

Abstract
Acute stress evokes the fight-or-flight reflex, which via release of the catecholamine hormones affects the function of every major organ. Although the reflex is transient, it has lasting consequences that produce an exaggerated response when stress is reexperienced. How this change is encoded is not known. We investigated whether the reflex affects the adrenal component of the sympathetic nervous system, a major branch of the stress response. Mice were briefly exposed to the cold-water forced swim test (FST) which evoked an increase in circulating catecholamines. Although this hormonal response was transient, the FST led to a long-lasting increase in the catecholamine secretory capacity measured amperometrically from chromaffin cells and in the expression of tyrosine hydroxylase. A variety of approaches indicate that these changes are regulated postsynaptically by neuropeptide Y (NPY), an adrenal cotransmitter. Using immunohistochemistry, RT-PCR, and NPY(GFP) BAC mice, we find that NPY is synthesized by all chromaffin cells. Stress failed to increase secretory capacity in NPY knock-out mice. Genetic or pharmacological interference with NPY and Y1 (but not Y2 or Y5) receptor signaling attenuated the stress-induced change in tyrosine hydroxylase expression. These results indicate that, under basal conditions, adrenal signaling is tonically inhibited by NPY, but stress overrides this autocrine negative feedback loop. Because acute stress leads to a lasting increase in secretory capacity in vivo but does not alter sympathetic tone, these postsynaptic changes appear to be an adaptive response. We conclude that the sympathetic limb of the stress response exhibits an activity-dependent form of long-lasting plasticity.
Introduction
Exposure to stress triggers a coordinated response involving two adrenal-dependent pathways: (1) the hypothalamic–pituitary–adrenal (HPA) axis and (2) the sympathetic-adrenal system (Sapolsky et al., 2000; Kvetnansky et al., 2009). HPA activation evokes cortisol secretion from the adrenal cortex, whereas increased activity in the sympathetic–adrenal system provokes elevated catecholamine release (primarily epinephrine) from chromaffin cells in the adrenal medulla. This hormone is a key component of the fight-or-flight response, altering blood pressure and circulating glucose levels (Cherrington et al., 1984; Mathar et al., 2010).
The systemic response to an acute stressor is usually transient. For example, severe hypoglycemia leads to increased release of epinephrine and cortisol (Watts and Donovan, 2010), and circulating levels of both hormones subsequently decline (Widmaier, 1989; Ritter et al., 2006). However, a transient stress can have lasting consequences. The fight-or-flight response is often described as a reflex, but the underlying circuits are plastic and can be modified by previous activity (Gordon and Bains, 2006). Stress can sensitize the HPA axis and increase the response to a repeated challenge (Figueiredo et al., 2003). How these changes are encoded is not fully understood, but multiple signaling pathways appear to be involved. Early life stress tonically increases corticosterone secretion and epigenetically regulates vasopressin gene expression (Murgatroyd et al., 2009). Immobilization and predator stress, which change HPA activity, induce presynaptic plasticity at the glutamatergic synaptic input that impinges on hypothalamic neurons in this pathway (Kuzmiski et al., 2010).
Stress also appears to produce a lasting change in the sympathetic–adrenal limb of the stress response (Konarska et al., 1989). Repeated hemorrhage, immobilization, and intermittent hypoxia can potentiate catecholamine release (Kvetnansky and Mikulaj, 1970; Lilly et al., 1986; Kuri et al., 2007; Souvannakitti et al., 2009), and multiple stressors, including restraint, cold, glucoprivation, exercise, and social stress (Chuang and Costa, 1974; Mormède et al., 1990; Nankova et al., 1994; Vietor et al., 1996; Levenson and Moore, 1998) alter the adrenal expression or activity of tyrosine hydroxylase (TH). However, within the sympathetic nervous system, the cellular mechanisms that encode the lasting effects of a transient exposure to stress are less clear.
To investigate this issue, we briefly exposed mice to a stressor and then 1 d later analyzed the secretory capacity of the sympathetic nervous system. In these experiments, we used an in vivo paradigm, the cold-water forced swim test (FST). This mixed stressor (Kvetnansky et al., 2009) induces synaptic plasticity in the CNS (Saal et al., 2003; Campioni et al., 2009). Using this approach, we find that a local peptidergic signaling pathway tonically suppresses adrenal catecholamine release but that acute stress overrides this negative feedback loop and leads to an increase in adrenal secretory capacity that lasts far longer than the initiating stimulus. This novel activity-dependent change in adrenal functioning appears to be an adaptive postsynaptic mechanism that selectively boosts sympathetic capacity while avoiding the pathological changes that are associated with tonic changes in circulating catecholamines (Jacobs et al., 1997). Thus, both the sympathetic and HPA limbs of the stress response exhibit plasticity, but different cellular mechanisms are involved.
Materials and Methods
Animals and stress paradigm.
C57BL/6J wild-type mice, neuropeptide Y (NPY) knock-out mice (129S–Npytm1Rpa/J; (Erickson et al., 1996), and NPY(GFP) mice [Npy–hrGFP; (van den Pol et al., 2009)] were purchased from The Jackson Laboratory. Male mice were used in the majority of experiments (female animals were used in some of the experimental groups shown in Fig. 6B,C). Mice (usually P21–P23) were individually housed for 3–5 d and then either exposed to the cold-water FST or left undisturbed. Experimental animals were placed in cold (4–5°C) water for 5–6 min (cold-water FST) as described previously (Saal et al., 2003; Campioni et al., 2009). After recovery under an infrared heat lamp for 15–25 min, the animals were returned to their home cage. Experimental and control animals were killed by decapitation, 1 d or 1 week later, as noted in the text. Littermates were used in all experiments and were matched for sex and weight. Experimental protocols were performed in parallel for each littermate in each individual experiment. In the text, littermates used in each experiment are described as “matched,” and the data points from these paired animals are connected by lines in each figure.
Figure 6.
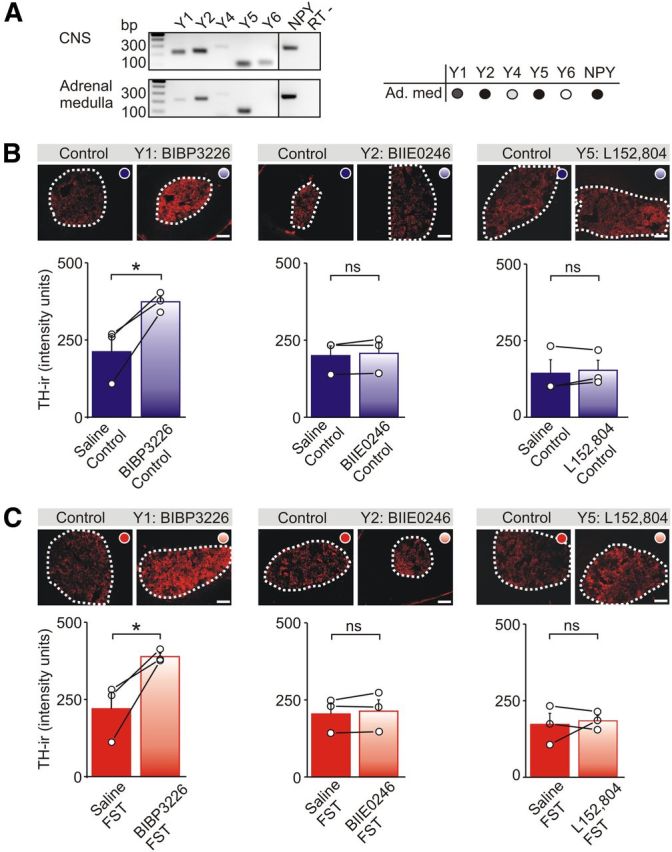
Y1 receptors tonically suppress TH expression in the adrenal medulla. A, RT-PCR of Y receptor expression in the adrenal medulla and CNS (positive control) of wild-type mice. Amplicons indicate the presence of mRNAs encoding Y1, Y2, Y4, and Y5 receptors and NPY in the adrenal medulla (Ad. med; results are tabulated on the right). The negative control (RT−) contained NPY primers but lacked reverse transcriptase. B, TH-IR in adrenal sections from saline-injected control mice or after injection with Y1, Y2, or Y5 antagonists. Group data shows that BIBP 3226 (a Y1 antagonist) significantly increased the basal level of TH-IR. Group data shows mean ± SEM, n = 3 independent experiments (7–10 sections from each animal). C, TH-IR in adrenal sections from mice exposed to the FST and injected with either saline or Y1, Y2, or Y5 antagonists. Group data shows that only BIBP 3226 significantly increased the level of TH-IR. Group data shows mean ± SEM, n = 3 independent experiments (7–10 sections from each animal). Scale bars, 100 μm. *p < 0.05.
Electrophysiology.
One day after the FST, paired control and experimental animals were killed, and adrenal chromaffin cells were isolated as described previously (Whim and Moss, 2001) with minor modifications. In brief, each medulla was isolated from cortex and digested for 15 min in saline (in mm: 138 NaCl, 5.3 KCl, 0.44 KH2PO4, 4 NaHCO3, 0.3 Na2HPO4, 20 HEPES, and 5.5 glucose, pH 7.25, with NaOH) containing 1 mg/ml collagenase type IA and 6 mg/ml BSA at 37°C. Tissue chunks were then incubated for 30 min in saline containing 1 mg/ml trypsin and 6 mg/ml BSA at 37°C, after which the tissue was mechanically dispersed using a fire-polished Pasteur pipette, and cells were plated in culture medium (DMEM/10% FCS) on culture dishes coated with poly-d-lysine. Each animal typically generated ∼8336 ± 1535 chromaffin cells (mean ± SEM, n = 3), and recordings were made within 20 h after isolation. Cells were superfused with extracellular solution (in mm: 135 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 11 glucose, pH 7.3, with NaOH) at 32–34°C. Amperometric and voltage-clamp recordings were made from chromaffin cells (5–9 pF) as described previously (Whim and Moss, 2001; Whim, 2006). Amperometric recordings used carbon fiber electrodes that were insulated using Sylgard and mounted on the head stage of a Multiclamp 700A amplifier (Molecular Devices). Electrodes were held at +700 mV, and fouling was minimized by repetitively switching the holding potential between 700 and −700 mV for ∼2 min after each recording. Sensitivity was determined for each electrode by measuring the change in holding current evoked by the bath application of 50 μm norepinephrine. Control experiments indicated that the mean amplitude of secretory events did not covary with electrode sensitivity (R2 = 0.0161, n = 113). Catecholamine secretion was evoked from single cells using two methods. In the first method, focal application of 100 μm nicotine from a patch pipette placed close to the recorded cell was used. Each application was controlled using a Picospritzer (10 s application repeated 10 times with a 50 s inter-application interval). In the second method, cells were voltage clamped in the whole-cell configuration, and secretion was evoked by a 5 Hz train of 200 depolarizing steps (each from −80 to 0 mV for 20 ms). In these experiments, the patch pipette solution contained the following (in mm): 120 Cs acetate, 15 CsCl, 5 NaCl, 10 HEPES, 4 MgATP, 0.3 NaGTP, and 1 cAMP, pH 7.2, with CsOH. Amperometric and membrane currents were sampled at 5 kHz and filtered at 2 kHz. Amperometric events were analyzed using pClamp 9 (Molecular Devices) using the threshold search method, and stimulation artifacts were removed by subtraction of a blank trace. Only events >10 pA with a rapid rising phase were selected. All electrophysiological studies were performed blind with respect to the experimental treatment until the analysis was complete.
Immunohistochemistry.
Adrenal glands were dissected free of fat, then fixed in 4% paraformaldehyde in PBS overnight at room temperature, and processed as described previously (Wang and Whim, 2013). The glands were washed with PBS, snap frozen in 2-methylbutane on dry ice, embedded in cryomatrix medium, and kept at −80°C until use. Frozen sections (30 μm) were washed with PBS, refixed with 4% paraformaldehyde for 20 min, and then permeabilized in 0.3% Triton X-100 in PBS for 15 min. Sections were subsequently incubated in 3% H2O2 for 45–60 min to quench endogenous peroxidase activity and then transferred to blocking reagent (in Tris-buffered saline; PerkinElmer Life and Analytical Sciences) for 30 min. Sections were incubated in primary antibody [rabbit anti-NPY (1:10,000; T-4070; Peninsula Labs); sheep anti-TH (1:100; AB1542; Millipore); rabbit anti-TH (1:100; AB152; Millipore); and rabbit anti-c-Fos (1:1000; PC38; Calbiochem)] at 4°C overnight. Sections were washed with PBS containing 0.05% Tween 20 and incubated with a secondary antibody conjugated to peroxidase (1:500 donkey anti-rabbit or donkey anti-sheep; Jackson ImmunoResearch) for 30 min at room temperature, washed with PBS containing 0.05% Tween 20, incubated in tyramide signal amplification (TSA)–FITC or TSA–Cy3 (PerkinElmer Life and Analytical Sciences) for 10 min, washed with PBS containing 0.05% Tween 20, rinsed with distilled water, and finally mounted in Vectashield.
In vitro cell culture and immunocytochemistry.
Chromaffin cell cultures were prepared from P24–P26 mice. Cells were fixed 2 h after plating and then stained primarily as described previously (Whim, 2006). Primary antibodies included those noted above (rabbit anti-NPY, 1:40,000; sheep anti-TH, 1:200; rabbit anti-TH, 1:200) plus guinea pig anti-phenylethanolamine N-methyltransferase (PNMT) (1:100; EUD7001; Acris). Secondary antibodies included donkey anti-rabbit Alexa Fluor 488 (1:200; Invitrogen), donkey anti-sheep Dylight 549, donkey anti-rabbit Dylight 549, and donkey anti-guinea pig DyLight 549 (all 1:100; Jackson ImmunoResearch).
Image analysis.
Images were obtained using a Nikon TE2000U microscope, 10× and 60× oil-immersion (1.4 numerical aperture) objectives, and a Retiga 1300 monochrome camera. Image-Pro Plus 5.1 (Media Cybernetics), Origin Pro7, Excel, and Minitab 15 were used for data analyses.
For quantification of immunoreactivity (IR) in tissue sections, an area of interest (AOI; the entire medulla minus any cell-free areas) was selected, and the mean pixel intensity was calculated. Images were then background subtracted using AOIs taken from the adrenal cortex, and 7–10 sections from each animal were measured. In each figure, open circles indicate the mean value from each animal, and black lines link the littermate control and stressed animals (“matched”) from each independent experiment.
When analyzing cells in vitro, images of single chromaffin cells were taken at the level of the nucleus to maintain a consistent plane of focus between experiments. The mean pixel intensity was calculated as above, but background subtraction was not used. In all staining experiments, slides were blinded until the analysis was complete.
Injection with Y receptor antagonists.
Mice received an intraperitoneal injection of BIBP 3226 (1 mg/kg), BIIE 0246 (1 mg/kg), or L152,804 (10 mg/kg) 15 min before the cold FST. These concentrations are expected to selectively block Y1, Y2, and Y5 receptors (Lundberg and Modin, 1995; Doods et al., 1996; Pedrazzini et al., 1998; Dumont et al., 2000; Kanatani et al., 2000; Malmström, 2001; Smith-White et al., 2001; Sorensen et al., 2012; Wang and Whim, 2013). Control animals were injected with the same volume (200 μl) of sterile saline.
Measurement of corticosterone and catecholamine levels.
To quantify plasma corticosterone levels, 500 μl of blood was collected from the body cavity of decapitated animals and centrifuged at 1000 × g at 4°C for 15 min. Plasma was collected in 1 mm EDTA-containing tubes and kept at −80°C until use. Corticosterone levels were measured using a commercial ELISA kit (Assay Designs). Urine (15–300 μl) was collected from live animals (by handling or brief housing in a Parafilm-lined cage), and catecholamine levels were quantified using commercial ELISA kits (RM Diagnostics and Abnova). Catecholamine measurements from four pairs of animals with anomalously high basal or stress-evoked levels of catecholamines were excluded (>7 SDs from the adjusted mean). All corticosterone and catecholamine samples were measured in duplicate. In later experiments, epinephrine was normalized to the levels of urine creatinine, measured using the Jaffe reaction (Cayman Chemicals).
RT-PCR.
mRNA was extracted from the adrenal medulla, whole brain, or pancreatic islets using TRIzol (Invitrogen) following the instructions of the manufacturer. Samples were treated with DNase as described previously (Ramamoorthy and Whim, 2008) and then purified using an RNeasy kit (Qiagen). cDNA from adrenal or whole brain was obtained by RT of 20 ng of mRNA and used in PCR reactions with primers for NPY Y1, Y2, Y5, and Y6 receptors, whereas 60 ng of mRNA was used with Y4 primers. Single-cell RT-PCR was performed essentially as described previously (Whim, 2011) except a touchdown protocol was used. Primers and PCR protocols were as follows: NPY1 (216 bp), 5′-CGGCGTTCAAGGACAAGTAT-3′ and 5′-TGATTCGCTTGGTCTCACTG-3′ [annealing melting temperature (Tm) of 55°C, 35 cycles]; NPY2 (233 bp), 5′-TGCCAATCTGGTTAGGGAAG-3′ and 5′-GGTGCCAACTCCTTGTTCTG-3′ (annealing Tm of 55°C, 35 cycles); NPY4 (293 bp), 5′-TTGCAGTTCTCTGGCTACCCCTG-3′ and 5′-CTTGCTACCCATCCTCATCGA-3′ (annealing Tm of 56°C, 35 cycles); NPY5 (111 bp), 5′-CAGATTAATCCAGCTGTTCTGC-3′ and 5′-GAAAACAGCCTTTATTTGACAATG-3′ (annealing Tm of 55°C, 35 cycles); NPY6 (126 bp), 5′-TCACTAAATAAGACCATCGGGTAG-3′ and 5′-GGGAGGTTTACCCTAGGAAATG-3′ (annealing Tm of 55°C, 10 cycles; 53°C, 30 cycles); and NPY (288 bp), 5′-GCTAGGTAACAAGCGAATGGGG-3′ and 5′-CACATGGAAGGGTCTTCAAGC-3′ (annealing Tm of 55°C, 35 cycles). Y2, Y5, and Y6 primers were described previously (Klenke et al., 2010).
For single-cell touchdown RT-PCR, the primers were NPY (see above) and TH: 5′-CCCCACCTGGAGTACTTTGTGC-3′ and 5′-TGTGCACTGAAACACACGGAAG (558 bp). In single-cell experiments, the starting annealing Tm (60°C) was decreased to 50°C by 1°C per cycle and then an additional 40 cycles at Tm of 50°C was used. In Figure 8A, cDNA was obtained by RT of 3 ng of mRNA from pancreatic islets and 7 ng of mRNA from adrenal medulla. Primers and PCR protocols were as follows: insulin (241 bp), 5′-TTTGTCAAGCAGCACCTTTG-3′ and 5′-GCTGGTAGAGGGAGCAGATG-3′; pancreatic polypeptide (PP) (196 bp), 5′-CACGATGCTAGGTAACAAG-3′ and 5′-CACATGGAAGGGTCTTCAAG-3′; and peptide YY (PYY) (260 bp), 5′-CACGATGCTAGGTAACAAG-3′ and 5′-CACATGGAAGGGTCTTCAAG-3′ (annealing Tm of 55°C, 35 cycles).
Figure 8.
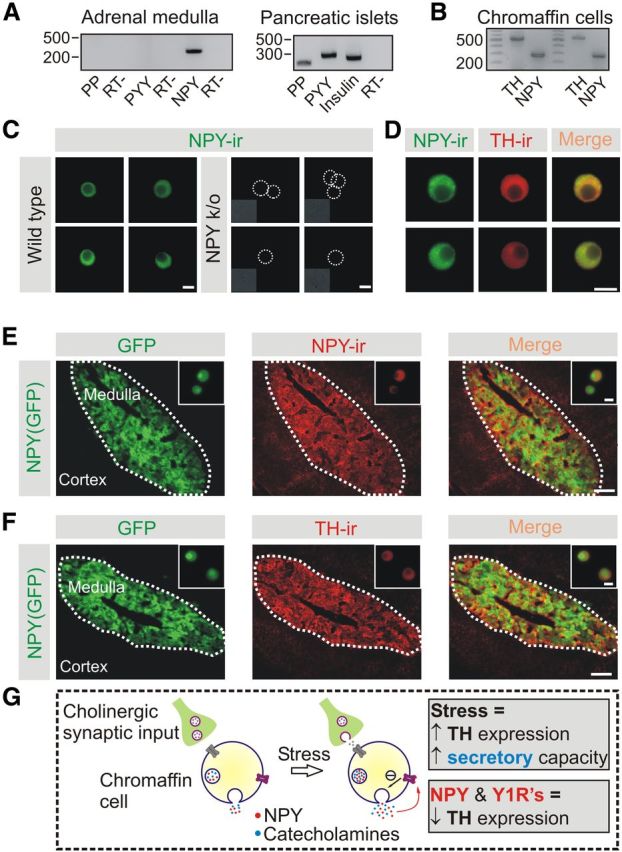
Adrenal chromaffin cells from wild-type and NPY(GFP) transgenic mice synthesize NPY and TH. A, RT-PCR shows the presence of NPY mRNA in the adrenal medulla, whereas PP and PYY are absent (left). Pancreatic islets were used as a positive control for PP and PYY expression. The negative control (RT−) contained insulin primers but lacked reverse transcriptase (right). B, Single-cell RT-PCR confirmed that individual chromaffin cells contain NPY and TH mRNAs. C, Examples of NPY-IR in chromaffin cells from wild-type mice (left). No NPY-IR is observed in chromaffin cells from NPY knock-out mice (right), confirming that the staining is specific. Insets are bright-field images. Scale bars, 5 μm. D, Cells from the adrenal medulla of wild-type mice are NPY and TH immunoreactive, confirming that NPY is expressed in the catecholaminergic chromaffin cells. Scale bars, 10 μm. E, In adrenal sections from NPY(GFP) BAC mice the GFP signal overlaps with NPY-IR. Thus, GFP is located in the chromaffin cells. Insets, Chromaffin cells in vitro from NPY(GFP) mice. Scale bars: 100 μm; inset, 10 μm. F, Colocalization of TH-IR and GFP fluorescence in adrenal sections from NPY(GFP) mice. Insets, Chromaffin cells in vitro. Thus, the GFP-expressing cells are catecholaminergic chromaffin cells. Scale bars: 100 μm; inset, 10 μm. G, Working model of the experimental results. After the FST, adrenal expression of TH and catecholamine secretory capacity is increased (shown as a change in the concentration of catecholamines in each dense core granule). For simplicity, the catecholamines and NPY are shown as colocalized [some granules are thought to contain both transmitters (Whim, 2006)]. NPY secretion activates Y1 receptors mediating an autocrine tonic inhibitory influence on TH expression. In the mouse adrenal gland, Y1 receptors are postsynaptic (Cavadas et al., 2006). Although NPY release inhibits TH expression, the net effect of acute stress is an increase in TH expression and an increase in the catecholamine secretory capacity. This suggests that the role of adrenal NPY over the medium to long term is to limit a stress-induced increase in adrenal output.
Data analyses.
Statistical significance was assessed using the paired Student's t test (raw data; immunohistochemistry and ELISA). Comparisons between three or more groups were made with a general linear model ANOVA (post hoc Tukey's paired comparison). Population distributions were fit using OriginPro 7. The Kolmogorov–Smirnov test was used for analyzing cumulative fraction datasets. Because the absolute level of staining varied between individual experiments, comparisons were made between paired control and experimental groups (which were processed in parallel and generated from littermates in each independent experiment).
Results
An acute stressor leads to a long-lasting increase in adrenal secretory capacity
Mice were exposed to the cold-water FST for 5 min, a mixed stressor (Kvetnansky et al., 2009) with cold, psychological and exercise components, all of which are expected to lead to activation of the sympathetic–adrenal system (Guidotti and Costa, 1974; Ulus and Wurtman, 1979; Jefferys and Funder, 1987). This paradigm has also been shown to lead to synaptic plasticity within the CNS (Saal et al., 2003; Campioni et al., 2009). To confirm that the FST activated adrenal chromaffin cells, mice were killed 1 h after exposure, and adrenal sections were stained for c-Fos, a marker of cellular activity. The number of c-Fos-immunoreactive cells in the adrenal medulla was higher in stressed versus control animals (Fig. 1A,B). Consistent with the activation of a stress response, the plasma levels of corticosterone and the urinary levels of the catecholamines were significantly elevated when sampled 30 min to 2 h after the FST (Fig. 1C–E). Circulating epinephrine is primarily derived from chromaffin cells in the adrenal medulla, whereas most norepinephrine is released from postganglionic sympathetic neurons (Kvetnansky et al., 1979).
Figure 1.

The cold FST activates the HPA axis and the adrenal stress response. A, Example of adrenal sections stained for c-Fos; insets are the regions outlined by the white squares. B, Group data shows that stress increased the number of c-Fos-immunoreactive cells in the medulla (mean ± SEM, n = 3) measured 1 h after the FST. Seven to 10 sections were quantified from each animal using multiple AOIs (white circles in A). Open circles are the mean value from each animal, and black lines link matched control and FST animals from each independent experiment. C, Plasma corticosterone levels were elevated when measured 30 min after the cold FST. D, E, Urine levels of norepinephrine (D) and epinephrine (E) were significantly higher from animals exposed to the cold FST compared with matched controls. Urine samples were taken 30–120 min after the cold FST. Open circles indicate the mean value from each animal, and black lines link the matched control and stressed animals from each independent experiment. *p < 0.05, **p < 0.01.
To determine whether acute stress had any lasting effects on the function of the adrenal medulla, we next measured the secretion of catecholamines from single chromaffin cells isolated from control and experimental animals 1 d after the FST. Catecholamine secretion was evoked by the application of 100 μm nicotine and quantified using amperometry (Fig. 2A). The amperometric events were larger from stressed versus control animals (Fig. 2B), and the amplitude distribution of amperometric events was shifted significantly to the right after stress (Fig. 2C). In contrast, there was no difference in the total number of amperometric events (1554 ± 246 and 1954 ± 59, control vs FST, mean ± SEM, n = 3 paired experiments, 22 cells per treatment).
Figure 2.

Acute stress leads to a lasting increase in the adrenal secretory capacity. A, Amperometric events evoked by 100 μm nicotine from typical chromaffin cells from control and stressed animals. Mice were killed 1d after exposure to the FST. B, Average amperometric events from a representative pair of control and experimental cells (control, 170 events; FST, 238 events). C, Cumulative amplitude distribution of amperometric events from control and stressed mice (control, 4662 events; FST, 5861 events; n = 22 cells per condition). **p < 0.01, Kolmogorov–Smirnov test.
Because chronic cold stress can alter nAChR expression in chromaffin cells (Colomer et al., 2010), we tested whether the potentiating effect of the FST was still present when catecholamine release was evoked by a method that bypassed nAChR activation. Chromaffin cells were voltage clamped, and secretion was elicited by a train of depolarizations (Fig. 3A,B). Significantly higher levels of release were found from cells from stressed animals as shown by the rightward shift in the cumulative amplitude distribution (Fig. 3C). A plot of the cube root of the amperometric charge (Q1/3) from both control and experimental animals was skewed and best described with the sum of two Gaussian distributions (Fig. 3D). This is consistent with the presence of two populations of dense core granules in mouse chromaffin cells, in agreement with previous studies (Grabner et al., 2005). Subtracting the amperometric charge curves confirmed that the distribution from the stressed animals was right-shifted compared with the controls, as expected if stress did indeed lead to an increase in secretion (Fig. 3D, FST - Control). In contrast, there was no significant difference in the total number of amperometric events or the amplitude of the depolarization-evoked calcium current between control and stressed animals (Fig. 3E,F). Because an increase in secretion could be attributable to a change in the probability of dense core granule fusion, we assessed release probability in two ways. In the first, the normalized cumulative release curves between stressed and control animals during the train of 200 depolarizations were compared. The two curves were very similar (Fig. 3G), and the near-linear time versus secretion relationship implies that, once release was triggered, it proceeded at a constant rate with no significant depletion. The only distinctive feature we observed was a slight increase in release rate during the first 10 depolarizations that was seen in the FST cells (Fig. 3G). In the second, we examined whether stress altered the temporal coupling between each depolarization and release. However, the cumulative release curves were very similar in cells from control and stressed mice (Fig. 3H). By plotting the fractional release during each sweep, it was evident that the timing of release was also not different in cells from control and stressed animals. The release rate was uniformly low at −80 mV but rose during the depolarizing step and decayed with an identical time course (Fig. 3I).
Figure 3.

Stress-induced plasticity is mediated postsynaptically at a site that is downstream from nAChR activation. A, Amperometric events evoked by a 5 Hz train of 200 voltage-clamp depolarizations from chromaffin cells from control and stressed mice. B, Excerpts from the regions indicated by the gray bars in A, showing the response to five depolarizing steps. Top trace, Amperometric electrode. Bottom trace, ICa. C, Cumulative amplitude distribution of amperometric events from control and stressed animals (control, 1151 events; FST, 1696 events; n = 17 cells per condition). D, Cube root of event charge (from data in C) fitted with two Gaussian distributions (solid lines). The relative residual distribution is the difference between the FST and control histograms. E, F, Number of amperometric events (E) and amplitude of voltage-dependent calcium current (F) from control and experimental cells (mean ± SEM, n = 3 paired experiments, 5–6 cells per treatment in each experiment). G, Normalized plot of the number of amperometric secretory events occurring during a train of 200 voltage-clamp depolarizations. The right is an expansion of the cumulative release curve from the first 10 depolarizations and indicates that proportionally higher levels of secretion occur early in the depolarizing train in chromaffin cells from the mice exposed to the FST. Ctrl, Control. H, Time of occurrence of the amperometric release events relative to each depolarizing voltage-clamp step. Each depolarization (indicated by the gray bar) was a 20 ms voltage step from −80 to 0 mV repeated 200 times at 5 Hz. I, Plot of the time of occurrence of the amperometric events indicates that the FST does not change the probability of release (quantified as the fraction of total events occurring during 1 ms time bins; mean ± SD, n = 3 separate experiments, 5–6 cells per treatment per experiment). Data in G–I are from the cells shown in C. J, Urine norepinephrine and epinephrine were not significantly different between control animals and those exposed to the FST when measured 1 d after exposure to stress. K, Urine epinephrine values were significantly elevated in mice exposed twice to the FST on consecutive days compared with a single exposure. Epinephrine was measured during a 60 min window after the final FST. Thus, the FST leads to a long-lasting increase in secretory capacity but does not alter the sympathetic tone. *p < 0.05, **p < 0.01; ns, not significant.
These experiments indicate that acute stress leads to a lasting increase in evoked quantal catecholamine secretion, and the effect is mediated at a point downstream from AChR activation and calcium entry. Because there was no significant change in event number or release probability, this suggests that stress leads to an increase in quantal size rather than quantal content (i.e., each granule contains more catecholamines after stress). However, these changes did not alter circulating catecholamines because, 1 d after the FST, the urinary levels of catecholamines were not different between control and stressed animals (Fig. 3J). Thus, stress does not lead to a lasting change in the basal sympathetic tone. Instead, it selectively alters the sympathetic secretory capacity. We next examined whether the electrophysiological changes were associated with altered adrenal responsiveness in vivo. We found that the levels of urine epinephrine were significantly higher in mice exposed to two episodes of the FST (given on consecutive days) compared with mice exposed to a single episode of stress (FST–FST, 167.5 ± 25.8 vs non-stressed control–FST, 83.5 ± 11.6 ng epinephrine/mg urine creatinine, mean ± SEM, n = 3, p = 0.028, paired t test). This result (Fig. 3K) indicates that acute stress can lead to a long-lasting plasticity in adrenal function in vivo and is consistent with the electrophysiological and biochemical changes (see below) that occur after the FST.
Acute stress increases the activity of the catecholamine–synthetic pathway
Although acute stress evokes the fight-or-flight response and significant catecholamine secretion, the releasable stores of hormone are evidently not depleted. In fact, the rightward shift in the cumulative amperometric amplitude distribution suggests that the stores become “overfilled.” What is the mechanism underlying this effect? TH, the rate limiting enzyme for catecholamine synthesis, is upregulated by multiple stress paradigms (Chuang and Costa, 1974; Mormède et al., 1990; Nankova et al., 1994; Vietor et al., 1996; Levenson and Moore, 1998) and is a possible mediator. We next quantified the levels of TH-IR in adrenal sections from stressed animals and matched controls. As expected, TH-IR was limited to the adrenal medulla (Fig. 4A). One day after exposure to the FST, the levels of TH-IR were significantly elevated in the stressed animals (Fig. 4A). This effect was reversible because, 1 week later, the levels of TH-IR had declined back almost to the control values (Fig. 4A,B).
Figure 4.

Acute stress leads to a lasting increase in the adrenal expression of enzymes in the catecholamine synthetic pathway. A, TH-IR in adrenal sections from a control animal and 1 d and 1 week after exposure to the FST. Scale bar, 100 μm. B, Group data (mean ± SEM, n = 3) shows that the stress-induced increase in TH-IR in the adrenal medulla that was evident 1 d after the FST had declined toward basal levels by 1 week. C, Examples of PNMT-IR in chromaffin cells from a control animal and 1 d after exposure to the FST. Scale bars, 10 μm. D, Frequency histograms of the levels of PNMT-IR in chromaffin cells from matched control and stressed animals (211 cells for each distribution, n = 3 separate experiments). The histograms were each fitted with two Gaussian distributions (green lines). E, Cumulative frequency distributions showing that the FST led to a significant increase in the levels of PNMT-IR (211 cells for each distribution; Kolmogorov–Smirnov test). F, TH-IR in adrenal sections from control (wild-type or NPY knock-out) mice and 1 d after exposure to the FST. Scale bars, 100 μm. G, In wild-type mice, the FST increased the level of TH-IR compared with matched controls. H, The FST significantly increased the level of TH-IR in the NPY knock-out (k/o) animals, but the increase was smaller than that seen in the wild-type animals. *p < 0.05, **p < 0.01.
To determine whether stress regulated the expression of other enzymes in the catecholamine biosynthetic pathway, we also quantified the levels of PNMT, the enzyme required for the conversion of norepinephrine to epinephrine. Because PNMT is expressed in a subset of chromaffin cells (Henion and Landis, 1990), we quantified PNMT-IR in chromaffin cells that were fixed within 2 h after isolation from control and stressed mice (Fig. 4C). The intensity distribution of PNMT-IR was bimodal (Fig. 4D), and stress also led to a significant increase in PNMT-IR (Fig. 4E).
NPY inhibits the expression of adrenal TH
What signaling pathways are responsible for regulating the expression of TH? We reasoned that, because the increase in TH expression was activity dependent, a molecule that was cosecreted with the catecholamines would be an efficient way to couple adrenal activity to secretory capacity. One possibility is NPY, which is synthesized in the adrenal medulla (Henion and Landis, 1990) and can be coreleased with the catecholamines (Whim, 2006). Previous studies have examined whether NPY can regulate TH expression in chromaffin cells, but the results have been contradictory, showing an increase, decrease, or no effect (Erdem et al., 2002; Cavadas et al., 2006; Luke and Hexum, 2008). To investigate this issue in vivo, we exposed NPY knock-out mice to the FST.
In these animals, a stress-induced increase in TH-IR in the adrenal medulla was still present (Fig. 4F,H; 17 ± 8% increase, mean ± SEM, n = 3). However, it was smaller than that induced in pair-matched wild-type animals (Fig. 4F,G; 94 ± 14% increase, mean ± SEM, n = 3).
We noted that the level of TH-IR in the unstressed NPY knock-out animals was elevated compared with the unstressed wild-type animals (compare the left, top, and bottom panels in Fig. 4F). Although a quantitative comparison between wild-type and NPY knock-out animals was not possible because the latter were maintained on a mixed background, it suggested that loss of NPY had removed a tonic inhibitory influence on TH expression. If this was correct, then the effect of stress on secretory capacity should be reduced in the NPY knock-out animals.
We tested this prediction by measuring the evoked release of the catecholamines from control and stressed NPY knock-out animals (Fig. 5A). The cumulative amplitude distribution of amperometric events was indeed not different in the two conditions (Fig. 5B). Subtraction of the charge distribution from control and stressed animals confirmed that the stress effect was not present in the NPY knock-out mice (Fig. 5C). There was also no difference in the amperometric event amplitude (Fig. 5D), the number of events, or the amplitude of the depolarization-evoked calcium current between cells from control and stressed NPY knock-out mice (Fig. 5E). Finally, we also measured the release probability using the same two approaches described above for the wild-type animals. There was no difference in the normalized cumulative release curves between stressed and control knock-out animals during the train of 200 depolarizations (Fig. 5F). Curiously, the change in release rate that was seen during the first 10 depolarizations in the wild-type cells was not present in the control cells that lacked NPY (compare the right panels in Figs. 3G, 5F). There was also no difference in the temporal coupling between each depolarizing step and release between control and stressed NPY knock-out animals (Fig. 5G,H). Thus, the loss of NPY specifically prevented the stress-induced change in secretory capacity. Cumulatively, these results indicate that NPY (1) regulates TH expression and (2) can modulate the long-lasting increase in catecholamine secretion that is induced by acute stress. In the next series of experiments, we examined which Y receptors were involved in mediating this effect.
Figure 5.

Loss of NPY eliminates the stress-induced increase in adrenal catecholamine secretion. A, Amperometric events evoked by a 5 Hz train of 200 voltage-clamp depolarizations from chromaffin cells from NPY knock-out (k/o) control and stressed mice. Right panel are excerpts from the regions indicated by the gray bars showing the response to five depolarizing steps. Top trace, Amperometric electrode. Bottom trace, ICa. B, Cumulative amplitude distribution of amperometric events from control and stressed NPY knock-out animals (control, 2113 events; FST, 2281 events; n = 18 cells per condition). C, Cube root of event charge (from data in B) fit with two Gaussian distributions. D, Average amperometric events from a representative pair of control and experimental cells (control, 43 events; FST, 144 events). E, Secretory event number and amplitude of voltage-dependent calcium current from control and experimental cells (mean ± SEM, n = 3 paired experiments, 6 cells per treatment in each experiment). F, Normalized plot of the number of amperometric secretory events occurring during a train of 200 voltage-clamp depolarizations. The right panel is an expansion of the cumulative release curve from the first 10 depolarizations. Compare this with the equivalent region of the cumulative release curve from wild-type animals (Fig. 3G). Cntl, Control. G, Time of occurrence of the amperometric release events relative to each depolarizing voltage-clamp step. Each depolarization (indicated by the gray bar) was a 20 ms voltage step from −80 to 0 mV repeated 200 times at 5 Hz. H, Plot of the time of occurrence of the amperometric events indicates that the FST does not change the probability of release in mice lacking NPY (quantified as the fraction of total events that occur during 1 ms time bins, mean ± SD, n = 3 separate experiments, 6 cells per treatment per experiment). Data in F–H are from cells shown in B. ns, Not significant.
Multiple subtypes of NPY receptors are expressed in the adrenal medulla, but only Y1 receptors regulate the expression of TH
Of the five cloned Y receptors, RT-PCR indicated the presence of Y1, Y2, Y4, and Y5 mRNAs in the adrenal medulla (Fig. 6A). Y6 was not detected, although it was present in CNS, which was used as a positive control (Fig. 6A). The Y4 receptor is preferentially activated by pancreatic peptide, whereas Y1, Y2, and Y5 can be activated by NPY and a related peptide, PYY (Michel et al., 1998).
To test which Y receptors were involved in regulating the expression of TH in vivo and to avoid possible developmental compensation that may have occurred in the NPY knock-out mice, antagonists of Y1, Y2, and Y5 receptors were injected intraperitoneally 15 min before the FST and animals were killed 1 d later. Control animals received vehicle or antagonist. These experiments showed that BIBP 3226, a selective antagonist of the Y1 receptor that is not thought to cross the blood–brain barrier (Doods et al., 1996), had two effects. First, the levels of TH-IR were significantly higher in the BIBP 3226-injected animals compared with those injected with saline (Fig. 6B). This suggests that NPY tonically inhibits TH expression. It is also consistent with the phenotype observed in Y1 knock-out mice (Cavadas et al., 2006) and with the high level of TH expression in the NPY knock-out animals (Fig. 4F–H). Second, in the stressed animals, the levels of TH-IR were higher in the BIBP 3226-injected animals (Fig. 6C). Because Y1-IR has been localized to chromaffin cells (Matsuda et al., 2002), the receptors that mediate the inhibition of TH are likely to be those expressed by chromaffin cells.
The involvement of additional Y receptors was investigated by injecting BIIE 0246 and L152,804, antagonists of the Y2 and Y5 receptors, respectively (Doods et al., 1999; Kanatani et al., 2000). In contrast to BIBP 3226, these blockers did not alter the basal levels of TH-IR (Fig. 6B) or the levels in the stressed animals (Fig. 6C). This indicates that NPY selectively inhibits the basal expression of TH via Y1 receptors and that Y2 and Y5 receptors are not involved. The lack of an effect of the Y2 and Y5 blockers was probably not attributable to a subthreshold dose of antagonist because we find that the same BIIE 0246 treatment elevates adrenal NPY expression, implicating a Y2-mediated regulation of peptide expression (Wang and Whim, 2013), and L152,804 at the concentration used can prevent a Y5-dependent modulation of behavioral hyperactivity (Sorensen et al., 2012). We could not determine whether Y receptors also regulated the stress-induced change in TH because the FST did not increase TH-IR in the saline-injected animals (compare raw control and FST values in Fig. 6B,C). This is probably because injection per se is stressful and can alter TH expression as shown previously (Okimoto et al., 2002). We next tested whether the BIBP 3226-induced increase in TH expression was associated with a change in catecholamine release in vivo. There was no significant difference in urine epinephrine levels after the FST in mice that had previously been injected with saline or BIBP 3226 (saline–FST, 118.2 ± 39.8 vs BIBP 3226–FST, 87.4 ± 12.8 ng epinephrine/mg urine creatinine measured during a 2 h window after the FST, mean ± SEM, n = 3, p = 0.59, paired t test). However, these levels were elevated compared with their respective controls, which were measured from the same animals 3 d after the FST (10.8 ± 2.6 vs 9.6 ± 1.5, mean ± SEM, n = 3). This could indicate that the stress-induced change in adrenal responsiveness in vivo is TH independent or that Y1 receptors regulate multiple components of the stress response. However, amperometry experiments show there is a change in catecholamine secretory capacity after BIBP 3226 injection (see below), which suggests that the difference may be attributable to the intrinsic sensitivities of the two types of techniques (which is likely higher for electrophysiology).
The data so far indicated that blocking Y1 receptors with BIBP 3226 injection increased TH expression and genetic loss of NPY eliminated the FST-induced increase in catecholamine secretory capacity. If a common mechanism underlies these effects, treatment with BIBP 3226 should prevent the ability of the FST to alter catecholamine secretory capacity (i.e., should mimic the NPY knock-out phenotype). To test this idea, we quantified the depolarization-evoked amperometric events in control and stressed mice after BIBP 3226 injection (Fig. 7A). The cumulative amplitude distribution of amperometric events was not different in the two conditions (Fig. 7B). There was also no difference in the total number of amperometric events or the size of the depolarization-evoked calcium current after the FST (Fig. 7C). These results mimic the effects seen in the NPY knock-outs.
Figure 7.

Y1 receptors regulate the stress-induced change in adrenal secretory capacity. A, Example of amperometric events evoked by a 5 Hz train of 200 voltage-clamp depolarizations from chromaffin cells from BIBP 3226-injected control and stressed mice. B, Cumulative amplitude distribution of amperometric events from control (Ctrl) and stressed BIBP 3226-injected wild-type animals (control, 1867 events; FST, 2223 events; n = 20 cells per condition). C, Secretory event number and amplitude of voltage-dependent calcium current from BIBP 3226-injected control and experimental animals (mean ± SEM, n = 3 paired experiments, 6–7 cells per treatment in each experiment). D, The same protocol as in A was used to evoke amperometric events from chromaffin cells from saline-injected control and stressed animals. E, Cumulative amplitude distribution of amperometric events shows a significant rightward shift from saline-injected, stressed animals (control, 1809 events; FST, 2040 events; n = 20 cells per condition). *p < 0.01. F, Secretory event number and amplitude of voltage-dependent calcium current from saline-injected control and experimental animals (mean ± SEM, n = 3 paired experiments, 6–7 cells per treatment in each experiment).
Although the effect of the FST was lost in the BIBP 3226-injected mice, we considered the possibility that this was attributable to the stress of the intraperitoneal injection occluding the effect of the FST (rather than an effect of BIBP 3226). However, we found that the FST still led to a significant increase in secretory capacity when we compared control mice that had been injected with saline versus littermates that were injected with saline and then exposed to the FST (Fig. 7D,E). There was no difference in the total number of events or the amplitude of depolarization-evoked calcium current between control and stressed animals (Fig. 7F). These results are consistent with idea that NPY release and Y1 activation regulate the long-lasting change in the adrenal response to stress.
NPY is widely expressed in the adrenal medulla, but related Y family peptides are absent
What is the source of NPY that regulates the adrenal expression of TH? Because chromaffin cells synthesize NPY (Henion and Landis, 1990), the adrenal itself is a likely candidate. However, it is not clear what proportion of chromaffin cells synthesize NPY and whether the related peptides, PP and PYY, are also present. This is important because PP and PYY can activate the same receptors as NPY (Michel et al., 1998), and some NPY antibodies cross-react with both of these peptides (Whim, 2011).
To address this issue, we used a range of approaches. First, RT-PCR experiments indicated the presence of NPY mRNA in the adrenal medulla, whereas PP and PYY were absent (Fig. 8A, left). Because pancreatic islets contain PP and PYY (Myrsén-Axcrona et al., 1997), these were used as a positive control (Fig. 8A, right). Single-cell RT-PCR was then used to confirm that individual chromaffin cells contained NPY (and TH) mRNA (Fig. 8B). Second, we found that all chromaffin cells in vitro were NPY-immunoreactive (Fig. 8C, left). The staining arose from authentic NPY because it was absent from NPY knock-out mice (Fig. 8C, right). As expected, all of the NPY-immunoreactive cells also costained with an antibody to TH, consistent with the coexpression of NPY and the catecholamines in chromaffin cells (Fig. 8D). Although previous studies have indicated that only a subset of chromaffin cells in rats are NPY immunoreactive (Henion and Landis, 1990), all the TH-immunoreactive cells in these adrenal cultures were stained, suggesting that, in mice, NPY is ubiquitously expressed. In these experiments, we also observed sparsely distributed NPY-immunoreactive fibers with en passsant boutons in the adrenal capsule as described previously (Maubert et al., 1993). Third, to examine the distribution of NPY in situ, we used an NPY(GFP) BAC transgenic mouse. In these animals, there was strong somatic GFP expression in the adrenal medulla but none in the adrenal cortex. The expression of GFP matched the distribution of NPY-IR both in situ and in vitro (Fig. 8E). Finally, adrenal GFP expression in the NPY(GFP) mice colocalized with TH-IR in vitro and in situ (Fig. 8F).
These experiments indicate that authentic NPY is expressed throughout the adrenal medulla and is present in all chromaffin cells (Wang and Whim, 2013). The source of NPY that regulates the expression of TH and the stress-induced, long-term change in catecholamine secretion is therefore likely to involve the adrenal chromaffin cells.
Discussion
Stress elicits a characteristic hormonal and neuronal response involving an activation of the HPA axis and increased sympathetic activity (the fight-or-flight response). Because these pathways are not under conscious control, they are often viewed as simple reflexes. However, the HPA axis can show habituation and sensitization and thus exhibits plasticity (Ma and Lightman, 1998; Figueiredo et al., 2003). Here, we investigated whether the brief exposure to a stressful event can lead to a maintained change in the functioning of the sympathetic nervous system, the other limb of the stress response.
The FST led to a significant increase in urine catecholamine levels, a commonly used measure of sympathetic activity (Gautam et al., 2006; Obst et al., 2006; Mathar et al., 2010). The rise in urine epinephrine and the increase in adrenal c-Fos-IR confirmed that this paradigm evoked a stress response that involved the chromaffin cells in the adrenal medulla. Thus, it seems reasonable to anticipate that the adrenal catecholamine stores must be subsequently replenished. Unexpectedly, however, we found that acute stress did not lead to a simple refilling of the stores but actually produced a sustained increase in the catecholamine secretory capacity measured both in vivo and in vitro. This is a form of long-term plasticity and presumably gives the system an increased ability to respond to a repeated challenge. As noted previously (Kvetnansky et al., 2009), the organism may not know whether a stressor will recur and therefore the elevated secretory capacity that we observed is probably as an adaptive response.
The nature of this adrenal plasticity was selective. A depolarizing train evoked more secretion from chromaffin cells from stressed mice, but there was no change in the number of amperometric events or the release probability. This is consistent with the idea that acute stress led to an increase in the amount of catecholamines packaged into each dense core granule rather than an increase in the number of granules. Although modulation of release capacity can involve a change in the number of vesicles in the releasable pool (Stevens and Sullivan, 1998; Sakaba and Neher, 2001; Zhao and Klein, 2002), previous studies have shown that the amount of transmitter contained within a dense core granule can also be regulated (Colliver et al., 2000; Montesinos et al., 2008). An increase in granular catecholamines could be mediated via a number of mechanisms, including a change in catecholamine synthesis, granule loading, or granule size (Meligeni et al., 1982; Bruns et al., 2000; Tabares et al., 2001). However, the activity-induced increase that we observed in the level of TH is consistent with altered synthesis playing a role.
How acute stress resulting from exposure to the FST leads to an increase in TH expression is not known. Using a variety of approaches, we found that NPY is an adrenal cotransmitter that is synthesized by all chromaffin cells and functions as a negative regulator of TH expression and catecholamine secretion. First, genetic loss of NPY eliminates the ability of acute stress to induce a long-lasting increase in catecholamine release. Second, inhibition of Y1 receptors, possibly those located within the medulla, leads to an increase in TH expression. Although the NPY that mediates these effects is likely to have been released from the neuroendocrine chromaffin cells, the adrenal medulla also contains a population of type I neurons that are NPY-IR (Holgert et al., 1996). Other peripheral sources include sympathetic postganglionic neurons that synthesize this peptide (Lundberg et al., 1983) and contribute to the plasma levels of NPY. It is also possible that central pathways could be involved. For example, NPY-synthesizing neurons in the arcuate nucleus have been shown to regulate sympathetic output via a Y1-mediated inhibition of TH expression in the paraventricular nucleus (Shi et al., 2013).
Curiously, although multiple subtypes of Y receptors are present in the adrenal and all Y subtypes are Gi/o-protein coupled and inhibit cAMP production (Michel et al., 1998), only Y1 receptors are involved in the peptidergic regulation of TH. Because NPY is a suppressor of adrenal signaling, it is theoretically possible that the stress-induced increase in catecholamine secretion and TH expression is actually mediated by an activity-dependent decrease in NPY release. However, it is more likely that the inhibitory effect of NPY is reduced by the actions of an antagonistic signal (presently unknown), leading to an overall facilitation of adrenal secretion and synthesis. Nevertheless, the release of NPY as an autocrine modulator clearly plays a significant role in regulating adrenal function. Although we did not investigate the inducing signal, it probably does not require crosstalk between the HPA axis and the sympathetic nervous system because a restraint stress-induced increase in TH expression is present in corticotrophin-releasing hormone knock-out mice and is not prevented by hypophysectomy (Nankova et al., 1994; Kvetnanský et al., 2008). Although splanchnic nerve transection has shown that synaptic input to the chromaffin cells is also not needed for an increase in TH mRNA levels in response to a single 2 h immobilization (Nankova et al., 1994), the increase in TH expression that is induced in response to a 6 h immobilization or insulin-induced hypoglycemia is substantially reduced in pituitary adenylate cyclase-activating polypeptide (PACAP) knock-out mice (Hamelink et al., 2002; Stroth and Eiden, 2010). Because PACAP is a peptidergic cotransmitter in the preganglionic neurons that innervate chromaffin cells (Hamelink et al., 2002), this suggests that PACAP may also be involved in the induction of the acute stress response (although probably not in its maintenance because this is a postsynaptic effect). Finally, although we did not investigate the underlying molecular mechanism, it is likely to be transcriptionally mediated because actinomycin D prevents the increase in TH activity that is evoked in response both to swimming stress and immobilization (Otten et al., 1973; Nankova et al., 1994). Using an NPY(GFP) reporter mouse line, we find that the FST also leads to an increase in adrenal NPY expression by a mechanism that appears to involve an increase in transcription (Wang and Whim, 2013).
Our results show that acute stress can lead to a long-term change in sympathetic capacity. Thus, both hormonal and neuronal limbs of the stress response exhibit plasticity (Kuzmiski et al., 2010; Wamsteeker and Bains, 2010). Activity-dependent and lasting changes in cellular signaling are characteristic features of the nervous system (Johansen et al., 2011; Lüscher and Malenka, 2011; Spitzer, 2012), but experience-dependent plasticity has also been described in the endocrine system (Hodson et al., 2012). Although the change in hormonal secretory capacity that we observed was triggered by a single brief stress exposure, this effect may be mechanistically related to other forms of adrenal memory. For example, repeated exposure to restraint stress leads to persistently elevated levels of TH (Nankova et al., 1994), and intermittent hypoxic exposure over several days is associated with an increase in catecholamine secretion (Kuri et al., 2007; Souvannakitti et al., 2009, 2010). In the latter case, when the exposure is confined to the neonatal stage, the effects can last well into adulthood (Nanduri et al., 2012).
What might be the functional consequences of the type of sympathetic plasticity we have described? The catecholamine hormones potently control many physiological parameters. A typical response to adrenal epinephrine release includes an increase in the plasma levels of glucose and elevated blood pressure (Bao et al., 2007; Verberne and Sartor, 2010). Thus, increased catecholamine synthesis may be beneficial as a short-term response to stress. However, over longer periods, this modulation could be maladaptive particularly if it leads to a chronic elevation in circulating catecholamines. Our results suggest that NPY plays an inhibitory role in adrenal signaling because disrupting NPY signaling with a Y1 antagonist or genetic removal of NPY lead to increased TH expression and eliminates the stress-induced increase in catecholamine secretory capacity. This might explain the somewhat paradoxical observation that, for stress to increase TH expression, it must override the apparently tonic inhibitory influence of NPY. However an adrenal-specific deletion of NPY will be required to determine whether acute stress alters catecholamine secretion in vivo by antagonizing a local effect of NPY. Our hypothesis is that adrenal NPY is part of an inhibitory feedback loop that prevents a pathological increase in TH levels (and thus epinephrine release) during the aftermath of the fight-or-flight response (Fig. 8G). In this way, it plays an analogous role to the inhibitory feedback actions of cortisol in the HPA axis (Di et al., 2003). Curiously, NPY is also involved in the suppression of the hypothalamic–pituitary–thyroid axis that occurs during the response to starvation (Vella et al., 2011), perhaps indicating a similar inhibitory role in other stress-activated pathways. Because metabolic and cardiovascular disorders are associated with altered sympathetic activity (Jacobs et al., 1997; Campanucci et al., 2010), it will of interest to determine whether these involve the same type of NPY-dependent synaptic plasticity we describe here.
Footnotes
This work was supported by National Institutes of Health Grant RO1DK080441 (M.D.W.). We thank Dr. June Liu for critically reading this manuscript and Drs. Timothy Foster, Ben Kelly, and Ya-Ping Tang for the use of equipment.
References
- Bao X, Lu CM, Liu F, Gu Y, Dalton ND, Zhu BQ, Foster E, Chen J, Karliner JS, Ross J, Jr, Simpson PC, Ziegler MG. Epinephrine is required for normal cardiovascular responses to stress in the phenylethanolamine N-methyltransferase knockout mouse. Circulation. 2007;116:1024–1031. doi: 10.1161/CIRCULATIONAHA.107.696005. [DOI] [PubMed] [Google Scholar]
- Bruns D, Riedel D, Klingauf J, Jahn R. Quantal release of serotonin. Neuron. 2000;28:205–220. doi: 10.1016/S0896-6273(00)00097-0. [DOI] [PubMed] [Google Scholar]
- Campanucci V, Krishnaswamy A, Cooper E. Diabetes depresses synaptic transmission in sympathetic ganglia by inactivating nAChRs through a conserved intracellular cysteine residue. Neuron. 2010;66:827–834. doi: 10.1016/j.neuron.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Campioni MR, Xu M, McGehee DS. Stress-induced changes in nucleus accumbens glutamate synaptic plasticity. J Neurophysiol. 2009;101:3192–3198. doi: 10.1152/jn.91111.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavadas C, Céfai D, Rosmaninho-Salgado J, Vieira-Coelho MA, Moura E, Busso N, Pedrazzini T, Grand D, Rotman S, Waeber B, Aubert JF, Grouzmann E. Deletion of the neuropeptide Y (NPY) Y1 receptor gene reveals a regulatory role of NPY on catecholamine synthesis and secretion. Proc Natl Acad Sci U S A. 2006;103:10497–10502. doi: 10.1073/pnas.0600913103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherrington AD, Fuchs H, Stevenson RW, Williams PE, Alberti KG, Steiner KE. Effect of epinephrine on glycogenolysis and gluconeogenesis in conscious overnight-fasted dogs. Am J Physiol. 1984;247:E137–E144. doi: 10.1152/ajpendo.1984.247.2.E137. [DOI] [PubMed] [Google Scholar]
- Chuang DM, Costa E. Biosynthesis of tyrosine hydroxylase in rat adrenal medulla after exposure to cold. Proc Natl Acad Sci U S A. 1974;71:4570–4574. doi: 10.1073/pnas.71.11.4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colliver TL, Pyott SJ, Achalabun M, Ewing AG. VMAT-mediated changes in quantal size and vesicular volume. J Neurosci. 2000;20:5276–5282. doi: 10.1523/JNEUROSCI.20-14-05276.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colomer C, Olivos-Orée LA, Vincent A, McIntosh JM, Artalejo AR, Guérineau NC. Functional characterization of α9-containing cholinergic nicotinic receptors in the rat adrenal medulla: implication in stress-induced functional plasticity. J Neurosci. 2010;30:6732–6742. doi: 10.1523/JNEUROSCI.4997-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Halmos KC, Tasker JG. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci. 2003;23:4850–4857. doi: 10.1523/JNEUROSCI.23-12-04850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doods HN, Wieland HA, Engel W, Eberlein W, Willim KD, Entzeroth M, Wienen W, Rudolf K. BIBP 3226, the first selective neuropeptide Y1 receptor antagonist: a review of its pharmacological properties. Regul Pept. 1996;65:71–77. doi: 10.1016/0167-0115(96)00074-2. [DOI] [PubMed] [Google Scholar]
- Doods H, Gaida W, Wieland HA, Dollinger H, Schnorrenberg G, Esser F, Engel W, Eberlein W, Rudolf K. BIIE0246: a selective and high affinity neuropeptide Y Y(2) receptor antagonist. Eur J Pharmacol. 1999;384:R3–R5. doi: 10.1016/S0014-2999(99)00650-0. [DOI] [PubMed] [Google Scholar]
- Dumont Y, Cadieux A, Doods H, Pheng LH, Abounader R, Hamel E, Jacques D, Regoli D, Quirion R. BIIE0246, a potent and highly selective non-peptide neuropeptide Y Y(2) receptor antagonist. Br J Pharmacol. 2000;129:1075–1088. doi: 10.1038/sj.bjp.0703162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdem SR, Broxson CS, Erdem A, Spar DS, Williams RT, Tümer N. The age-related discrepancy in the effect of neuropeptide Y on select catecholamine biosynthetic enzymes in the adrenal medulla and hypothalamus in rats. Neuropharmacology. 2002;43:1280–1288. doi: 10.1016/S0028-3908(02)00293-9. [DOI] [PubMed] [Google Scholar]
- Erickson JC, Clegg KE, Palmiter RD. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature. 1996;381:415–421. doi: 10.1038/381415a0. [DOI] [PubMed] [Google Scholar]
- Figueiredo HF, Bodie BL, Tauchi M, Dolgas CM, Herman JP. Stress integration after acute and chronic predator stress: differential activation of central stress circuitry and sensitization of the hypothalamo-pituitary-adrenocortical axis. Endocrinology. 2003;144:5249–5258. doi: 10.1210/en.2003-0713. [DOI] [PubMed] [Google Scholar]
- Gautam D, Gavrilova O, Jeon J, Pack S, Jou W, Cui Y, Li JH, Wess J. Beneficial metabolic effects of M3 muscarinic acetylcholine receptor deficiency. Cell Metab. 2006;4:363–375. doi: 10.1016/j.cmet.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Gordon GR, Bains JS. Can homeostatic circuits learn and remember? J Physiol. 2006;576:341–347. doi: 10.1113/jphysiol.2006.110270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabner CP, Price SD, Lysakowski A, Fox AP. Mouse chromaffin cells have two populations of dense core vesicles. J Neurophysiol. 2005;94:2093–2104. doi: 10.1152/jn.00316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti A, Costa E. Association between increase in cyclic AMP and subsequent induction of tyrosine hydroxylase in rat adrenal medulla. Experiments with swimming stress. Naunyn Schmiedebergs Arch Pharmacol. 1974;282:217–221. doi: 10.1007/BF00499036. [DOI] [PubMed] [Google Scholar]
- Hamelink C, Tjurmina O, Damadzic R, Young WS, Weihe E, Lee HW, Eiden LE. Pituitary adenylate cyclase-activating polypeptide is a sympathoadrenal neurotransmitter involved in catecholamine regulation and glucohomeostasis. Proc Natl Acad Sci U S A. 2002;99:461–466. doi: 10.1073/pnas.012608999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henion PD, Landis SC. Asynchronous appearance and topographic segregation of neuropeptide-containing cells in the developing rat adrenal medulla. J Neurosci. 1990;10:2886–2896. doi: 10.1523/JNEUROSCI.10-09-02886.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodson DJ, Schaeffer M, Romanò N, Fontanaud P, Lafont C, Birkenstock J, Molino F, Christian H, Lockey J, Carmignac D, Fernandez-Fuente M, Le Tissier P, Mollard P. Existence of long-lasting experience-dependent plasticity in endocrine cell networks. Nat Commun. 2012;3:605. doi: 10.1038/ncomms1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holgert H, Dagerlind A, Hökfelt T. Phenotype of intraadrenal ganglion neurons during postnatal development in rat. J Comp Neurol. 1996;371:603–620. doi: 10.1002/(SICI)1096-9861(19960805)371:4<603::AID-CNE9>3.3.CO%3B2-S. [DOI] [PubMed] [Google Scholar]
- Jacobs MC, Lenders JW, Willemsen JJ, Thien T. Adrenomedullary secretion of epinephrine is increased in mild essential hypertension. Hypertension. 1997;29:1303–1308. doi: 10.1161/01.HYP.29.6.1303. [DOI] [PubMed] [Google Scholar]
- Jefferys D, Funder JW. Glucocorticoids, adrenal medullary opioids, and the retention of a behavioral response after stress. Endocrinology. 1987;121:1006–1009. doi: 10.1210/endo-121-3-1006. [DOI] [PubMed] [Google Scholar]
- Johansen JP, Cain CK, Ostroff LE, LeDoux JE. Molecular mechanisms of fear learning and memory. Cell. 2011;147:509–524. doi: 10.1016/j.cell.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanatani A, Ishihara A, Iwaasa H, Nakamura K, Okamoto O, Hidaka M, Ito J, Fukuroda T, MacNeil DJ, Van der Ploeg LH, Ishii Y, Okabe T, Fukami T, Ihara M. L-152,804: orally active and selective neuropeptide Y Y5 receptor antagonist. Biochem Biophys Res Commun. 2000;272:169–173. doi: 10.1006/bbrc.2000.2696. [DOI] [PubMed] [Google Scholar]
- Klenke U, Constantin S, Wray S. Neuropeptide Y directly inhibits neuronal activity in a subpopulation of gonadotropin-releasing hormone-1 neurons via Y1 receptors. Endocrinology. 2010;151:2736–2746. doi: 10.1210/en.2009-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarska M, Stewart RE, McCarty R. Sensitization of sympathetic-adrenal medullary responses to a novel stressor in chronically stressed laboratory rats. Physiol Behav. 1989;46:129–135. doi: 10.1016/0031-9384(89)90245-X. [DOI] [PubMed] [Google Scholar]
- Kuri BA, Khan SA, Chan SA, Prabhakar NR, Smith CB. Increased secretory capacity of mouse adrenal chromaffin cells by chronic intermittent hypoxia: involvement of protein kinase C. J Physiol. 2007;584:313–319. doi: 10.1113/jphysiol.2007.140624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmiski JB, Marty V, Baimoukhametova DV, Bains JS. Stress-induced priming of glutamate synapses unmasks associative short-term plasticity. Nat Neurosci. 2010;13:1257–1264. doi: 10.1038/nn.2629. [DOI] [PubMed] [Google Scholar]
- Kvetnansky R, Mikulaj L. Adrenal and urinary catecholamines in rats during adaptation to repeated immobilization stress. Endocrinology. 1970;87:738–743. doi: 10.1210/endo-87-4-738. [DOI] [PubMed] [Google Scholar]
- Kvetnansky R, Weise VK, Thoa NB, Kopin IJ. Effects of chronic guanethidine treatment and adrenal medullectomy on plasma levels of catecholamines and corticosterone in forcibly immobilized rats. J Pharmacol Exp Ther. 1979;209:287–291. [PubMed] [Google Scholar]
- Kvetnanský R, Krizanova O, Tillinger A, Sabban EL, Thomas SA, Kubovcakova L. Regulation of gene expression of catecholamine biosynthetic enzymes in dopamine-beta-hydroxylase- and CRH-knockout mice exposed to stress. Ann N Y Acad Sci. 2008;1148:257–268. doi: 10.1196/annals.1410.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvetnansky R, Sabban EL, Palkovits M. Catecholaminergic systems in stress: structural and molecular genetic approaches. Physiol Rev. 2009;89:535–606. doi: 10.1152/physrev.00042.2006. [DOI] [PubMed] [Google Scholar]
- Levenson CW, Moore JB. Response of rat adrenal neuropeptide Y and tyrosine hydroxylase mRNA to acute stress is enhanced by long-term voluntary exercise. Neurosci Lett. 1998;242:177–179. doi: 10.1016/S0304-3940(98)00064-0. [DOI] [PubMed] [Google Scholar]
- Lilly MP, Engeland WC, Gann DS. Adrenal medullary responses to repeated hemorrhage in conscious dogs. Am J Physiol. 1986;251:R1193–R1199. doi: 10.1152/ajpregu.1986.251.6.R1193. [DOI] [PubMed] [Google Scholar]
- Luke TM, Hexum TD. Tyrosine hydroxylase phosphorylation increases in response to ATP and neuropeptide Y co-stimulation of ERK2 phosphorylation. Pharmacol Res. 2008;58:52–57. doi: 10.1016/j.phrs.2008.06.010. [DOI] [PubMed] [Google Scholar]
- Lundberg JM, Modin A. Inhibition of sympathetic vasoconstriction in pigs in vivo by the neuropeptide Y-Y1 receptor antagonist BIBP 3226. Br J Pharmacol. 1995;116:2971–2982. doi: 10.1111/j.1476-5381.1995.tb15952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg JM, Terenius L, Hökfelt T, Goldstein M. High levels of neuropeptide Y in peripheral noradrenergic neurons in various mammals including man. Neurosci Lett. 1983;42:167–172. doi: 10.1016/0304-3940(83)90401-9. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, Lightman SL. The arginine vasopressin and corticotrophin-releasing hormone gene transcription responses to varied frequencies of repeated stress in rats. J Physiol. 1998;510:605–614. doi: 10.1111/j.1469-7793.1998.605bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmström RE. Vascular pharmacology of BIIE0246, the first selective non-peptide neuropeptide Y Y(2) receptor antagonist, in vivo. Br J Pharmacol. 2001;133:1073–1080. doi: 10.1038/sj.bjp.0704171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathar I, Vennekens R, Meissner M, Kees F, Van der Mieren G, Camacho Londoño JE, Uhl S, Voets T, Hummel B, van den Bergh A, Herijgers P, Nilius B, Flockerzi V, Schweda F, Freichel M. Increased catecholamine secretion contributes to hypertension in TRPM4-deficient mice. J Clin Invest. 2010;120:3267–3279. doi: 10.1172/JCI41348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda H, Brumovsky PR, Kopp J, Pedrazzini T, Hökfelt T. Distribution of neuropeptide Y Y1 receptors in rodent peripheral tissues. J Comp Neurol. 2002;449:390–404. doi: 10.1002/cne.10303. [DOI] [PubMed] [Google Scholar]
- Maubert E, Dupouy JP, Bernet F. Effect of adrenal demedullation on neuropeptide Y content of the capsule/glomerulosa zone of the rat adrenal gland. Neurosci Lett. 1993;156:5–8. doi: 10.1016/0304-3940(93)90425-K. [DOI] [PubMed] [Google Scholar]
- Meligeni JA, Haycock JW, Bennett WF, Waymire JC. Phosphorylation and activation of tyrosine hydroxylase mediate the cAMP-induced increase in catecholamine biosynthesis in adrenal chromaffin cells. J Biol Chem. 1982;257:12632–12640. [PubMed] [Google Scholar]
- Michel MC, Beck-Sickinger A, Cox H, Doods HN, Herzog H, Larhammar D, Quirion R, Schwartz T, Westfall T. XVI. International Union of Pharmacology recommendations for the nomenclature of neuropeptide Y, peptide YY, and pancreatic polypeptide receptors. Pharmacol Rev. 1998;50:143–150. [PubMed] [Google Scholar]
- Montesinos MS, Machado JD, Camacho M, Diaz J, Morales YG, Alvarez de la Rosa D, Carmona E, Castañeyra A, Viveros OH, O'Connor DT, Mahata SK, Borges R. The crucial role of chromogranins in storage and exocytosis revealed using chromaffin cells from chromogranin A null mouse. J Neurosci. 2008;28:3350–3358. doi: 10.1523/JNEUROSCI.5292-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormède P, Lemaire V, Castanon N, Dulluc J, Laval M, Le Moal M. Multiple neuroendocrine responses to chronic social stress: interaction between individual characteristics and situational factors. Physiol Behav. 1990;47:1099–1105. doi: 10.1016/0031-9384(90)90358-B. [DOI] [PubMed] [Google Scholar]
- Murgatroyd C, Patchev AV, Wu Y, Micale V, Bockmühl Y, Fischer D, Holsboer F, Wotjak CT, Almeida OF, Spengler D. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci. 2009;12:1559–1566. doi: 10.1038/nn.2436. [DOI] [PubMed] [Google Scholar]
- Myrsén-Axcrona U, Ekblad E, Sundler F. Developmental expression of NPY, PYY and PP in the rat pancreas and their coexistence with islet hormones. Regul Pept. 1997;68:165–175. doi: 10.1016/S0167-0115(96)02113-1. [DOI] [PubMed] [Google Scholar]
- Nanduri J, Makarenko V, Reddy VD, Yuan G, Pawar A, Wang N, Khan SA, Zhang X, Kinsman B, Peng YJ, Kumar GK, Fox AP, Godley LA, Semenza GL, Prabhakar NR. Epigenetic regulation of hypoxic sensing disrupts cardiorespiratory homeostasis. Proc Natl Acad Sci U S A. 2012;109:2515–2520. doi: 10.1073/pnas.1120600109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nankova B, Kvetnanský R, McMahon A, Viskupic E, Hiremagalur B, Frankle G, Fukuhara K, Kopin IJ, Sabban EL. Induction of tyrosine hydroxylase gene expression by a nonneuronal nonpituitary-mediated mechanism in immobilization stress. Proc Natl Acad Sci U S A. 1994;91:5937–5941. doi: 10.1073/pnas.91.13.5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obst M, Tank J, Plehm R, Blumer KJ, Diedrich A, Jordan J, Luft FC, Gross V. NO-dependent blood pressure regulation in RGS2-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1012–R1019. doi: 10.1152/ajpregu.00288.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okimoto DK, Blaus A, Schmidt M, Gordon MK, Dent GW, Levine S. Differential expression of c-fos and tyrosine hydroxylase mRNA in the adrenal gland of the infant rat: evidence for an adrenal hyporesponsive period. Endocrinology. 2002;143:1717–1725. doi: 10.1210/en.143.5.1717. [DOI] [PubMed] [Google Scholar]
- Otten U, Paravicini U, Oesch F, Thoenen H. Time requirement for the single steps of trans-synaptic induction of tyrosine hydroxylase in the peripheral sympathetic nervous system. Naunyn Schmiedeberg Arch Pharmacol. 1973;280:117–127. doi: 10.1007/BF00499173. [DOI] [Google Scholar]
- Pedrazzini T, Seydoux J, Künstner P, Aubert JF, Grouzmann E, Beermann F, Brunner HR. Cardiovascular response, feeding behavior and locomotor activity in mice lacking the NPY Y1 receptor. Nat Med. 1998;4:722–726. doi: 10.1038/nm0698-722. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy P, Whim MD. Trafficking and fusion of neuropeptide Y-containing dense-core granules in astrocytes. J Neurosci. 2008;28:13815–13827. doi: 10.1523/JNEUROSCI.5361-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter S, Dinh TT, Li AJ. Hindbrain catecholamine neurons control multiple glucoregulatory responses. Physiol Behav. 2006;89:490–500. doi: 10.1016/j.physbeh.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/S0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Preferential potentiation of fast-releasing synaptic vesicles by cAMP at the calyx of Held. Proc Natl Acad Sci U S A. 2001;98:331–336. doi: 10.1073/pnas.98.1.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89. doi: 10.1210/er.21.1.55. [DOI] [PubMed] [Google Scholar]
- Shi YC, Lau J, Lin Z, Zhang H, Zhai L, Sperk G, Heilbronn R, Mietzsch M, Weger S, Huang XF, Enriquez RF, Baldock PA, Zhang L, Sainsbury A, Herzog H, Lin S. Arcuate NPY controls sympathetic output and BAT function via a relay of tyrosine hydroxylase neurons in the PVN. Cell Metab. 2013;17:236–248. doi: 10.1016/j.cmet.2013.01.006. [DOI] [PubMed] [Google Scholar]
- Smith-White MA, Hardy TA, Brock JA, Potter EK. Effects of a selective neuropeptide Y Y2 receptor antagonist, BIIE0246, on Y2 receptors at peripheral neuroeffector junctions. Br J Pharmacol. 2001;132:861–868. doi: 10.1038/sj.bjp.0703879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen G, Jensen M, Weikop P, Dencker D, Christiansen SH, Loland CJ, et al. Neuropeptide Y Y5 receptor antagonism attenuates cocaine-induced effects in mice. Psychopharmacology (Berl) 2012;222:565–577. doi: 10.1007/s00213-012-2651-y. [DOI] [PubMed] [Google Scholar]
- Souvannakitti D, Kumar GK, Fox A, Prabhakar NR. Neonatal intermittent hypoxia leads to long-lasting facilitation of acute hypoxia-evoked catecholamine secretion from rat chromaffin cells. J Neurophysiol. 2009;101:2837–2846. doi: 10.1152/jn.00036.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souvannakitti D, Nanduri J, Yuan G, Kumar GK, Fox AP, Prabhakar NR. NADPH oxidase-dependent regulation of T-type Ca2+ channels and ryanodine receptors mediate the augmented exocytosis of catecholamines from intermittent hypoxia-treated neonatal rat chromaffin cells. J Neurosci. 2010;30:10763–10772. doi: 10.1523/JNEUROSCI.2307-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer NC. Activity-dependent neurotransmitter respecification. Nat Rev Neurosci. 2012;13:94–106. doi: 10.1038/nrn3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Sullivan JM. Regulation of the readily releasable vesicle pool by protein kinase C. Neuron. 1998;21:885–893. doi: 10.1016/S0896-6273(00)80603-0. [DOI] [PubMed] [Google Scholar]
- Stroth N, Eiden LE. Stress hormone synthesis in mouse hypothalamus and adrenal gland triggered by restraint is dependent on pituitary adenylate cyclase-activating polypeptide signaling. Neuroscience. 2010;165:1025–1030. doi: 10.1016/j.neuroscience.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabares L, Alés E, Lindau M, Alvarez de Toledo G. Exocytosis of catecholamine (CA)-containing and CA-free granules in chromaffin cells. J Biol Chem. 2001;276:39974–39979. doi: 10.1074/jbc.M106498200. [DOI] [PubMed] [Google Scholar]
- Ulus IH, Wurtman RJ. Selective response of rat peripheral sympathetic nervous system to various stimuli. J Physiol. 1979;293:513–523. doi: 10.1113/jphysiol.1979.sp012904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Yao Y, Fu LY, Foo K, Huang H, Coppari R, Lowell BB, Broberger C. Neuromedin B and gastrin-releasing peptide excite arcuate nucleus neuropeptide Y neurons in a novel transgenic mouse expressing strong Renilla green fluorescent protein in NPY neurons. J Neurosci. 2009;29:4622–4639. doi: 10.1523/JNEUROSCI.3249-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella KR, Ramadoss P, Lam FS, Harris JC, Ye FD, Same PD, O'Neill NF, Maratos-Flier E, Hollenberg AN. NPY and MC4R signaling regulate thyroid hormone levels during fasting through both central and peripheral pathways. Cell Metab. 2011;14:780–790. doi: 10.1016/j.cmet.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verberne AJ, Sartor DM. Rostroventrolateral medullary neurons modulate glucose homeostasis in the rat. Am J Physiol Endocrinol Metab. 2010;299:E802–E807. doi: 10.1152/ajpendo.00466.2010. [DOI] [PubMed] [Google Scholar]
- Vietor I, Rusnak M, Viskupic E, Blazicek P, Sabban EL, Kvetnansky R. Glucoprivation by insulin leads to trans-synaptic increase in rat adrenal tyrosine hydroxylase mRNA levels. Eur J Pharmacol. 1996;313:119–127. doi: 10.1016/0014-2999(96)00508-0. [DOI] [PubMed] [Google Scholar]
- Wamsteeker JI, Bains JS. A synaptocentric view of the neuroendocrine response to stress. Eur J Neurosci. 2010;32:2011–2021. doi: 10.1111/j.1460-9568.2010.07513.x. [DOI] [PubMed] [Google Scholar]
- Wang Q, Whim MD. Stress-induced changes in adrenal neuropeptide Y expression are regulated by a negative feedback loop. J Neurochem. 2013;125:16–25. doi: 10.1111/jnc.12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts AG, Donovan CM. Sweet talk in the brain: glucosensing, neural networks, and hypoglycemic counterregulation. Front Neuroendocrinol. 2010;31:32–43. doi: 10.1016/j.yfrne.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whim MD. Near simultaneous release of classical and peptide cotransmitters from chromaffin cells. J Neurosci. 2006;26:6637–6642. doi: 10.1523/JNEUROSCI.5100-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whim MD. Pancreatic beta cells synthesize neuropeptide Y and can rapidly release peptide co-transmitters. PLoS One. 2011;6:e19478. doi: 10.1371/journal.pone.0019478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whim MD, Moss GW. A novel technique that measures peptide secretion on a millisecond timescale reveals rapid changes in release. Neuron. 2001;30:37–50. doi: 10.1016/S0896-6273(01)00261-6. [DOI] [PubMed] [Google Scholar]
- Widmaier EP. Development in rats of the brain-pituitary-adrenal response to hypoglycemia in vivo and in vitro. Am J Physiol. 1989;257:E757–E763. doi: 10.1152/ajpendo.1989.257.5.E757. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Klein M. Modulation of the readily releasable pool of transmitter and of excitation-secretion coupling by activity and by serotonin at Aplysia sensorimotor synapses in culture. J Neurosci. 2002;22:10671–10679. doi: 10.1523/JNEUROSCI.22-24-10671.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]