–1832. doi: 10.1152/jn.00463.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groat WC, Krier J. The central control of the lumbar sympathetic pathway to the large intestine of the cat. J Physiol. 1979;289:449–468. doi: 10.1113/jphysiol.1979.sp012746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercoli J, Miolan JP, Niel JP, Quinson N. Presynaptic GABA-A receptors prevent depression of nicotinic transmission in rabbit coeliac ganglion neurones. Eur J Neurosci. 2007;25:1307–1318. doi: 10.1111/j.1460-9568.2007.05382.x. [DOI] [PubMed] [Google Scholar]
- Ermilov LG, Schmalz PF, Miller SM, Szurszewski JH. PACAP modulation of the colon-inferior mesenteric ganglion reflex in the guinea pig. J Physiol. 2004;560:231–247. doi: 10.1113/jphysiol.2004.070060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Bereguiaín MA, Samhan-Arias AK, Martín-Romero FJ, Gutiérrez-Merino C. Hydrogen sulfide raises cytosolic calcium in neurons through activation of L-type Ca2+ channels. Antioxid Redox Signal. 2008;10:31–42. doi: 10.1089/ars.2007.1656. [DOI] [PubMed] [Google Scholar]
- Gibbins IL, Jobling P, Teo EH, Matthew SE, Morris JL. Heterogeneous expression of SNAP-25 and synaptic vesicle proteins by central and peripheral inputs to sympathetic neurons. J Comp Neurol. 2003;459:25–43. doi: 10.1002/cne.10527. [DOI] [PubMed] [Google Scholar]
- Hosoki R, Matsuki N, Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun. 1997;237:527–531. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- Kabil O, Banerjee R. Redox biochemistry of hydrogen sulfide. J Biol Chem. 2010;285:21903–21907. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata A, Ishiki T, Nagasawa K, Yoshida S, Maeda Y, Takahashi T, Sekiguchi F, Wada T, Ichida S, Nishikawa H. Hydrogen sulfide as a novel nociceptive messenger. Pain. 2007;132:74–81. doi: 10.1016/j.pain.2007.01.026. [DOI] [PubMed] [Google Scholar]
- Kimura H. Hydrogen sulfide induces cyclic AMP and modulates the NMDA receptor. Biochem Biophys Res Commun. 2000;267:129–133. doi: 10.1006/bbrc.1999.1915. [DOI] [PubMed] [Google Scholar]
- Kombian SB, Reiffenstein RJ, Colmers WF. The actions of hydrogen sulfide on dorsal raphe serotonergic neurons in vitro. J Neurophysiol. 1993;70:81–96. doi: 10.1152/jn.1993.70.1.81. [DOI] [PubMed] [Google Scholar]
- Krueger D, Foerster M, Mueller K, Zeller F, Slotta-Huspenina J, Donovan J, Grundy D, Schemann M. Signaling mechanisms involved in the intestinal pro-secretory actions of hydrogen sulfide. Neurogastroenterol Motil. 2010;22:1224–1231. doi: 10.1111/j.1365-2982.2010.01571.x. [DOI] [PubMed] [Google Scholar]
- Lagoutte E, Mimoun S, Andriamihaja M, Chaumontet C, Blachier F, Bouillaud F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim Biophys Acta. 2010;1797:1500–1511. doi: 10.1016/j.bbabio.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Lee SW, Hu YS, Hu LF, Lu Q, Dawe GS, Moore PK, Wong PT, Bian JS. Hydrogen sulphide regulates calcium homeostasis in microglial cells. Glia. 2006;54:116–124. doi: 10.1002/glia.20362. [DOI] [PubMed] [Google Scholar]
- Levitt MD, Furne J, Springfield J, Suarez F, DeMaster E. Detoxification of hydrogen sulfide and methanethiol in the cecal mucosa. J Clin Invest. 1999;104:1107–1114. doi: 10.1172/JCI7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DR, Sha L, Mazzone A, Stoltz GJ, Bernard CE, Furne JK, Levitt MD, Farrugia G, Szurszewski JH. Production of the gaseous signal molecule hydrogen sulfide in mouse tissues. J Neurochem. 2008;106:1577–1585. doi: 10.1111/j.1471-4159.2008.05502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DR, Levitt MD, Farrugia G, Szurszewski JH. Endogenous production of H2S in the gastrointestinal tract: still in search of a physiologic function. Antioxid Redox Signal. 2010;12:1135–1146. doi: 10.1089/ars.2009.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DR, Furne J, Stoltz GJ, Abdel-Rehim MS, Levitt MD, Szurszewski JH. Sulphide quinone reductase contributes to hydrogen sulphide metabolism in murine peripheral tissues but not in the CNS. Br J Pharmacol. 2012;165:2178–2190. doi: 10.1111/j.1476-5381.2011.01681.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y, Aoki Y, Sekiguchi F, Matsunami M, Takahashi T, Nishikawa H, Kawabata A. Hyperalgesia induced by spinal and peripheral hydrogen sulfide: evidence for involvement of Cav3.2 T-type calcium channels. Pain. 2009;142:127–132. doi: 10.1016/j.pain.2008.12.021. [DOI] [PubMed] [Google Scholar]
- Matsunami M, Tarui T, Mitani K, Nagasawa K, Fukushima O, Okubo K, Yoshida S, Takemura M, Kawabata A. Luminal hydrogen sulfide plays a pronociceptive role in mouse colon. Gut. 2009;58:751–761. doi: 10.1136/gut.2007.144543. [DOI] [PubMed] [Google Scholar]
- Miller SM, Szurszewski JH. Colonic mechanosensory afferent input to neurons in the mouse superior mesenteric ganglion. Am J Physiol. 1997;272:G357–G366. doi: 10.1152/ajpgi.1997.272.2.G357. [DOI] [PubMed] [Google Scholar]
- Miller SM, Szurszewski JH. Relationship between colonic motility and cholinergic mechanosensory afferent synaptic input to mouse superior mesenteric ganglion. Neurogastroenterol Motil. 2002;14:339–348. doi: 10.1046/j.1365-2982.2002.00338.x. [DOI] [PubMed] [Google Scholar]
- Mimoun S, Andriamihaja M, Chaumontet C, Atanasiu C, Benamouzig R, Blouin JM, Tomé D, Bouillaud F, Blachier F. Detoxification of H(2)S by differentiated colonic epithelial cells: implication of the sulfide oxidizing unit and of the cell respiratory capacity. Antioxid Redox Signal. 2012;17:1–10. doi: 10.1089/ars.2011.4186. [DOI] [PubMed] [Google Scholar]
- Nagai Y, Tsugane M, Oka J, Kimura H. Hydrogen sulfide induces calcium waves in astrocytes. FASEB J. 2004;18:557–559. doi: 10.1096/fj.03-1052fje. [DOI] [PubMed] [Google Scholar]
- Pagliusi SR, Gomes C, Leite JR, Trolin G. Aminooxyacetic acid induced accumulation of GABA in the rat brain. Interaction with GABA receptors and distribution in compartments. Naunyn Schmiedebergs Arch Pharmacol. 1983;322:210–215. doi: 10.1007/BF00500767. [DOI] [PubMed] [Google Scholar]
- Parkman HP, Stapelfeldt WH, Williams CL, Lennon VA, Szurszewski JH. Enteric GABA-containing nerves projecting to the guinea-pig inferior mesenteric ganglion modulate acetylcholine release. J Physiol. 1993;471:191–207. doi: 10.1113/jphysiol.1993.sp019897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patacchini R, Santicioli P, Giuliani S, Maggi CA. Hydrogen sulfide (H2S) stimulates capsaicin-sensitive primary afferent neurons in the rat urinary bladder. Br J Pharmacol. 2004;142:31–34. doi: 10.1038/sj.bjp.0705764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarna SK. Physiology and pathophysiology of colonic motor activity (2) Dig Dis Sci. 1991;36:998–1018. doi: 10.1007/BF01297155. [DOI] [PubMed] [Google Scholar]
- Sawynok J, LaBella FS. On the involvement of GABA in the analgesia produced by baclofen, muscimol and morphine. Neuropharmacology. 1982;21:397–403. doi: 10.1016/0028-3908(82)90022-3. [DOI] [PubMed] [Google Scholar]
- Schicho R, Krueger D, Zeller F, Von Weyhern CW, Frieling T, Kimura H, Ishii I, De Giorgio R, Campi B, Schemann M. Hydrogen sulfide is a novel prosecretory neuromodulator in the Guinea-pig and human colon. Gastroenterology. 2006;131:1542–1552. doi: 10.1053/j.gastro.2006.08.035. [DOI] [PubMed] [Google Scholar]
- Sha L, Farrugia G, Linden DR, Szurszewski JH. The transwall gradient across the mouse colonic circular muscle layer is carbon monoxide dependent. FASEB J. 2010;24:3840–3849. doi: 10.1096/fj.10-156232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapelfeldt WH, Parkman HP, Szurszewski JH. The electrophysiological effects of endogenous GABA in the guinea-pig inferior mesenteric ganglion. J Physiol. 1993;471:175–189. doi: 10.1113/jphysiol.1993.sp019896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Wang Y. Facilitation and depression at single central synapses. Neuron. 1995;14:795–802. doi: 10.1016/0896-6273(95)90223-6. [DOI] [PubMed] [Google Scholar]
- Szurszewski JH, Linden DR. Physiology of prevertebral sympathetic ganglia. In: Johnson LR, Ghishan FK, Kaunitz JD, Merchant JL, Hamid SM, Wood JD, editors. Physiology of the gastrointestinal tract. Boston: Academic; 2012. pp. 583–627. [Google Scholar]
- Tan BH, Wong PT, Bian JS. Hydrogen sulfide: a novel signaling molecule in the central nervous system. Neurochem Int. 2010;56:3–10. doi: 10.1016/j.neuint.2009.08.008. [DOI] [PubMed] [Google Scholar]
- Wadel K, Neher E, Sakaba T. The coupling between synaptic vesicles and Ca2+ channels determines fast neurotransmitter release. Neuron. 2007;53:563–575. doi: 10.1016/j.neuron.2007.01.021. [DOI] [PubMed] [Google Scholar]
- Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- Wood JD. Neurotransmission at the interface of sympathetic and enteric divisions of the autonomic nervous system. Chin J Physiol. 1999;42:201–210. [PubMed] [Google Scholar]
- Wu LG, Borst JG. The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron. 1999;23:821–832. doi: 10.1016/S0896-6273(01)80039-8. [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong QC, Choo CH, Tan BH, Low CM, Bian JS. Effect of hydrogen sulfide on intracellular calcium homeostasis in neuronal cells. Neurochem Int. 2010;56:508–515. doi: 10.1016/j.neuint.2009.12.011. [DOI] [PubMed] [Google Scholar]