

Abstract
Mitogen-activated protein (MAP) kinase signaling cascades orchestrate diverse cellular activities with common molecular players. To achieve specific cellular outcomes in response to specific signals, scaffolding proteins play an important role. Here we investigate the role of the scaffolding protein JNK interacting protein-1 (JIP1) in neuronal signaling by a conserved axonal MAP kinase kinase kinase, known as Wallenda (Wnd) in Drosophila and dual leucine kinase (DLK) in vertebrates and Caenorhabditis elegans. Recent studies in multiple model organisms suggest that Wnd/DLK regulates both regenerative and degenerative responses to axonal injury. Here we report a new role for Wnd in regulating synaptic structure during development, which implies that Wnd is also active in uninjured neurons. This synaptic role of Wnd can be functionally separated from the role of Wnd in axonal regeneration and injury signaling by the requirement for the JIP1 scaffold and the p38b MAP kinase. JIP1 mediates the synaptic function of Wnd via p38, which is not required for injury signaling or new axonal growth after injury. Our results indicate that Wnd regulates multiple independent pathways in Drosophila motoneurons and that JIP1 scaffolds a specific downstream cascade required for the organization of presynaptic microtubules during synaptic development.
Introduction
Neurons use mitogen-activated protein kinase (MAPK) signaling cascades to regulate many different processes, including synaptic development and plasticity, axonal growth, and survival. A conserved upstream regulator of MAPK signaling, named Wallenda (Wnd) in Drosophila and dual leucine kinase (DLK) in vertebrates and Caenorhabditis elegans, has received much recent attention for its roles in regulating neuronal responses to axonal injury. This kinase becomes activated by axonal injury and mediates different downstream responses depending on the cell type and context: regenerative axonal growth, cell death, axonal degeneration, and protection from degeneration (for review, see Tedeschi and Bradke, 2013).
In contrast to these postdevelopmental roles, previous studies have suggested that Wnd/DLK is highly regulated during development and that this regulation is important for neuronal migration, axon termination, apoptosis, and synaptic development (Chen and Tan, 2000; Collins et al., 2006; Hirai et al., 2006; Bloom et al., 2007; Grill et al., 2007; Ghosh et al., 2011).
How can a single kinase mediate such diverse and dichotomous functions in neurons? Wnd is a member of the mixed lineage family of kinases, which has been shown to function as upstream regulators of the stress-activated MAPKs, c-Jun NH2-terminal kinase (JNK) and p38 (Fan et al., 1996). Toward understanding the mechanism for the multiple functions of Wnd, we investigated the role of scaffolding proteins. By coordinating interactions between specific MAPKs and their activators, inactivators, or substrates, scaffolding proteins can influence when and where MAPKs become activated, as well as the downstream consequences of their activation (Morrison and Davis, 2003; Dhanasekaran et al., 2007).
We focused our study on the JNK interacting proteins (JIPs), which have been implicated in a number of JNK-regulated processes in neurons (Whitmarsh, 2006; Koushika, 2008). The Drosophila genome encodes two JIP proteins: JIP1 (APLIP1) and JIP3, also known as Sunday Driver (Syd). In both vertebrate and Drosophila cells, it has been shown that JIP1 mediates the activation of JNK by Wnd/DLK (Whitmarsh et al., 1998; Nihalani et al., 2001; Whitmarsh, 2006; Horiuchi et al., 2007). Both JIP1 and JIP3/Syd are carried by kinesin motors in axons (Verhey et al., 2001) and influence the process of axonal transport (Bowman et al., 2000; Byrd et al., 2001; Taru et al., 2002; Horiuchi et al., 2005). Because functional axonal transport machinery is required for injury signaling (Abe and Cavalli, 2008; Xiong et al., 2010), we needed to consider the relationship between JIP1 and the roles of Wnd in axonal transport and signaling.
Our findings suggest that these processes can be functionally separated. Characterization of jip1 null mutants has revealed a new role for Wnd in regulating the structure of synaptic microtubules during development of the Drosophila neuromuscular junction (NMJ). This developmental role, which requires JIP1 and the downstream p38b MAPK, is distinct from the roles of Wnd in axonal transport and injury signaling, which do not require p38. Hence, the JIP1 scaffold promotes a specific synaptic function for Wnd and MAPK signaling.
Materials and Methods
Generation of jip1 mutant.
The jip1ex allele was created by the imprecise excision of the P-element insertion P-Aplip1DG20707, which lies in the 3′ UTR of JIP1/Aplip1. Approximately 260 independent lines were screened by PCR to uncover one deletion that removed the entire JIP1/Aplip1 locus.
Genetics.
The following strains were used in this study: Canton S [wild-type (WT)], puc–lacZE69 (Martín-Blanco et al., 1998), BG380–Gal4 (Budnik et al., 1996), m12–Gal4 [P(Gal4)5053A] (Ritzenthaler et al., 2000), RRa(eve)–Gal4 (Fujioka et al., 2003), OK6–Gal4 (Aberle et al., 2002), wnd1, wnd2, wnd3 (Collins et al., 2006), hiwND8 (Wan et al., 2000), hiwΔN (Wu et al., 2005), UAS–FosDN (Eresh et al., 1997), UAS–Bsk(Jnk)DN (Weber et al., 2000), Δp38a (Craig et al., 2004), p38bΔ45, p38bΔ25;Δp38a (Vrailas-Mortimer et al., 2011), UAS–p38bDN (Adachi-Yamada et al., 1999), sydZ4, syd2H (Bowman et al., 2000), and jip1ek4, UAS–JIP1ΔKBD, genomic JIP1 (Horiuchi et al., 2005). Df(3L)ED229 (wnd), Df(3L)Fpa2 (jip1), Df(2L)b80e3 (p38b), UAS–bsk–RNAi (TRiP–JF01275), UAS–p38b–RNAi (TRiP–JF03341), UAS–wnd–RNAi (TRiP–JF02675), p38bKG01337, P-Aplip1DG20707, and FutschEP(x)1419 were obtained from the Bloomington Stock Center. UAS–wnd–RNAi (26910) was acquired from the Vienna RNAi Center (Dietzl et al., 2007). UAS–Dcr2 was a gift from Stefan Thor (Linkoping University, Linkoping, Sweden). GeneSwitch elav–Gal4 driver (GSelav) was used to control temporal expression of UAS transgenes in neurons (Osterwalder et al., 2001). To activate the GSelav driver, flies were reared on standard food that contained 20 μg/ml RU-486 (11β-[p-(dimethylamino)phenyl]-17β-hydroxy-17-(1-propynyl)estra-4,9-dien-3-one), a nonlethal dose of the drug. Male larvae were used for all experiments using the BG380–Gal4 driver. For other experiments, larvae of both sexes were used.
Immunocytochemistry.
Larvae were dissected in PBS and fixed in either 4% paraformaldehyde in PBS or Bouin's fixative for 15–30 min, depending on the antibodies used. Antibodies were used at the following dilutions in PBS with 0.3% Triton X-100 and 5% normal goat serum: mouse anti-Futsch, 1:100 (22c10; Developmental Studies Hybridoma Bank); rabbit anti-Drosophila vesicular glutamate transporter (DVGLUT) (Daniels et al., 2004), 1:5000; rat anti-elav (7E8A10; Developmental Studies Hybridoma Bank), 1:50; mouse anti-acetylated tubulin (Sigma-Aldrich), 1:100; Cy3 goat anti-HRP (Jackson ImmunoResearch), 1:500; Cy5 goat anti-HRP (Jackson ImmunoResearch), 1:100; and Alexa Fluor 488 rabbit anti-GFP (Invitrogen), 1:1000. For secondary antibodies, Cy3 and Alexa Fluor 488-conjugated goat anti-rabbit, anti-mouse, and anti-rat (Invitrogen) were used at 1:1000.
Imaging and quantification.
Confocal images were collected at room temperature on an Improvision spinning-disk confocal system (PerkinElmer Life and Analytical Sciences). All imaging and quantification were conducted with Volocity software (PerkinElmer Life and Analytical Sciences). Similar settings were used to collect all compared genotypes and conditions.
To quantify the mean intensity of puc–lacZ expression, we used the protocol described previously (Xiong et al., 2010).
Futsch bundling was quantified as described previously (Viquez et al., 2006). Briefly, larvae were stained with Futsch and DVGLUT antibodies to label both the cytoskeleton and the synaptic boutons, respectively. Futsch staining that colocalized with DVGLUT was classified as either unbundled (looped, splayed, punctate, or missing) or bundled Futsch (tightly wound filamentous Futsch staining). The synaptic area of unbundled and bundled Futsch were measured, and the area of unbundled Futsch was divided by the total Futsch area.
The axonal transport severity index was ranked by qualitative assessment of the number and size of axonal accumulations for the synaptic vesicle marker DVGLUT, while blind to genotype. Individual nerves were given a score of 0 to 4 depending on severity, with 4 being the greatest amount of axonal accumulations.
The regeneration ratio was quantified as the fraction of injured axons that exhibited sprouting (at least five branches) per genotype, while blind to the genotype, as described by Xiong et al. (2010).
Nerve crush assay.
The segmental nerves of third-instar larvae were subjected to nerve crush injury as described previously (Xiong et al., 2010).
Results
Comparison of JIP1 and JIP3 in axonal transport and injury signaling
To study the role of JIP1/APLIP1 in Wnd signaling, we generated a null allele via imprecise excision of the P-Aplip1DG20707 transposon. jip1ex removes the entire jip1 coding region, leaving the flanking genes LysX and mwh intact (Fig. 1A). Unlike the larval lethality observed for mutations in the JIP3 homolog Syd (Bowman et al., 2000), jip1ex/jip1ex and jip1ex/Df mutants develop into fully viable adults. Defects in axonal transport, as measured by accumulations of the synaptic vesicle marker DVGLUT in segmental nerves, were also less severe for jip1ex mutants compared with jip3 mutants (Fig. 1B,C). The axonal transport defect of jip1ex can be rescued by the presence of a transgene containing one copy of genomic jip1 (Fig. 1C).
Figure 1.
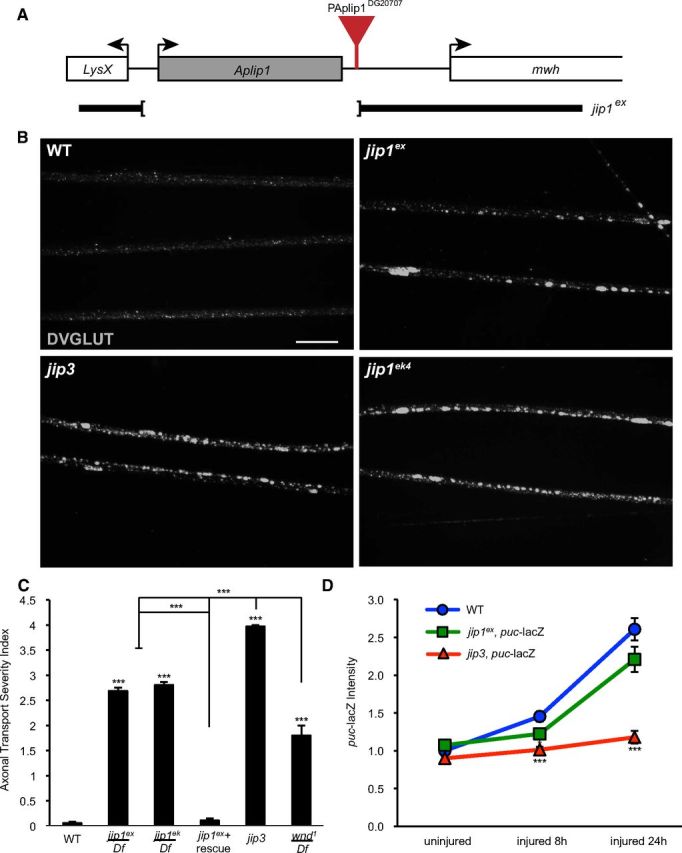
Comparison of JIP1 and JIP3 in injury signaling and axonal transport. A, Schematic of the JIP1/APLIP1 genomic region depicting the jip1ex excision deletion mutation. The jip1/Aplip1 locus with flanking genes LysX and mwh. The jip1ex null allele was created by imprecise excision of P-Aplip1DG20707. B, Segmental nerves from third-instar larvae immunostained with DVGLUT. Small punctae of DVGLUT are observed in WT nerves. jip1ex null mutation caused accumulations of DVGLUT in nerves consistent with defective axonal transport. A similar amount of accumulations were observed in the jip1ek4 hypomorphic allele. jip3 mutants (sydZ4/syd2H) displayed an increase in the number of DVGLUT accumulations compared with jip1 mutants. C, Quantification of the axonal transport severity index. The severity of the DVGLUT accumulation phenotype was ranked by qualitative assessment of the number and size of axonal accumulations (see Materials and Methods). Note that defects in jip1 and jip3 mutants are stronger than the strongest observed phenotype in wnd mutants. D, Quantification of puc–lacZ. The mean intensity of puc–lacZ is measured as described in Materials and Methods for the dorsal midline neurons. At 8 h after injury, puc–lacZ intensity is increased in WT animals. puc–lacZ intensity for jip1 (jip1ex/jip1Df) and jip3 (sydZ4/syd2H) mutants is significantly decreased compared with control animals. By 24 h after injury jip1ex, puc–lacZ intensity increases to near WT levels, but puc–lacZ intensity in jip3 mutants is comparable with uninjured control animals. Error bars indicate mean ± SEM. *p ≤ 0.01; ***p ≤ 0.0001. Scale bars, 10 μm.
Wnd regulates a transcriptional response to axonal injury, which can be measured by the induction of the puc–lacZ reporter (Martín-Blanco et al., 1998) in response to a larval segmental nerve crush (Xiong et al., 2010). Injury signaling in jip1ex mutant animals is slightly reduced at 8 h but reaches levels similar to WT animals within 24 h after injury (Fig. 1D). In contrast, injury signaling was dramatically inhibited in the jip3 mutant animals at both 8 and 24 h after injury (Fig. 1D). These results suggest that JIP3/Syd plays an essential role in the injury signaling mechanism, whereas JIP1 is dispensable. It has been shown previously that general inhibition of axonal transport can diminish the induction of puc–lacZ (Xiong et al., 2010); therefore, the divergent phenotypes for jip1 and jip3 in injury signaling may be an indirect consequence of their different effects on axonal transport (Fig. 1B,C). Because axonal transport plays a fundamental role in neurons and its disruption would affect many cellular pathways, this was an important concern in understanding the function of JIP1. Therefore, we asked whether JIP1 played other roles in neurons that could be functionally or phenotypically separated from its role in axonal transport.
JIP1 promotes the development of presynaptic boutons
The most striking phenotype observed for jip1ex mutants was the enlargement of the synaptic boutons at the larval NMJ (Fig. 2A,B). The most proximal boutons were particularly enlarged, showing an approximately twofold increase in diameter compared with WT synapses (Fig. 2B). We also measured a greater than twofold increase in the total number of boutons that exceeded 5 μm/NMJ: WT animals had an average of one bouton per NMJ that reached this size, but jip1 mutants had an average of three boutons per NMJ that were >5 μm (SEM of 0.54, p = 0.03) (data not shown). In contrast to the enlarged bouton sizes, we observed no differences in the overall number of boutons or branches (see Fig. 6B).
Figure 2.
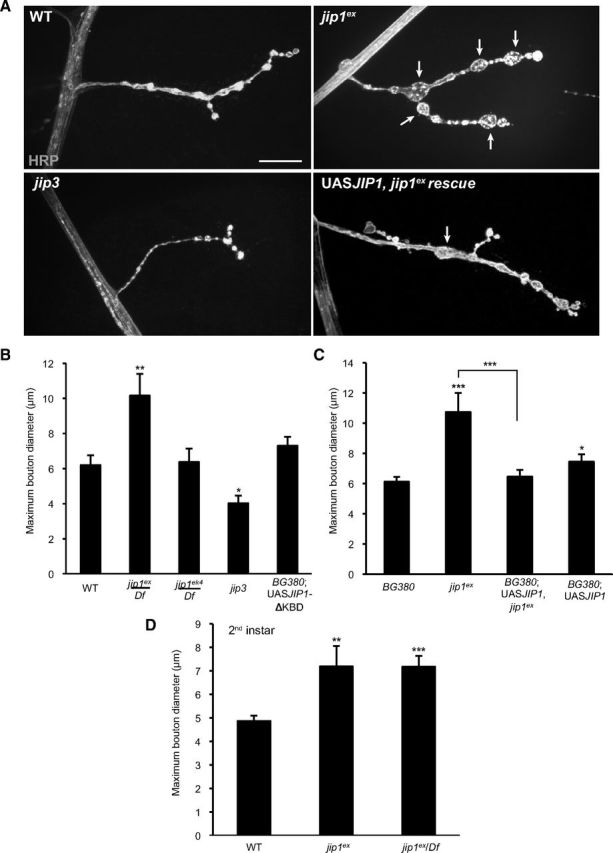
jip1 mutants have a synaptic NMJ phenotype. A, The axonal membrane at muscle 4 NMJ synapses is labeled by immunostaining with anti-HRP antibodies for WT, jip1 mutants (jip1ex/jip1ex), jip3 mutants (sydZ4/syd2H), and jip1 rescue animals (BG380–Gal4; UAS–JIP1/+; jip1ex/jip1ex). jip1ex mutants display enlarged boutons, and this phenotype can be rescued by neuronal expression of a JIP1 transgene. Arrows indicate the enlarged boutons ≥5 μm. B, C, Quantification of maximum bouton diameter. B, jip1ex mutants have larger [and a greater number of oversized (data not shown)] boutons compared with control animals. jip1ek4 synapses look similar to WT animals. jip3 synapses display smaller boutons compared with controls. Expression of the jip1ΔKBD transgene lacking the kinesin binding domain (UAS–JIP1ΔKBD) did not result in an enlarged bouton phenotype. C, Rescue of the jip1ex bouton phenotype. Neuronal expression of a JIP1 transgene results in synapses with slightly larger boutons compared with WT animals, but rescues the enlarged boutons observed in jip1ex mutants. D, Quantification of maximum bouton diameter in second-instar larvae. Boutons are enlarged in jip1ex and jip1ex/Df mutants even at this earlier developmental stage. Error bars indicate mean ± SEM. *p ≤ 0.01, **p ≤ 0.001, ***p ≤ 0.0001. Scale bars, 10 μm.
Figure 6.
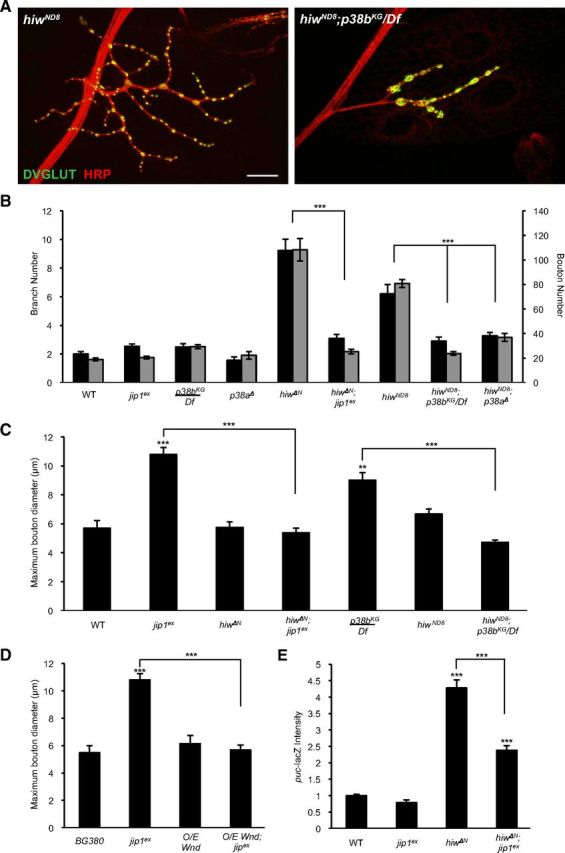
Wnd regulates synaptic structure via the p38 MAPK and JIP1 scaffold. A, NMJ synapses at muscle 4 stained with the neuronal membrane marker HRP (red) and synaptic vesicle marker DVGLUT (green) of hiw (hiwND8) mutants and double mutants for hiw and p38b (hiwND8; p38bKG/Df). B, Quantification of suppression of the hiw synaptic morphology phenotype including branch number (black bars) and bouton number (gray bars). p38a, p38b, and jip1 can all rescue the synaptic overgrowth phenotype of hiw mutants. C, D, Quantification of suppression of maximum bouton diameter. hiw and neuronally expressed Wnd (BG380–Gal4;UAS-wnd/+) can rescue the enlarged bouton phenotype in jip1 and p38b (p38bKG/Df) mutants. E, Quantification of puc–lacZ. jip1 is required for the induction of puc–lacZ in hiw mutants. Error bars indicate mean ± SEM. **p ≤ 0.001, ***p ≤ 0.0001. Scale bar, 10 μm. O/E, Overexpressed.
Surprisingly, jip1ek4 mutants, which carry a point mutation within the kinesin binding domain (Horiuchi et al., 2005) and displayed comparable defects in axonal transport as jip1ex (Fig. 1B,C), did not affect bouton morphology (Fig. 2B). This observation suggested that the role of JIP1 in controlling synaptic morphology may be separable from its role in axonal transport. Similarly, neuronal expression of a JIP1 transgene lacking the kinesin binding domain (UAS–JIP1ΔKBD) also had no effect on bouton morphology (Fig. 2B,C), although these animals display strong defects in axonal transport (Horiuchi et al., 2005). In jip3 mutants, which exhibit the strongest defect in axonal transport, the bouton diameter was actually slightly decreased compared with control synapses (Fig. 2A,B). Because the synaptic phenotypes did not correlate with the axonal transport phenotypes, they suggest an independent function for JIP1 in the regulation of synaptic structure.
To verify that the enlarged bouton phenotype was specific for JIP1, we used the UAS/Gal4 system to drive expression of a UAS–JIP1 transgene using the neuronal BG380–Gal4 driver in a jip1ex mutant background. This rescued the enlarged bouton phenotype (Fig. 2A,C). Expression of the JIP1 transgene alone in a WT background also resulted in a slight but significant increase in the maximum bouton diameter (Fig. 2C). The similarity between gain-of-function and loss-of-function phenotypes for jip1 was observed previously for defects in axonal transport (Horiuchi et al., 2005), and, given the hypothesized role of JIP1 as a scaffolding protein (Whitmarsh and Davis, 1998; Whitmarsh et al., 1998), these results are not unexpected.
To determine whether the enlarged boutons in jip1 mutant animals resulted from a failure to maintain synaptic structure or whether JIP1 played a role in synaptic development, we examined the synaptic morphology of jip1 mutant synapses in younger animals. We found that second-instar animals, similar to third-instar animals, had an average of 2.5 enlarged boutons (≥5 μm) per NMJ, but these enlarged boutons were slightly smaller in diameter (Fig. 2D and data not shown) than in third-instar larvae (Fig. 2B). We interpret that the abnormal boutons form early in NMJ development and become larger with time. These observations implicate a role for JIP1 in regulating the development of synaptic boutons at the NMJ.
JIP1 is required for microtubule organization in synaptic boutons
It is well established that synaptic morphology depends on cytoskeletal organization (Jin and Garner, 2008; Goellner and Aberle, 2012) and that vertebrate JIP1 participates in controlling microtubule dynamics in neurons (Chang et al., 2003; Tararuk et al., 2006). Therefore, we wanted to determine whether the increased bouton size reflected changes in the synaptic microtubules. A particularly useful marker for synaptic microtubules is the neuronal-specific microtubule binding protein Futsch (homologous to MAP1B), which plays a critical role in microtubule organization at the Drosophila NMJ (Hummel et al., 2000; Roos et al., 2000). Another indicator of microtubule stability is the presence of posttranslational modifications such as acetylated tubulin (Conde and Cáceres, 2009; Janke and Kneussel, 2010). At the larval NMJ, both Futsch and acetylated tubulin form a tightly bundled cable that runs through most of the NMJ (Ruiz-Cañada and Budnik, 2006; Fig. 3A,B). In jip1 mutants, this cable is disorganized (for Futsch) and broken (for acetylated tubulin), particularly in the largest boutons. Futsch staining becomes splayed and unbundled, whereas acetylated tubulin accumulated in a punctate, fragmented pattern, suggesting a breakdown or misregulation of the microtubule cytoskeleton (Fig. 3A,B). This disruption of the microtubule cytoskeleton is not observed in either jip1ek4 mutants or animals neuronally expressing the JIP1 transgene lacking the kinesin binding domain (UAS–JIP1ΔKBD); hence, is not simply a consequence of defects in axonal transport (Fig. 3C). Importantly, both the unbundling of Futsch and the fragmentation of acetylated tubulin are rescued by the presence of a transgene containing one copy of genomic jip1 (Fig. 3A–C).
Figure 3.

JIP1 is required for microtubule organization and stability. A, WT, jip1ex/jip1ex, and jip1ex genomic rescue synapses immunostained with HRP (red) and Futsch (green). Although microtubules normally appear bundled within WT boutons, in the enlarged boutons (≥5 μm) of jip1 mutants, splaying of the microtubules is observed. This unbundling of the microtubules can be rescued with the expression of one copy of a genomic JIP1 transgene. B, Acetylated tubulin (green) is localized in a tightly bundled cable that extends through the NMJ synapse of WT animals. Within enlarged boutons of jip1 mutants, acetylated tubulin has a discontinuous, punctate pattern, which can be rescued with one copy of a genomic JIP1 transgene. C, Quantification of the percentage of unbundled Futsch for different genotypes (see Materials and Methods). C, Error bars indicate mean ± SEM. ***p ≤ 0.0001. Scale bars, 10 μm.
Mutations in futsch give rise to enlarged boutons (Roos et al., 2000); hence, the enlarged boutons in jip1 mutants may be the result of misregulated microtubules. We tested whether the enlarged bouton phenotype in jip1 mutants could be suppressed by increasing the expression of Futsch. Although overexpression of Futsch (using the BG380–Gal4 driver) did not significantly alter NMJ morphology in the WT background, it led to a full rescue of the enlarged bouton phenotype in jip1 mutants (Fig. 4A,B). These observations suggest that the bouton morphology defect in jip1 mutants reflects a role for JIP1 in the organization of synaptic microtubules.
Figure 4.

Overexpression of Futsch rescues the jip1 synaptic defect but not the axonal transport defect. A, Representative muscle 4 NMJs were costained with anti-HRP (red) and anti-Futsch (green) for WT (Canton S), jip1 mutants (jip1ex/jip1ex), neuronally expressed Futsch (BG380–Gal4, FutschEP/+), and neuronally expressed Futsch in a jip1 mutant background (BG380–Gal4, FutschEP/+; jip1ex/jip1ex). B, Quantification of maximum bouton diameter and number of boutons ≥5 μm. Neuronal expression of Futsch in a jip1 mutant background can rescue the enlarged bouton phenotype. Neuronal expression of Futsch alone does not alter the synaptic bouton size. C, Peripheral nerves from third-instar larvae immunostained with DVGLUT. Overexpression of Futsch in a jip1 mutant background is unable to rescue the axonal transport defect. D, Quantification of the axonal transport severity index. There is no difference in axonal trafficking defects between jip1 mutants and jip1 mutants with Futsch overexpressed in neurons. Error bars indicate mean ± SEM. **p ≤ 0.001, ***p ≤ 0.0001. Scale bar, 10 μm. O/E, Overexpressed.
In contrast to the synaptic phenotype, overexpression of Futsch failed to rescue the axonal transport defect of jip1 mutants (Fig. 4C,D). These observations further suggest that JIP1 plays at least two independent roles in motoneurons: (1) one in the regulation of synaptic structure and (2) another in axonal transport.
The Wnd MAP kinase kinase kinase regulates synaptic morphology
To understand the mechanism for the synaptic function of JIP1, we considered the possible role of the Wnd MAP kinase kinase kinase (MAPKKK) in regulating synaptic morphology. Studies in vertebrate and Drosophila cells suggest that JIP1 functions as a scaffolding protein for Wnd/DLK signaling (Whitmarsh et al., 1998; Nihalani et al., 2001; Horiuchi et al., 2005; Whitmarsh, 2006) and that Wnd/DLK signaling can influence microtubule structure (Eto et al., 2010; Bounoutas et al., 2011; Hirai et al., 2011; Ghosh-Roy et al., 2012). Previous characterization of wnd mutants found no obvious defects in synaptic morphology (Collins et al., 2006), but closer examination of wnd mutant genotypes (wnd1/wnd2, wnd1/Df, and wnd3/Df; Fig. 5A,B), as well as neuronal-specific RNAi knockdown (Fig. 5D,E), revealed an enlarged bouton phenotype similar to jip1 mutants. This enlarged bouton phenotype for wnd, which is not as strong as the jip1 phenotype, may not have been noticed in a previous study (Collins et al., 2006) because of differences in the measurement method (see Materials and Methods). The larger boutons in wnd mutants also displayed Futsch unbundling (Fig. 5A) similar to jip1 mutants. These observations suggest that JIP1 and Wnd may function together and suggest a new role for Wnd in regulating microtubule structure at synapses.
Figure 5.
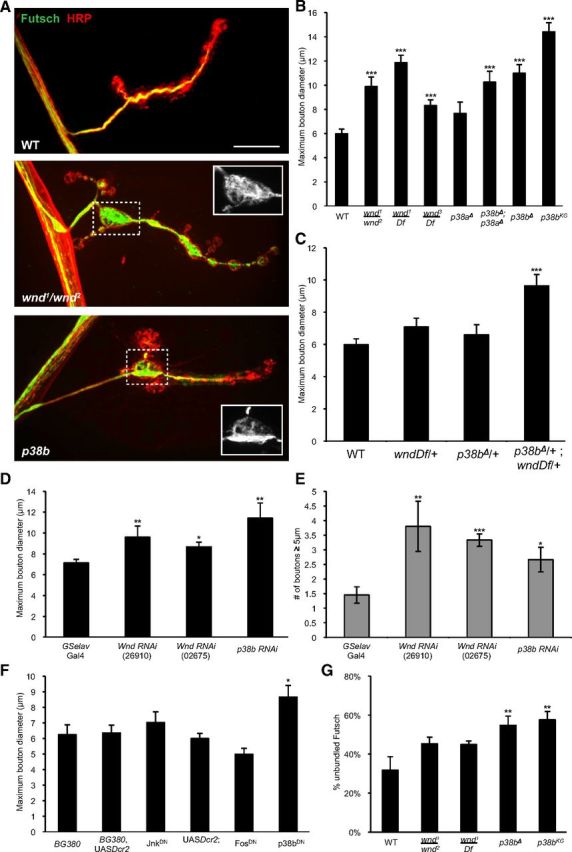
Regulation of synaptic morphology and cytoskeleton by the Wnd/DLK MAPKKK and p38b MAPK. A, WT (Canton S), wnd mutant (wnd1/wnd2), and p38b mutant (p38bΔ) muscle 4 synapses immunostained with HRP (red) and Futsch (green). Both wnd and p38b mutants display enlarged boutons compared with WT animals. In the large boutons, Futsch staining becomes unbundled (see inset). B–F, Quantification of maximum bouton diameter and number of boutons ≥5 μm. B, Three different allelic combinations for wnd display an increase in maximum bouton diameter and the number of boutons ≥5 μm (data not shown). p38a null mutants do not have a synaptic morphology defect. p38b mutants have an enlarged bouton phenotype that is similar to both wnd and jip1 mutants. p38a and p38b double mutants display enlarged boutons similar to the p38b null mutant alone. C, Trans-heterozygous genetic interaction between wndDf/+ and p38bΔ/+. D, E, Neuronal expression of either wnd or p38b RNAi knockdown constructs resulted in animals with significantly larger boutons compared with control animals (GSelav–Gal4/+). F, Neuronal expression of either DN transgenes or RNAi knockdown of JNK or Fos does not result in animals that have larger boutons. Expression of a DN transgene for p38b resulted in animals with enlarged boutons. G, Quantification of the percentage of unbundled Futsch. Both p38b null and p38bKG01337 insertion animals have a significant increase in the amount of unbundled Futsch. wnd mutants display unbundled Futsch in large boutons, but the total amount of unbundled Futsch is unchanged compared with controls. Error bars indicate mean ± SEM. *p ≤ 0.01, **p ≤ 0.001, ***p ≤ 0.0001. Scale bar, 10 μm.
The p38b MAPK regulates synaptic morphology
Previously characterized roles for Wnd, which include the promotion of synaptic overgrowth and injury signaling (Collins et al., 2006; Xiong et al., 2010), involve downstream activation of the JNK MAPK and transcription factor Fos. We tested whether the role of Wnd in regulating the synaptic cytoskeleton could also involve signaling through these same downstream components. Inhibition of JNK and Fos, by either strong expression of dominant-negative (DN) transgenes (JNKDN and FosDN) or RNAi targeted knockdown, did not result in any changes to bouton morphology (Fig. 5F), suggesting that other factors may function downstream of Wnd and JIP1 at synapses.
Therefore, we tested the role of the p38 MAPKs, because it has been reported previously that Wnd/DLK signals through p38 in C. elegans (Hammarlund et al., 2009; Nix et al., 2011). Also, JIP family members have been shown to scaffold p38 MAPKs as well as JNK MAPKs in vertebrate cells (Schoorlemmer and Goldfarb, 2001; Buchsbaum et al., 2002; Lee et al., 2002; Kelkar et al., 2005). In Drosophila, the two p38 MAPK genes p38a and p38b (Han et al., 1998; Adachi-Yamada et al., 1999; Suzanne et al., 1999; Zhuang et al., 2006), have been extensively studied in relation to stress (Inoue et al., 2001; Craig et al., 2004; Sano et al., 2005; Cully et al., 2010; Vrailas-Mortimer et al., 2011) and the fly immune system (Han and Ip, 1999; Davis et al., 2008; Ha et al., 2009; Shinzawa et al., 2009). p38 has also been shown to participate in developmental processes, including axial polarity during oogenesis, intestinal stem cell proliferation and differentiation, and dpp regulated stem cell morphogenesis (Adachi-Yamada et al., 1999; Suzanne et al., 1999; Park et al., 2009).
Using a null mutation of p38a (p38aΔ) that is deleted for the entire p38a locus (Craig et al., 2004), we found no significant role for p38a in the regulation of bouton size (Fig. 5B). However, additional genetic interactions with Wnd (Fig. 6) still imply a function for p38a at the synapse (discussed further below). To examine the role of p38b, we used two different alleles: (1) a null allele that removes most of the coding region (p38bΔ) (Vrailas-Mortimer et al., 2011) and (2) a transposon insertion allele (p38bKG01337). In contrast to p38a, p38b mutants displayed a prominent bouton morphology defect that resembled both jip1 and wnd mutants in the increase in maximum bouton size (Fig. 5A,B), as well as the number of boutons exceeding 5 μm (Fig. 5A and data not shown). Double mutants for p38aΔ and p38bΔ did not enhance this phenotype. Inhibition of p38b, by either RNAi knockdown (Fig. 5D,E) or the expression of a DN allele (p38bDN), specifically in neurons also resulted in enlarged boutons (Fig. 5F), indicating a cell-autonomous role for p38b in regulating bouton morphology. The loss of p38b also resulted in an increase in the overall percentage of unbundled Futsch (Fig. 5A,G), similar to what was observed in jip1 mutants (Fig. 3C). These findings signify a new role for p38b in regulating bouton morphology and microtubule structure at Drosophila synapses.
To further probe the hypothesis that Wnd and p38 regulate synaptic boutons through a common pathway, we asked whether wnd and p38b genetically interact. Figure 5C shows that, although animals missing one copy of either wnd or p38b have no phenotype, animals missing one copy of both wnd and p38b display enlarged boutons (and misregulated cytoskeleton; data not shown) similar to complete loss-of-function mutations in wnd and p38b. Additional genetic interactions are described further below.
JIP1 and p38 mediate synaptic growth and nuclear signaling downstream of Hiw
One of the most striking documented regulators of synaptic growth is Hiw, a conserved E3 ubiquitin ligase (Schaefer et al., 2000; Wan et al., 2000; Zhen et al., 2000). At the Drosophila NMJ, mutations in hiw cause a dramatic increase in the number of synaptic boutons and branches at the larval NMJ (Wan et al., 2000; Collins et al., 2006; Fig. 6A,B). This synaptic overgrowth phenotype is caused by an increased activity of Wnd, whose levels in axons and synapses is regulated by Hiw (Collins et al., 2006). We found that this synaptic gain-of-function phenotype for wnd could be suppressed by mutations in either jip1 or p38b (Fig. 6A,B). In addition, p38a mutants, which did not have a loss-of-function phenotype on their own at the NMJ (Fig. 5B), suppressed the hiw synaptic overgrowth phenotype (Fig. 6B). These observations revise a previous conclusion based on DN constructs for p38 (Collins et al., 2006) and imply a role for both p38a and p38b in synaptic growth. This role may function independently of the regulation of microtubules and bouton size. Alternatively, the function of p38a may be specific to situations when Wnd signaling levels are high. These findings further support the model that JIP1 and the p38 MAPK function together with Wnd to regulate the morphology of the presynaptic axon terminus.
Of note, the genetic interactions revealed reciprocal suppression of phenotypes for hiw, jip1, and p38: not only did mutations in either jip1 or p38 suppress the hiw synaptic overgrowth phenotype (Fig. 6A,B), mutations in hiw also suppressed the enlarged bouton (Fig. 6A,C) and Futsch unbundling (data not shown) phenotypes of jip1 and p38 mutants. Similarly, overexpression of Wnd also suppressed the bouton morphology phenotype of jip1 mutants (Fig. 6D). This suppression interaction is consistent with the hypothesized role of JIP1 as a scaffold for Wnd signaling: its function in assisting the activation of Wnd can be overcome if Wnd levels are increased. However, the suppression of p38b by hiw suggests that the relationships are more complex than a simple linear pathway. One possibility is that, when Wnd levels are high, then p38a can substitute for p38b. Overall, these genetic interactions suggest that Wnd, JIP1, and p38 function together to regulate synaptic morphology.
Previous studies of Hiw indicate that the synaptic overgrowth phenotype is mediated by a Wnd-regulated nuclear signaling cascade, which is overactive in hiw mutants. This leads to a strong induction of the puc–lacZ reporter (Xiong et al., 2010; Fig. 6E), which also becomes induced after axonal injury (Xiong et al., 2010; Fig. 1D). In both injured neurons and hiw mutants, the induction of puc–lacZ is mediated by Wnd (Xiong et al., 2010). Although jip1 is not required for the induction of puc–lacZ after injury (Fig. 1D), we found that it is partially required for the induction of puc–lacZ in hiw mutants (Fig. 6E). The differences in requirement for JIP1 suggest that there may be multiple mechanisms for activating Wnd in neurons. Axonal injury activates Wnd signaling through a mechanism that does not require JIP1 (Fig. 1D). In contrast, in uninjured neurons, Wnd regulates a signaling pathway that controls the structure of synaptic boutons, and this pathway is significantly diminished in the absence of JIP1 (Fig. 6E).
Wnd regulates synaptic structure and injury responses through independent signaling mechanisms
Our findings support the model that Wnd regulates multiple functions in neurons via independent downstream signaling pathways. To further test this model, we asked whether p38a and p38b, which are required for the synaptic roles of Wnd (Fig. 6), are required for axonal transport, injury signaling, and axonal regeneration after injury. In contrast to wnd, p38aΔ and p38bΔ, as well as p38aΔ, p38bΔ double mutants, displayed only mild defects in axonal transport (Fig. 7A). Also in contrast to Wnd, neither p38a nor p38b are required for the induction of puc–lacZ after injury (Fig. 7B). Similarly, mutations in p38a or p38b did not impair the ability of injured axons to form new axonal branches after injury (Fig. 7C,D). This contrasts to the essential roles for Wnd and JNK in controlling this regenerative sprouting response to injury (Xiong et al., 2010). These findings imply that different downstream functions of Wnd depend on different and functionally separable downstream mechanisms.
Figure 7.
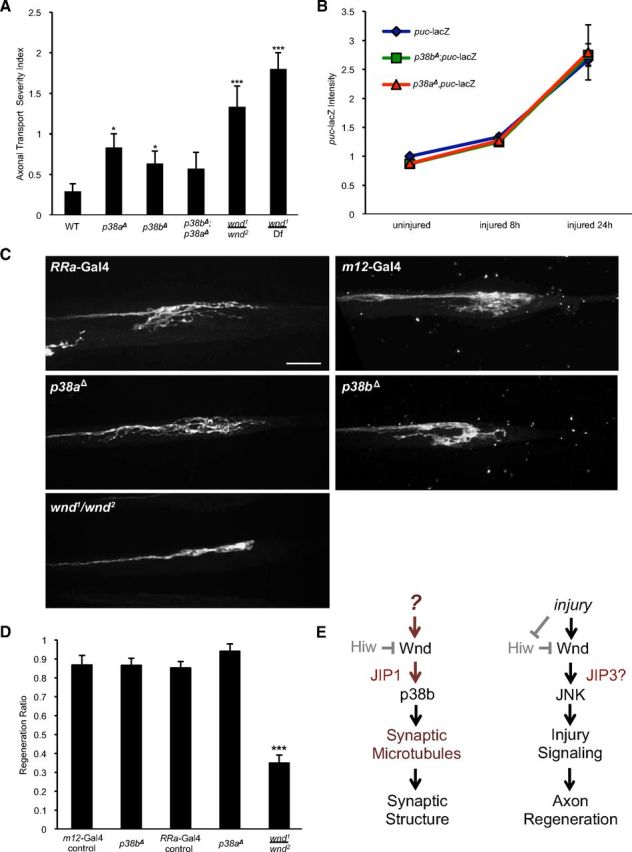
Different downstream actions of Wnd require different downstream signaling components. A, Quantification of axonal transport severity index. p38a and p38b null mutants display very minor, but statistically significant, defects in axonal transport. B, Quantification of puc–lacZ intensity after injury. Both p38a and p38b mutants show increased levels of puc–lacZ intensity similar to controls. C, Axons are labeled by driving expression of UAS–mCD8–RFP with RRa(eve)–Gal4 or UAS–mCD8–GFP with m12–Gal4. In WT animals, the proximal stump forms extensive new branches by 15 h after injury. Sprouting after injury is inhibited in a wnd mutant background (wnd1/wnd2). New sprouting forms at the proximal stumps in both p38b and p38a mutants similar to control axons. D, Quantification of regeneration ratio 15 h after injury. The fraction of injured axons that displayed sprouting was measured while blinded to the genotype. E, Model: Wnd promotes the development of synaptic structure and injury signaling through separate pathways that differ in their requirement for p38b and the JIP1 scaffolding protein. Error bars indicate mean ± SEM. *p ≤ 0.01, ***p ≤ 0.0001. Scale bar, 10 μm.
Discussion
Requirement for the JIP1 scaffold reveals independent pathways downstream of Wnd
Wnd/DLK signaling regulates multiple processes in neurons, including axonal transport, neuronal migration, developmental apoptosis, axonal regeneration, axonal degeneration, and cell death after injury (Hirai et al., 2006; Bloom et al., 2007; Horiuchi et al., 2007; Chen et al., 2008; Hammarlund et al., 2009; Miller et al., 2009; Yan et al., 2009; Xiong et al., 2010; Ghosh et al., 2011; Itoh et al., 2011; Xiong and Collins, 2012; Watkins et al., 2013; Welsbie et al., 2013). Our data suggest the existence of a new role for Wnd in regulating the structure of synaptic microtubules. A major question is how a single MAPKKK can achieve these different and often dichotomous functions in both neuronal development and maintenance. We find that the role of Wnd in synaptic development can be functionally separated from its role in responding to axonal injury. The regulation by Wnd of synaptic development requires both the scaffolding protein JIP1 as well as the downstream MAPK p38. In contrast, p38 is not required for the role of Wnd in injury signaling and the promotion of new axonal growth after injury. Therefore, in Drosophila motoneurons, Wnd regulates at least two independent pathways: (1) one that promotes responses to axonal damage and (2) another that regulates synaptic morphology in uninjured neurons. JIP1 plays an essential role in the second pathway but not the first (Fig. 7E).
It is intriguing to note that jip3/Syd mutants have a complementary phenotype to jip1 mutants: JIP3 is required for injury signaling but not for regulating synaptic microtubules. The model that JIP3 scaffolds the injury signaling pathway is supported by studies of vertebrate JIP3/Syd, which interacts with phosphorylated JNK in axons, and is retrogradely transported in response to axonal injury (Cavalli et al., 2005). Therefore, the JIP1 and JIP3 scaffolds can mediate independent roles for Wnd through distinct downstream signaling mechanisms.
Wnd/DLK regulates synaptic microtubules via p38
A number of studies of Wnd/DLK homologs in both C. elegans and vertebrate neurons suggest that this kinase may regulate the microtubule cytoskeleton via both JNK (Hirai et al., 2002; Eto et al., 2010; Hirai et al., 2011) and p38 (Lewcock et al., 2007; Bounoutas et al., 2011; Ghosh-Roy et al., 2012) signaling, both of which are known to have microtubule-associated substrates, including Tau, MAP1B, MAP2B, and stathmin (Gelderblom et al., 2004; Corrêa and Eales, 2012). Moreover, the JIP1 scaffold is known to play an important role in the regulation of microtubules by MAPK signaling (Goedert et al., 1997; Reynolds et al., 1997a,b; Chang et al., 2003; Gdalyahu et al., 2004; Tararuk et al., 2006). Although the direct downstream effectors of Wnd/DLKs actions on synaptic microtubules remains to be fully characterized, important functional consequences may include the facilitation of axon formation during the early stages of neuronal polarization (Eto et al., 2010; Hirai et al., 2011), regulation of a transcriptional response to depolymerized microtubules (Bounoutas et al., 2011), and regulation of microtubule dynamics within injured axons (Ghosh-Roy et al., 2012), which are important for an injured axon to initiate regenerative growth (Gordon-Weeks, 2004; Erez et al., 2007; Stone et al., 2010; Chen et al., 2012; Hur et al., 2012).
In C. elegans, p38 appears to play a role in all the known functions of DLK, including both synapse formation (Nakata et al., 2005; Grill et al., 2007) and regeneration after injury (Hammarlund et al., 2009; Yan et al., 2009; Nix et al., 2011). In contrast, we observed that, in Drosophila, p38b mediates a synaptic role for Wnd, but p38a and p38b are not required for injury signaling and axonal sprouting after injury. Although downstream signaling pathways and the mechanisms of activation may diverge in evolution, we acknowledge that the assay for the requirement of p38 in axonal regeneration is more stringent in C. elegans than in our sprouting assay after nerve crush in Drosophila, because the sprouting axons in Drosophila nerves fail to reach their final target (Xiong et al., 2010). Therefore, it remains possible that p38 will be required for steps in axonal regeneration that could not be addressed in our current assay. The puc–lacZ induction likely reports a specific aspect of Wnd pathway activation, and this is useful for teasing apart multiple downstream events.
In C. elegans, an additional MAPKKK, mixed-lineage kinase-1 (MLK-1), functions in parallel to DLK to promote axonal regeneration (Nix et al., 2011). The Drosophila homolog of MLK-1, Slpr (Stronach and Perrimon, 2002; Sathyanarayana et al., 2003), is not required for the induction of puc–lacZ after injury (Xiong et al., 2010), but this does not rule out other potential functions for Slpr in neurons. Because the synaptic phenotype of jip1 mutants is more severe than the phenotype of wnd mutants, a potential role for additional MAPK regulators, such as Slpr, in the regulation of synaptic microtubules should also be considered.
Separating roles in signaling from roles in axonal transport
The JIP scaffolding proteins interact with both the kinesin-I and dynein motors (Verhey et al., 2001; Cavalli et al., 2005; Horiuchi et al., 2005; Kelkar et al., 2005; Nguyen et al., 2005; Arimoto et al., 2011; Sun et al., 2011) and may play a role in mediating the regulation of these motors by MAPK signaling (Morfini et al., 2006, 2009; Stagi et al., 2006; Horiuchi et al., 2007). Indeed, loss-of-function studies of JIP1/APLIP1 and JIP3/Syd suggest that both play roles in axonal transport (Bowman et al., 2000; Byrd et al., 2001; Taru et al., 2002; Horiuchi et al., 2005). Because JIPs are physically carried by kinesin and dynein motors, the converse relationship may also be true: motor proteins may regulate the signaling complexes that are scaffolded by JIPs, by delivering the signaling complexes to specific subcellular locations. This appears to be the case for Wnd signaling, because the downstream cascades for both injury signaling and synaptic growth appear to depend on functional axonal transport machinery. The localization of JIP1 to the axon terminus requires Kinesin-1 (Verhey et al., 2001; Reed et al., 2006), and we propose that this localization mediates a specific role for Wnd signaling at the synapse. Conversely, the interaction of JIP3/Syd with dynein is thought to mediate retrograde signaling in response to axonal injury (Cavalli et al., 2005).
An essential role for the axonal transport machinery within neurons makes it difficult to delineate the precise function for any individual molecule involved in this process. A mutant that exhibits axonal transport defects may affect multiple signaling pathways, which may rely either directly or indirectly on the axonal transport machinery. Therefore, it is remarkable that the jip1 mutants exhibit such a specific synaptic phenotype given their axonal transport impairment. Of the many other known mutations in Drosophila that inhibit axonal transport in motoneurons, including subunits of kinesin-1, kinesin-3, dynactin, and dynein, as well as jip3/Syd, none display the enlarged bouton phenotype observed for jip1 mutants (Fig. 2A,B; Hurd and Saxton, 1996; Eaton et al., 2002; Pack-Chung et al., 2007). We were further able to dissociate a role for JIP1 signaling in synaptic development from axonal transport, because the enlarged bouton phenotype of jip1 mutants could be suppressed independently of the axonal transport defect. Although we expect that the roles of JIP1 in both axonal transport and synaptic development are intimately linked, they can nevertheless be genetically separated.
A role for Wnd/DLK in uninjured synapses
Our studies of JIP1 have led to the discovery of a new role for Wnd signaling in regulating synaptic development via JIP1 and p38b. Previous studies in Drosophila and C. elegans have failed to detect such a function for Wnd/DLK. Instead, the previously described synaptic phenotypes were gain-of-function because of the loss of regulation by the Hiw ubiquitin ligase. Since the discovery of the role of Wnd/DLK in axonal regeneration (Hammarlund et al., 2009; Yan et al., 2009; Ghosh-Roy et al., 2010; Xiong et al., 2010; Nix et al., 2011; Shin et al., 2012; Xiong and Collins, 2012), it has been hypothesized that its main function in neurons is to detect axonal injury. The current data now suggest otherwise. JIP1 promotes the activation of a signaling cascade that specifically regulates the structure of presynaptic boutons. This further suggests that Wnd becomes activated in uninjured neurons by unknown upstream factors. Because Wnd and JIP1 can localize to presynaptic boutons (Verhey et al., 2001; Muresan and Muresan, 2005; Collins et al., 2006; data not shown), they may potentially act locally to regulate presynaptic events during synaptic development and/or plasticity. Consistent with a synaptic function for Wnd, recent behavioral studies of hiw mutants imply that the regulation of Wnd in mushroom body neurons is important for constraining the formation of long-term memories (Huang et al., 2012). An important future direction will be to identify the mechanisms that mediate and regulate the function of Wnd/JIP1/p38b signaling at synapses.
Notes
Supplemental material for this article is available at http://labs.mcdb.lsa.umich.edu/labs/collins/files/SupplementaryFigures.pdf. Figure 1 shows the quantification of the number of boutons >5 μm for different alleles of jip1. Figure 2 shows the quantification of the number of boutons >5 μm for different alleles of wnd, p38a, and p38b. This material has not been peer reviewed.
Footnotes
This work was supported by National Science Foundation Grant IOS-0842701 and National Institutes of Health Grant NS069844. We thank Pavan Bhat, Ronny Ewanek, Mary Sprader, Jennifer Diep, Travis Washington, Nicolette Ognjanovski, Dhwani Joshi, and Dayna Menken for technical assistance and Aaron DiAntonio for logistical support in the early stages of this project. We thank Bill Saxton and Subhabrata Sanyal for Drosophila lines. Additionally, we thank the Bloomington Stock Center (Indiana University), the Vienna Drosophila RNAi Center, and the Developmental Studies Hybridoma Bank (University of Iowa).
The authors declare no competing financial interests.
References
- Abe N, Cavalli V. Nerve injury signaling. Curr Opin Neurobiol. 2008;18:276–283. doi: 10.1016/j.conb.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhães TR, Goodman CS. wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron. 2002;33:545–558. doi: 10.1016/S0896-6273(02)00589-5. [DOI] [PubMed] [Google Scholar]
- Adachi-Yamada T, Nakamura M, Irie K, Tomoyasu Y, Sano Y, Mori E, Goto S, Ueno N, Nishida Y, Matsumoto K. p38 mitogen-activated protein kinase can be involved in transforming growth factor beta superfamily signal transduction in Drosophila wing morphogenesis. Mol Cell Biol. 1999;19:2322–2329. doi: 10.1128/mcb.19.3.2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimoto M, Koushika SP, Choudhary BC, Li C, Matsumoto K, Hisamoto N. The Caenorhabditis elegans JIP3 protein UNC-16 functions as an adaptor to link kinesin-1 with cytoplasmic dynein. J Neurosci. 2011;31:2216–2224. doi: 10.1523/JNEUROSCI.2653-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom AJ, Miller BR, Sanes JR, DiAntonio A. The requirement for Phr1 in CNS axon tract formation reveals the corticostriatal boundary as a choice point for cortical axons. Genes Dev. 2007;21:2593–2606. doi: 10.1101/gad.1592107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bounoutas A, Kratz J, Emtage L, Ma C, Nguyen KC, Chalfie M. Microtubule depolymerization in Caenorhabditis elegans touch receptor neurons reduces gene expression through a p38 MAPK pathway. Proc Natl Acad Sci U S A. 2011;108:3982–3987. doi: 10.1073/pnas.1101360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman AB, Kamal A, Ritchings BW, Philp AV, McGrail M, Gindhart JG, Goldstein LS. Kinesin-dependent axonal transport is mediated by the sunday driver (SYD) protein. Cell. 2000;103:583–594. doi: 10.1016/S0092-8674(00)00162-8. [DOI] [PubMed] [Google Scholar]
- Buchsbaum RJ, Connolly BA, Feig LA. Interaction of Rac exchange factors Tiam1 and Ras-GRF1 with a scaffold for the p38 mitogen-activated protein kinase cascade. Mol Cell Biol. 2002;22:4073–4085. doi: 10.1128/MCB.22.12.4073-4085.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik V, Koh YH, Guan B, Hartmann B, Hough C, Woods D, Gorczyca M. Regulation of synapse structure and function by the Drosophila tumor suppressor gene dlg. Neuron. 1996;17:627–640. doi: 10.1016/S0896-6273(00)80196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd DT, Kawasaki M, Walcoff M, Hisamoto N, Matsumoto K, Jin Y. UNC-16, a JNK-signaling scaffold protein, regulates vesicle transport in C. elegans. Neuron. 2001;32:787–800. doi: 10.1016/S0896-6273(01)00532-3. [DOI] [PubMed] [Google Scholar]
- Cavalli V, Kujala P, Klumperman J, Goldstein LS. Sunday Driver links axonal transport to damage signaling. J Cell Biol. 2005;168:775–787. doi: 10.1083/jcb.200410136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Jones Y, Ellisman MH, Goldstein LS, Karin M. JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Dev Cell. 2003;4:521–533. doi: 10.1016/S1534-5807(03)00094-7. [DOI] [PubMed] [Google Scholar]
- Chen L, Stone MC, Tao J, Rolls MM. Axon injury and stress trigger a microtubule-based neuroprotective pathway. Proc Natl Acad Sci U S A. 2012;109:11842–11847. doi: 10.1073/pnas.1121180109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Rzhetskaya M, Kareva T, Bland R, During MJ, Tank AW, Kholodilov N, Burke RE. Antiapoptotic and trophic effects of dominant-negative forms of dual leucine zipper kinase in dopamine neurons of the substantia nigra in vivo. J Neurosci. 2008;28:672–680. doi: 10.1523/JNEUROSCI.2132-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YR, Tan TH. The c-Jun N-terminal kinase pathway and apoptotic signaling (review) Int J Oncol. 2000;16:651–662. doi: 10.3892/ijo.16.4.651. [DOI] [PubMed] [Google Scholar]
- Collins CA, Wairkar YP, Johnson SL, DiAntonio A. Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron. 2006;51:57–69. doi: 10.1016/j.neuron.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Conde C, Cáceres A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 2009;10:319–332. doi: 10.1038/nrn2631. [DOI] [PubMed] [Google Scholar]
- Corrêa SA, Eales KL. The role of p38 MAPK and its substrates in neuronal plasticity and neurodegenerative disease. J Signal Transduct. 2012;2012:649079. doi: 10.1155/2012/649079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig CR, Fink JL, Yagi Y, Ip YT, Cagan RL. A Drosophila p38 orthologue is required for environmental stress responses. EMBO Rep. 2004;5:1058–1063. doi: 10.1038/sj.embor.7400282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cully M, Genevet A, Warne P, Treins C, Liu T, Bastien J, Baum B, Tapon N, Leevers SJ, Downward J. A role for p38 stress-activated protein kinase in regulation of cell growth via TORC1. Mol Cell Biol. 2010;30:481–495. doi: 10.1128/MCB.00688-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, Krantz DE, DiAntonio A. Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J Neurosci. 2004;24:10466–10474. doi: 10.1523/JNEUROSCI.3001-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MM, Primrose DA, Hodgetts RB. A member of the p38 mitogen-activated protein kinase family is responsible for transcriptional induction of Dopa decarboxylase in the epidermis of Drosophila melanogaster during the innate immune response. Mol Cell Biol. 2008;28:4883–4895. doi: 10.1128/MCB.02074-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanasekaran DN, Kashef K, Lee CM, Xu H, Reddy EP. Scaffold proteins of MAP-kinase modules. Oncogene. 2007;26:3185–3202. doi: 10.1038/sj.onc.1210411. [DOI] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, Couto A, Marra V, Keleman K, Dickson BJ. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- Eaton BA, Fetter RD, Davis GW. Dynactin is necessary for synapse stabilization. Neuron. 2002;34:729–741. doi: 10.1016/S0896-6273(02)00721-3. [DOI] [PubMed] [Google Scholar]
- Eresh S, Riese J, Jackson DB, Bohmann D, Bienz M. A CREB-binding site as a target for decapentaplegic signalling during Drosophila endoderm induction. EMBO J. 1997;16:2014–2022. doi: 10.1093/emboj/16.8.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erez H, Malkinson G, Prager-Khoutorsky M, De Zeeuw CI, Hoogenraad CC, Spira ME. Formation of microtubule-based traps controls the sorting and concentration of vesicles to restricted sites of regenerating neurons after axotomy. J Cell Biol. 2007;176:497–507. doi: 10.1083/jcb.200607098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto K, Kawauchi T, Osawa M, Tabata H, Nakajima K. Role of dual leucine zipper-bearing kinase (DLK/MUK/ZPK) in axonal growth. Neurosci Res. 2010;66:37–45. doi: 10.1016/j.neures.2009.09.1708. [DOI] [PubMed] [Google Scholar]
- Fan G, Merritt SE, Kortenjann M, Shaw PE, Holzman LB. Dual leucine zipper-bearing kinase (DLK) activates p46SAPK and p38mapk but not ERK2. J Biol Chem. 1996;271:24788–24793. doi: 10.1074/jbc.271.40.24788. [DOI] [PubMed] [Google Scholar]
- Fujioka M, Lear BC, Landgraf M, Yusibova GL, Zhou J, Riley KM, Patel NH, Jaynes JB. Even-skipped, acting as a repressor, regulates axonal projections in Drosophila. Development. 2003;130:5385–5400. doi: 10.1242/dev.00770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gdalyahu A, Ghosh I, Levy T, Sapir T, Sapoznik S, Fishler Y, Azoulai D, Reiner O. DCX, a new mediator of the JNK pathway. EMBO J. 2004;23:823–832. doi: 10.1038/sj.emboj.7600079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelderblom M, Eminel S, Herdegen T, Waetzig V. c-Jun N-terminal kinases (JNKs) and the cytoskeleton—functions beyond neurodegeneration. Int J Dev Neurosci. 2004;22:559–564. doi: 10.1016/j.ijdevneu.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Ghosh AS, Wang B, Pozniak CD, Chen M, Watts RJ, Lewcock JW. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J Cell Biol. 2011;194:751–764. doi: 10.1083/jcb.201103153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh-Roy A, Wu Z, Goncharov A, Jin Y, Chisholm AD. Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J Neurosci. 2010;30:3175–3183. doi: 10.1523/JNEUROSCI.5464-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh-Roy A, Goncharov A, Jin Y, Chisholm AD. Kinesin-13 and tubulin posttranslational modifications regulate microtubule growth in axon regeneration. Dev Cell. 2012;23:716–728. doi: 10.1016/j.devcel.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Hasegawa M, Jakes R, Lawler S, Cuenda A, Cohen P. Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases. FEBS Lett. 1997;409:57–62. doi: 10.1016/S0014-5793(97)00483-3. [DOI] [PubMed] [Google Scholar]
- Goellner B, Aberle H. The synaptic cytoskeleton in development and disease. Dev Neurobiol. 2012;72:111–125. doi: 10.1002/dneu.20892. [DOI] [PubMed] [Google Scholar]
- Gordon-Weeks PR. Microtubules and growth cone function. J Neurobiol. 2004;58:70–83. doi: 10.1002/neu.10266. [DOI] [PubMed] [Google Scholar]
- Grill B, Bienvenut WV, Brown HM, Ackley BD, Quadroni M, Jin Y. C. elegans RPM-1 regulates axon termination and synaptogenesis through the Rab GEF GLO-4 and the Rab GTPase GLO-1. Neuron. 2007;55:587–601. doi: 10.1016/j.neuron.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Ha EM, Lee KA, Seo YY, Kim SH, Lim JH, Oh BH, Kim J, Lee WJ. Coordination of multiple dual oxidase-regulatory pathways in responses to commensal and infectious microbes in Drosophila gut. Nat Immunol. 2009;10:949–957. doi: 10.1038/ni.1765. [DOI] [PubMed] [Google Scholar]
- Hammarlund M, Nix P, Hauth L, Jorgensen EM, Bastiani M. Axon regeneration requires a conserved MAP kinase pathway. Science. 2009;323:802–806. doi: 10.1126/science.1165527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SJ, Choi KY, Brey PT, Lee WJ. Molecular cloning and characterization of a Drosophila p38 mitogen-activated protein kinase. J Biol Chem. 1998;273:369–374. doi: 10.1074/jbc.273.1.369. [DOI] [PubMed] [Google Scholar]
- Han ZS, Ip YT. Interaction and specificity of Rel-related proteins in regulating Drosophila immunity gene expression. J Biol Chem. 1999;274:21355–21361. doi: 10.1074/jbc.274.30.21355. [DOI] [PubMed] [Google Scholar]
- Hirai S, Kawaguchi A, Hirasawa R, Baba M, Ohnishi T, Ohno S. MAPK-upstream protein kinase (MUK) regulates the radial migration of immature neurons in telencephalon of mouse embryo. Development. 2002;129:4483–4495. doi: 10.1242/dev.129.19.4483. [DOI] [PubMed] [Google Scholar]
- Hirai S, Cui de F, Miyata T, Ogawa M, Kiyonari H, Suda Y, Aizawa S, Banba Y, Ohno S. The c-Jun N-terminal kinase activator dual leucine zipper kinase regulates axon growth and neuronal migration in the developing cerebral cortex. J Neurosci. 2006;26:11992–12002. doi: 10.1523/JNEUROSCI.2272-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai S, Banba Y, Satake T, Ohno S. Axon formation in neocortical neurons depends on stage-specific regulation of microtubule stability by the dual leucine zipper kinase-c-Jun N-terminal kinase pathway. J Neurosci. 2011;31:6468–6480. doi: 10.1523/JNEUROSCI.5038-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi D, Barkus RV, Pilling AD, Gassman A, Saxton WM. APLIP1, a kinesin binding JIP-1/JNK scaffold protein, influences the axonal transport of both vesicles and mitochondria in Drosophila. Curr Biol. 2005;15:2137–2141. doi: 10.1016/j.cub.2005.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi D, Collins CA, Bhat P, Barkus RV, Diantonio A, Saxton WM. Control of a kinesin-cargo linkage mechanism by JNK pathway kinases. Curr Biol. 2007;17:1313–1317. doi: 10.1016/j.cub.2007.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Zheng X, Zhao H, Li M, Wang P, Xie Z, Wang L, Zhong Y. A permissive role of mushroom body alpha/beta core neurons in long-term memory consolidation in Drosophila. Curr Biol. 2012;22:1981–1989. doi: 10.1016/j.cub.2012.08.048. [DOI] [PubMed] [Google Scholar]
- Hummel T, Krukkert K, Roos J, Davis G, Klämbt C. Drosophila Futsch/22C10 is a MAP1B-like protein required for dendritic and axonal development. Neuron. 2000;26:357–370. doi: 10.1016/S0896-6273(00)81169-1. [DOI] [PubMed] [Google Scholar]
- Hur EM, Saijilafu, Zhou FQ. Growing the growth cone: remodeling the cytoskeleton to promote axon regeneration. Trends Neurosci. 2012;35:164–174. doi: 10.1016/j.tins.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd DD, Saxton WM. Kinesin mutations cause motor neuron disease phenotypes by disrupting fast axonal transport in Drosophila. Genetics. 1996;144:1075–1085. doi: 10.1093/genetics/144.3.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Tateno M, Fujimura-Kamada K, Takaesu G, Adachi-Yamada T, Ninomiya-Tsuji J, Irie K, Nishida Y, Matsumoto K. A Drosophila MAPKKK, D-MEKK1, mediates stress responses through activation of p38 MAPK. EMBO J. 2001;20:5421–5430. doi: 10.1093/emboj/20.19.5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh A, Horiuchi M, Wakayama K, Xu J, Bannerman P, Pleasure D, Itoh T. ZPK/DLK, a mitogen-activated protein kinase kinase kinase, is a critical mediator of programmed cell death of motoneurons. J Neurosci. 2011;31:7223–7228. doi: 10.1523/JNEUROSCI.5947-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Kneussel M. Tubulin post-translational modifications: encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 2010;33:362–372. doi: 10.1016/j.tins.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Jin Y, Garner CC. Molecular mechanisms of presynaptic differentiation. Annu Rev Cell Dev Biol. 2008;24:237–262. doi: 10.1146/annurev.cellbio.23.090506.123417. [DOI] [PubMed] [Google Scholar]
- Kelkar N, Standen CL, Davis RJ. Role of the JIP4 scaffold protein in the regulation of mitogen-activated protein kinase signaling pathways. Mol Cell Biol. 2005;25:2733–2743. doi: 10.1128/MCB.25.7.2733-2743.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koushika SP. “JIP”ing along the axon: the complex roles of JIPs in axonal transport. Bioessays. 2008;30:10–14. doi: 10.1002/bies.20695. [DOI] [PubMed] [Google Scholar]
- Lee CM, Onésime D, Reddy CD, Dhanasekaran N, Reddy EP. JLP: a scaffolding protein that tethers JNK/p38MAPK signaling modules and transcription factors. Proc Natl Acad Sci U S A. 2002;99:14189–14194. doi: 10.1073/pnas.232310199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewcock JW, Genoud N, Lettieri K, Pfaff SL. The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron. 2007;56:604–620. doi: 10.1016/j.neuron.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Martín-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM, Martinez-Arias A. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 1998;12:557–570. doi: 10.1101/gad.12.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, DiAntonio A. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat Neurosci. 2009;12:387–389. doi: 10.1038/nn.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini GA, You YM, Pollema SL, Kaminska A, Liu K, Yoshioka K, Björkblom B, Coffey ET, Bagnato C, Han D, Huang CF, Banker G, Pigino G, Brady ST. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat Neurosci. 2009;12:864–871. doi: 10.1038/nn.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Szebenyi G, You Y, Pollema S, Brady ST. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat Neurosci. 2006;9:907–916. doi: 10.1038/nn1717. [DOI] [PubMed] [Google Scholar]
- Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- Muresan Z, Muresan V. Coordinated transport of phosphorylated amyloid-beta precursor protein and c-Jun NH2-terminal kinase-interacting protein-1. J Cell Biol. 2005;171:615–625. doi: 10.1083/jcb.200502043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata K, Abrams B, Grill B, Goncharov A, Huang X, Chisholm AD, Jin Y. Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell. 2005;120:407–420. doi: 10.1016/j.cell.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Nguyen Q, Lee CM, Le A, Reddy EP. JLP associates with kinesin light chain 1 through a novel leucine zipper-like domain. J Biol Chem. 2005;280:30185–30191. doi: 10.1074/jbc.M505499200. [DOI] [PubMed] [Google Scholar]
- Nihalani D, Meyer D, Pajni S, Holzman LB. Mixed lineage kinase-dependent JNK activation is governed by interactions of scaffold protein JIP with MAPK module components. EMBO J. 2001;20:3447–3458. doi: 10.1093/emboj/20.13.3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nix P, Hisamoto N, Matsumoto K, Bastiani M. Axon regeneration requires coordinate activation of p38 and JNK MAPK pathways. Proc Natl Acad Sci U S A. 2011;108:10738–10743. doi: 10.1073/pnas.1104830108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterwalder T, Yoon KS, White BH, Keshishian H. A conditional tissue-specific transgene expression system using inducible GAL4. Proc Natl Acad Sci U S A. 2001;98:12596–12601. doi: 10.1073/pnas.221303298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pack-Chung E, Kurshan PT, Dickman DK, Schwarz TL. A Drosophila kinesin required for synaptic bouton formation and synaptic vesicle transport. Nat Neurosci. 2007;10:980–989. doi: 10.1038/nn1936. [DOI] [PubMed] [Google Scholar]
- Park JS, Kim YS, Yoo MA. The role of p38b MAPK in age-related modulation of intestinal stem cell proliferation and differentiation in Drosophila. Aging. 2009;1:637–651. doi: 10.18632/aging.100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed NA, Cai D, Blasius TL, Jih GT, Meyhofer E, Gaertig J, Verhey KJ. Microtubule acetylation promotes kinesin-1 binding and transport. Curr Biol. 2006;16:2166–2172. doi: 10.1016/j.cub.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Reynolds CH, Utton MA, Gibb GM, Yates A, Anderton BH. Stress-activated protein kinase/c-jun N-terminal kinase phosphorylates tau protein. J Neurochem. 1997a;68:1736–1744. doi: 10.1046/j.1471-4159.1997.68041736.x. [DOI] [PubMed] [Google Scholar]
- Reynolds CH, Nebreda AR, Gibb GM, Utton MA, Anderton BH. Reactivating kinase/p38 phosphorylates tau protein in vitro. J Neurochem. 1997b;69:191–198. doi: 10.1046/j.1471-4159.1997.69010191.x. [DOI] [PubMed] [Google Scholar]
- Ritzenthaler S, Suzuki E, Chiba A. Postsynaptic filopodia in muscle cells interact with innervating motoneuron axons. Nat Neurosci. 2000;3:1012–1017. doi: 10.1038/79833. [DOI] [PubMed] [Google Scholar]
- Roos J, Hummel T, Ng N, Klämbt C, Davis GW. Drosophila Futsch regulates synaptic microtubule organization and is necessary for synaptic growth. Neuron. 2000;26:371–382. doi: 10.1016/S0896-6273(00)81170-8. [DOI] [PubMed] [Google Scholar]
- Ruiz-Cañada C, Budnik V. Synaptic cytoskeleton at the neuromuscular junction. Int Rev Neurobiol. 2006;75:217–236. doi: 10.1016/S0074-7742(06)75010-3. [DOI] [PubMed] [Google Scholar]
- Sano Y, Akimaru H, Okamura T, Nagao T, Okada M, Ishii S. Drosophila activating transcription factor-2 is involved in stress response via activation by p38, but not c-Jun NH(2)-terminal kinase. Mol Biol Cell. 2005;16:2934–2946. doi: 10.1091/mbc.E04-11-1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathyanarayana P, Barthwal MK, Lane ME, Acevedo SF, Skoulakis EM, Bergmann A, Rana A. Drosophila mixed lineage kinase/slipper, a missing biochemical link in Drosophila JNK signaling. Biochim Biophys Acta. 2003;1640:77–84. doi: 10.1016/S0167-4889(03)00022-3. [DOI] [PubMed] [Google Scholar]
- Schaefer AM, Hadwiger GD, Nonet ML. rpm-1, a conserved neuronal gene that regulates targeting and synaptogenesis in C. elegans. Neuron. 2000;26:345–356. doi: 10.1016/S0896-6273(00)81168-X. [DOI] [PubMed] [Google Scholar]
- Schoorlemmer J, Goldfarb M. Fibroblast growth factor homologous factors are intracellular signaling proteins. Curr Biol. 2001;11:793–797. doi: 10.1016/S0960-9822(01)00232-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Cho Y, Beirowski B, Milbrandt J, Cavalli V, DiAntonio A. Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron. 2012;74:1015–1022. doi: 10.1016/j.neuron.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinzawa N, Nelson B, Aonuma H, Okado K, Fukumoto S, Miura M, Kanuka H. p38 MAPK-dependent phagocytic encapsulation confers infection tolerance in Drosophila. Cell Host Microbe. 2009;6:244–252. doi: 10.1016/j.chom.2009.07.010. [DOI] [PubMed] [Google Scholar]
- Stagi M, Gorlovoy P, Larionov S, Takahashi K, Neumann H. Unloading kinesin transported cargoes from the tubulin track via the inflammatory c-Jun N-terminal kinase pathway. FASEB J. 2006;20:2573–2575. doi: 10.1096/fj.06-6679fje. [DOI] [PubMed] [Google Scholar]
- Stone MC, Nguyen MM, Tao J, Allender DL, Rolls MM. Global up-regulation of microtubule dynamics and polarity reversal during regeneration of an axon from a dendrite. Mol Biol Cell. 2010;21:767–777. doi: 10.1091/mbc.E09-11-0967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stronach B, Perrimon N. Activation of the JNK pathway during dorsal closure in Drosophila requires the mixed lineage kinase, slipper. Genes Dev. 2002;16:377–387. doi: 10.1101/gad.953002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Zhu C, Dixit R, Cavalli V. Sunday Driver/JIP3 binds kinesin heavy chain directly and enhances its motility. EMBO J. 2011;30:3416–3429. doi: 10.1038/emboj.2011.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzanne M, Irie K, Glise B, Agnès F, Mori E, Matsumoto K, Noselli S. The Drosophila p38 MAPK pathway is required during oogenesis for egg asymmetric development. Genes Dev. 1999;13:1464–1474. doi: 10.1101/gad.13.11.1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tararuk T, Ostman N, Li W, Björkblom B, Padzik A, Zdrojewska J, Hongisto V, Herdegen T, Konopka W, Courtney MJ, Coffey ET. JNK1 phosphorylation of SCG10 determines microtubule dynamics and axodendritic length. J Cell Biol. 2006;173:265–277. doi: 10.1083/jcb.200511055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taru H, Kirino Y, Suzuki T. Differential roles of JIP scaffold proteins in the modulation of amyloid precursor protein metabolism. J Biol Chem. 2002;277:27567–27574. doi: 10.1074/jbc.M203713200. [DOI] [PubMed] [Google Scholar]
- Tedeschi A, Bradke F. The DLK signalling pathway-a double-edged sword in neural development and regeneration. EMBO Rep. 2013 doi: 10.1038/embor.2013.64. doi: 10.1038/embor.2013.64. Advance online publication. Retrieved May 29, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey KJ, Meyer D, Deehan R, Blenis J, Schnapp BJ, Rapoport TA, Margolis B. Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J Cell Biol. 2001;152:959–970. doi: 10.1083/jcb.152.5.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viquez NM, Li CR, Wairkar YP, DiAntonio A. The B′ protein phosphatase 2A regulatory subunit well-rounded regulates synaptic growth and cytoskeletal stability at the Drosophila neuromuscular junction. J Neurosci. 2006;26:9293–9303. doi: 10.1523/JNEUROSCI.1740-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrailas-Mortimer A, del Rivero T, Mukherjee S, Nag S, Gaitanidis A, Kadas D, Consoulas C, Duttaroy A, Sanyal S. A muscle-specific p38 MAPK/Mef2/MnSOD pathway regulates stress, motor function, and life span in Drosophila. Dev Cell. 2011;21:783–795. doi: 10.1016/j.devcel.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan HI, DiAntonio A, Fetter RD, Bergstrom K, Strauss R, Goodman CS. Highwire regulates synaptic growth in Drosophila. Neuron. 2000;26:313–329. doi: 10.1016/S0896-6273(00)81166-6. [DOI] [PubMed] [Google Scholar]
- Watkins TA, Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, Eastham-Anderson J, Modrusan Z, Kaminker JS, Tessier-Lavigne M, Lewcock JW. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc Natl Acad Sci U S A. 2013;110:4039–4044. doi: 10.1073/pnas.1211074110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber U, Paricio N, Mlodzik M. Jun mediates Frizzled-induced R3/R4 cell fate distinction and planar polarity determination in the Drosophila eye. Development. 2000;127:3619–3629. doi: 10.1242/dev.127.16.3619. [DOI] [PubMed] [Google Scholar]
- Welsbie DS, Yang Z, Ge Y, Mitchell KL, Zhou X, Martin SE, Berlinicke CA, Hackler L, Jr, Fuller J, Fu J, Cao LH, Han B, Auld D, Xue T, Hirai S, Germain L, Simard-Bisson C, Blouin R, Nguyen JV, Davis CH, et al. Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proc Natl Acad Sci U S A. 2013;110:4045–4050. doi: 10.1073/pnas.1211284110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmarsh AJ. The JIP family of MAPK scaffold proteins. Biochem Soc Trans. 2006;34:828–832. doi: 10.1042/BST0340828. [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ, Davis RJ. Structural organization of MAP-kinase signaling modules by scaffold proteins in yeast and mammals. Trends Biochem Sci. 1998;23:481–485. doi: 10.1016/S0968-0004(98)01309-7. [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ, Cavanagh J, Tournier C, Yasuda J, Davis RJ. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science. 1998;281:1671–1674. doi: 10.1126/science.281.5383.1671. [DOI] [PubMed] [Google Scholar]
- Wu C, Wairkar YP, Collins CA, DiAntonio A. Highwire function at the Drosophila neuromuscular junction: spatial, structural, and temporal requirements. J Neurosci. 2005;25:9557–9566. doi: 10.1523/JNEUROSCI.2532-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Collins CA. A conditioning lesion protects axons from degeneration via the Wallenda/DLK MAP kinase signaling cascade. J Neurosci. 2012;32:610–615. doi: 10.1523/JNEUROSCI.3586-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Wang X, Ewanek R, Bhat P, Diantonio A, Collins CA. Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J Cell Biol. 2010;191:211–223. doi: 10.1083/jcb.201006039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Wu Z, Chisholm AD, Jin Y. The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell. 2009;138:1005–1018. doi: 10.1016/j.cell.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen M, Huang X, Bamber B, Jin Y. Regulation of presynaptic terminal organization by C. elegans RPM-1, a putative guanine nucleotide exchanger with a RING-H2 finger domain. Neuron. 2000;26:331–343. doi: 10.1016/S0896-6273(00)81167-8. [DOI] [PubMed] [Google Scholar]
- Zhuang ZH, Zhou Y, Yu MC, Silverman N, Ge BX. Regulation of Drosophila p38 activation by specific MAP2 kinase and MAP3 kinase in response to different stimuli. Cell Signal. 2006;18:441–448. doi: 10.1016/j.cellsig.2005.05.013. [DOI] [PubMed] [Google Scholar]