

Abstract
Tau, a microtubule-associated protein, is implicated in the pathogenesis of Alzheimer's Disease (AD) in regard to both neurofibrillary tangle formation and neuronal network hyperexcitability. The genetic ablation of tau substantially reduces hyperexcitability in AD mouse lines, induced seizure models, and genetic in vivo models of epilepsy. These data demonstrate that tau is an important regulator of network excitability. However, developmental compensation in the genetic tau knock-out line may account for the protective effect against seizures. To test the efficacy of a tau reducing therapy for disorders with a detrimental hyperexcitability profile in adult animals, we identified antisense oligonucleotides that selectively decrease endogenous tau expression throughout the entire mouse CNS—brain and spinal cord tissue, interstitial fluid, and CSF—while having no effect on baseline motor or cognitive behavior. In two chemically induced seizure models, mice with reduced tau protein had less severe seizures than control mice. Total tau protein levels and seizure severity were highly correlated, such that those mice with the most severe seizures also had the highest levels of tau. Our results demonstrate that endogenous tau is integral for regulating neuronal hyperexcitability in adult animals and suggest that an antisense oligonucleotide reduction of tau could benefit those with epilepsy and perhaps other disorders associated with tau-mediated neuronal hyperexcitability.
Introduction
As a member of the microtubule-associated protein family (Weingarten et al., 1975), the protein tau is enriched in axons of mature and growing neurons (Kempf et al., 1996). However, under certain conditions, tau can become hyperphosphorylated and accumulate into oligomeric species and neurofibrillary tangles, resulting in a group of disorders known collectively as tauopathies (Billingsley and Kincaid, 1997; Lovestone and Reynolds, 1997; Buée and Delacourte, 1999) with the most common being Alzheimer's disease (AD).
Although the role of tau in proteinaceous aggregates has long been studied (Iqbal et al., 1975; Brion et al., 1985), a new role has emerged that implicates tau as a regulator of neuronal hyperexcitability. Tau knock-out (tau−/−) mice demonstrate substantially reduced seizure severity in models of chemically induced seizures (Roberson et al., 2007, 2011; Ittner et al., 2010) and genetic models of severe epilepsy (Holth et al., 2013). These data suggest that tau plays a role in neuronal hyperexcitability and provide evidence that a tau-reducing therapy may be beneficial for those with seizure disorders. In addition, amyloid precursor protein overexpression/amyloid-β-depositing mouse lines show increased baseline neuronal hyperexcitability and spontaneous seizures. When placed onto a tau−/− background, these AD mouse models show both decreased seizure frequency and improved learning and memory (Roberson et al., 2007, 2011; Ittner et al., 2010), suggesting that tau-linked neuronal hyperexcitability may be an important component of AD pathophysiology.
However, whether reducing tau levels in an adult animal will modulate neuronal hyperexcitability similar to genetic deletion remains unknown. For example, developmental compensation could contribute to the protective effect of tau−/−, such as the reported increase in microtubule-associated protein 1A (Harada et al., 1994). Here, we tested directly the effect of reducing tau in adult nontransgenic mice by reducing endogenous tau levels and subsequently analyzing the effects on baseline behavior and induced seizure severity. We reduced murine endogenous tau levels using antisense oligonucleotides (ASOs) delivered directly to the CSF (DeVos and Miller, 2013a). Recent data demonstrating safety of CSF-delivered ASOs in humans (Miller et al., 2013) suggests that the strategy used here may be translated into therapy for seizures and possibly other neurodegenerative disorders.
Materials and Methods
Animals.
All ASO-treated mice were C57BL/6J nontransgenic mice ordered directly from The Jackson Laboratory. Tau−/− mice containing a GFP-encoding cDNA integrated into exon 1 of MAPT gene (Tucker et al., 2001) were obtained from The Jackson Laboratory and maintained on a C57BL/6J background. Characterization and behavioral experiments were performed using gender-balanced groups age 2–4 months (Figs. 1,2,3,4,5). Seizure experiments were performed using males age 3–5 months (Figs. 6,7,8). Mice had access to food and water ad libitum and were housed on a 12 h light/dark cycle. All animal protocols were approved by the institutional animal care and use committee at Washington University.
Figure 1.

ASOs reduce tau mRNA and protein. A, Saline, scrambled, or tau ASOs were infused into the right hippocampus of NT mice (n = 4–6 per group) and the area around the injection site was analyzed 1 week later for total tau mRNA levels. TauASO-3 was very potent at reducing total tau mRNA. One-way ANOVA and Bonferroni post hoc analysis were used. B, C, Saline, scrambled, or TauASO-3 was infused intracerebroventricularly (ICV) into NT mice at 100 μg/d for 1 month (n = 4–7). The right parietal cortex was analyzed for total tau mRNA levels (B) and the right ventral white matter was analyzed for total tau protein levels by ELISA (C). One-way ANOVA and Bonferroni post hoc analysis were used. D, Saline or TauASO-3 was delivered via ICV infusion with increasing concentrations of TauASO-3 (n = 3–13). After 1 month, total tau mRNA levels were measured. One-way ANOVA and Bonferroni post hoc analysis were used. E, Saline or TauASO-3 was delivered via ICV infusion at 25 μg/d for 1 month. Total tau mRNA levels were analyzed at 4, 8, 12, and 16 weeks after pump implantation (n = 3–5). Two-way ANOVA and Bonferroni post hoc analysis were used. *p < 0.05; **p < 0.01; ***p < 0.001. Error bars represent SEM.
Figure 2.

ASOs reduce tau mRNA and protein throughout the mouse CNS. A, Saline or TauASO-3 was delivered by intracerebroventricular infusion at 25 μg/d into NT mice for 1 month. In mice collected 8 weeks after pump implantation, brain tissue was costained with a total tau antibody (green), an ASO antibody (red), and counterstained with DAPI (blue). Three brain regions—the hippocampus, frontal cortex, and cerebellum—on the contralateral side of the brain from the catheter (left hemisphere) were analyzed. Scale bars, 500 μm. B–D, Using the same treatment paradigm as in A, brains were dissected into regions as shown in B (n = 4–6). Lateral ventricles are highlighted in blue and the catheter position in yellow. Tau mRNA (C) and protein levels (D) were measured in each region. #p < 0.1, *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed t test. Error bars represent SEM.
Figure 3.

Total tau protein in the brain ISF is decreased. A, Experimental paradigm. NT mice (n = 4–6) were treated with saline or 75 μg/d TauASO-3 via intracerebroventricular infusion for 1 month and the catheters then removed. After 14 d, a guide cannula and microdialysis probe were placed in the left hippocampus. ISF was collected for 48 h. Each ISF fraction comprised a 90 min collection time. B, Total brain tau protein levels in the left hippocampus were measured, confirming that brain tau was reduced. Two-tailed t test was used. C, Total tau protein levels were measured in several ISF fractions. Two-way ANOVA and Bonferroni post hoc analysis were used. D, Fractions from 18–34 h were pooled for each animal to measure total tau protein levels and lactate as a control for probe function. The concentrations were calculated for the 1 μl/min flow rate. There was no significant decrease in lactate levels, demonstrating adequate probe function. Two-tailed t test was used. *p < 0.05; **p < 0.01; ***p < 0.001. Error bars represent SEM.
Figure 4.

CSF tau protein levels are decreased and correlate with brain tau levels. A, B, NT mice were treated with saline or 25 μg/d TauASO-3 via intracerebroventricular infusion for 1 month. At 4 and 8 weeks after pump implantation, total tau protein levels in the brain (n = 4–7; A) and CSF (n = 8–10; B) were measured. One-way ANOVA and Bonferroni post hoc analysis were used. C, D, Total brain tau from the right (C) and left (D) sides of the brain were correlated with CSF tau levels for each mouse (n = 8). The best-fit line and 95% confidence bands were generated using linear regression analysis. #p < 0.1; **p < 0.01; ***p < 0.001. Error bars represent SEM.
Figure 5.
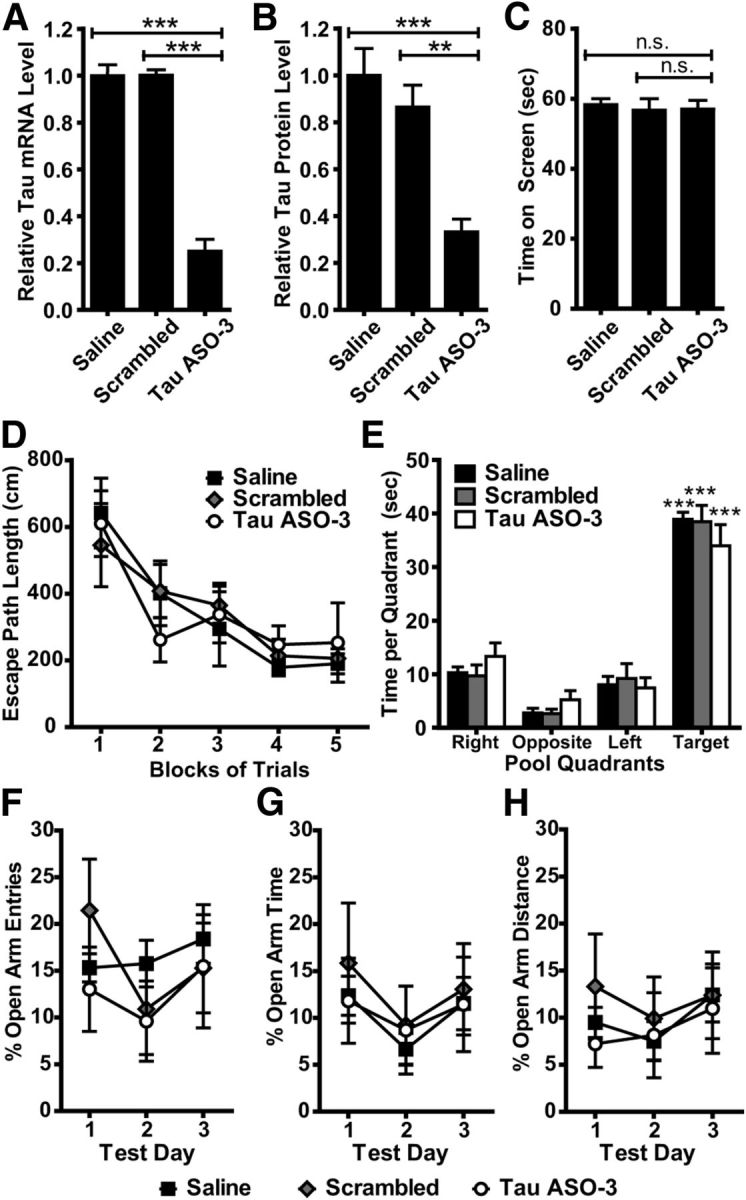
Tau reduction does not alter baseline behavior in vivo. A, B, NT mice (n = 7–8) were treated with saline, 25 μg/d scrambled ASO, or 25 μg/d TauASO-3 via intracerebroventricular infusion for 1 month. Pumps were removed and mice sent for a battery of behavioral tasks lasting a total of 1.5 months. Immediately after behavior testing, brain total tau mRNA (A) and protein (B) levels were measured. One-way ANOVA and Bonferroni post hoc analysis were used. C, All three treatment groups performed similarly on the inverted screen task from the sensorimotor battery. One-way ANOVA and Bonferroni post hoc analysis were used. D, E, No significant differences were observed among the three treatment groups with regard to performance on the place trials in the Morris water maze (D) or spatial bias for the target quadrant during the probe trial (E). Two-way rmANOVA and Bonferroni post hoc analysis were used. F–H The three treatment groups also performed similarly on the EPM in terms of open arm entry percentage (F), percentage time spent in open arms (G), and open arm distance percentage (H), suggesting that tau reduction does not result in anxiety-related behavioral performance deficits. Two-way rmANOVA and Bonferroni post hoc analysis were used. **p < 0.01; ***p < 0.001; n = 7–8. Error bars represent SEM.
Figure 6.

Tau reduction is protective against PTX-induced hyperexcitability in the hippocampus. A, Increasing doses of PTX were infused into the left hippocampus of NT male mice treated with saline, 25 μg/d scrambled ASO, or 12–25 μg/d TauASO-3 and into NT and tau−/− male mice with no catheter. Representative EEG traces from both baseline and 500 μm PTX are shown for NT/saline, scrambled ASO, Tau−/−, and TauASO-3 mice. B, The spike frequency during the last hour of PTX infusion for each dose was calculated and normalized to baseline EEG. Untreated NT and saline-treated mice were combined due to no significant difference between the groups (n = 4–8). Two-way rmANOVA and Bonferroni post hoc analysis were used. C, For those mice treated with saline, scrambled, and TauASO-3 intracerebroventricular pumps, total tau levels in the left hippocampus were plotted against normalized spike frequency at the 500 μm PTX concentration (Spearman r = 0.670, p = 0.0087; n = 14). The best-fit line and 95% confidence bands were generated using linear regression analysis. ***p < 0.001. Error bars represent SEM.
Figure 7.

Tau reduction is protective against PTZ-induced global seizures. A, Experimental paradigm. NT male mice were treated with saline, 25 μg/d scrambled ASO, or 25 μg/d TauASO-3 for 1 month. Pumps were then removed and seizures induced 21 d later. Brains were collected immediately after the seizure. B, PTZ (55 mg/kg) was given by intraperitoneal injection to saline-, scrambled ASO-, and TauASO-3-treated mice and final seizure stage was scored blinded. Kruskal–Wallis and Dunn's post hoc analysis were used. C, D, Total tau mRNA (D) and protein (E) levels were confirmed to be down in only the TauASO-3-treated group. One-way ANOVA and Bonferroni post hoc analysis were used. E–G, Final seizure stage for each treated mouse plotted against the total tau protein levels. (saline: Spearman r = 0.589, p = 0.002; scrambled: Spearman r = 0.680, p = 0.0007; TauASO-3: Spearman r = 0.504, p = 0.024). The best-fit line and 95% confidence bands were generated using linear regression analysis. *p < 0.05; **p < 0.01; ***p < 0.001; n = 18–26. Error bars represent SEM.
Figure 8.

Total tau protein is inversely correlated with seizure latency. A, A cohort of NT male mice (n = 5–9) were injected intraperitoneally with either saline or 80 mg/kg PTZ. Brains were collected immediately after mice reached a stage 8 seizure and total tau protein levels in right ventral white matter were measured. Two-tailed t test was used. B, For each PTZ-injected mouse, the time to reach a stage 8 seizure was plotted against total tau protein (Spearman r = −0.887, p = 0.003). The best-fit line and 95% confidence bands were generated using linear regression analysis. Error bars represent SEM.
ASOs.
The ASOs had the following modifications: 5 nucleotides on the 5′- and 3′-termini containing 2′-O-methoxyethyl modifications and 10 unmodified central oligodeoxynucleotides (DeVos and Miller, 2013b) to support RNaseH activity and a phosphorothioate backbone to improve nuclease resistance and promote cellular uptake (Bennett and Swayze, 2010). ASOs were synthesized as described previously (McKay et al., 1999; Cheruvallath et al., 2003) and solubilized in 0.9% sterile saline immediately before use. ASO sequences were as follows: TauASO-1: 5′-GCAGGAGTTCTTAGATGTCT-3′; TauASO-2: 5′-AAGCAGGTTAGGTGACAAGC-3′; TauASO-3: 5′-ATCACTGATTTTGAAGTCCC-3′; Scrambled ASO: 5′-CCTTCCCTGAAGGTTCCTCC-3′.
Surgical placement of intracerebroventricular pumps and tissue collection.
As described previously (Smith et al., 2006; DeVos and Miller, 2013a), mice were anesthetized with isoflurane and 28 d osmotic intracerebroventricular (ICV) pumps (ALZET) with ASO were implanted in a subcutaneous pocket that was formed on the back of the mouse. The catheter was placed in the right lateral ventricle using the following coordinates based on bregma: −0.5 mm posterior, −1.1 mm Lateral (right), −2.5 mm ventral. For CSF collection, mice were placed on a heating pad and anesthetized with isoflurane. CSF was drawn through the cisterna magna as described previously and immediately frozen on dry ice (Barten et al., 2011). For tissue collection, mice were anesthetized with isoflurane and perfused using chilled PBS-heparin. Brain and spinal cords were rapidly removed and either snap-frozen in liquid nitrogen and stored at −80°C or postfixed in 4% paraformaldehyde at 4°C and transferred to 30% sucrose 24 h later.
Quantitative real-time PCR.
RNA analyses were performed using qRT-PCR. Total RNA was extracted from brain tissue using a QIAGEN RNeasy Kit. For total tau analyses, RNA was reverse transcribed and amplified using the EXPRESS One-Step Superscript qRT-PCR Universal Kit (Invitrogen). For Nsmaf mRNA levels, RNA was reverse transcribed and amplified with the Power SYBR Green RT-to-Ct 1-Step Kit (Invitrogen). The qRT-PCRs were run and analyzed on the ABI PRISM 7500 Fast Real-Time PCR System (Applied Biosystems). Total tau and Nsmaf mRNA expression levels were normalized to GAPDH mRNA levels and analyzed using the ΔΔCt method for relative expression analysis. The primer/probe sequences were as follows: total tau: forward 5′-GAA CCA CCA AAA TCC GGA GA-3′; reverse 5′-CTC TTA CTA GCT GAT GGT GAC-3′; Probe 5′-/56-FAM/CC AAG AAG GTG GCA GTG GTC C/3IABkFQ/-3′, Nsmaf: forward 5′-CTTACTGCTTCTCAACTTGGA-3′; reverse 5′-GAACATATCTTTAAGGAGCCTC-3′, GAPDH: forward 5′-TGC CCC CAT GT TGT GAT G-3′; reverse 3′-TGT GGT CAT GAG CCC TTC C-3′; and Probe 5′/56-FAM/AAT GCA TCC TGC ACC ACC AAC TGC TT/3AHBkFQ/3′ (IDT).
Tau protein analysis.
Tissues were weighed and homogenized in 10× volume RAB buffer containing the following (in mm): 100 MES, 1 EDTA, 0.5 MgSO4, 750 NaCl, 20 NaF, and 1 Na3VO4, supplemented with protease inhibitor (Complete Protease Inhibitor; Roche) and phosphatase inhibitor (Sigma). Homogenate was spun at 21,000 × g on a tabletop centrifuge for 10 min at 4°C. Supernatant was collected and protein concentration measured using a Pierce BCA Protein Assay Kit (Thermo Scientific). For tau protein quantification in brain, interstitial fluid (ISF), and CSF, tau concentrations were analyzed using the published Tau5-BT2 sandwich ELISA (Yamada et al., 2011). Briefly, 96-half-well plates (Nunc) were coated with the Tau-5 antibody (Millipore) overnight at 4°C. Plates were blocked with 4% BSA for 60 min at 37°C, brain homogenate, ISF, or CSF diluted in standard buffer (0.25% BSA, 300 mm Tris, 0.05% azide, and 1× protease inhibitor in PBS) was added and incubated overnight at 4°C. For the standard curve, the longest mouse tau isoform recombinant protein was used (mTau40). The detection antibody biotinylated BT-2 (Pierce) was added the next day, followed by streptavidin poly-horseradish peroxidase-40 (Fitzgerald). Plates were developed using Super Slow ELISA TMB (Sigma) and read on an Epoch Microplate Spectrophotometer (BioTek).
Immunofluorescence.
Brains postfixed in 4% paraformaldehyde were sliced at 50 μm on a freezing microtome. Brain slices were treated with Citra Plus antigen retrieval (BioGenex) before antibody application. Brains were incubated with the primary antibodies Tau46 (1:300; Cell Signaling Technology) and Pan-ASO (1:1000; Isis) in 3% horse serum overnight at 4°C, followed by a 1 h incubation at room temperature with fluorescent-conjugated secondary antibodies (1:3000, DyLight; Thermo Scientific). Fluorescent images were captured using the Olympus Nanozoomer 2.0-HT (Hamamatsu) and processed using the NDP viewer software (Hamamatsu).
In vivo microdialysis for ISF collection.
In vivo microdialysis experiments to assess brain ISF tau levels from awake and freely moving mice were developed with modifications of our previously described method (Yamada et al., 2011). A guide cannula (Eicom Microdialysis) was stereotaxically implanted in the left hippocampus under isoflurane anesthesia and cemented. After implantation of the cannula and dummy probes (Eicom Microdialysis), mice were habituated to microdialysis cages for one more day. After this recovery period, a 2 mm 1000 kDa cutoff AtmosLM microdialysis probe (Eicom Microdialysis) was inserted through the guide cannula. A probe was connected to a microdialysis peristaltic pump with two channels (MAB20; SciPro), which was operated in a push pull mode. The perfusion buffer, 25% human albumin solution (Gemini Bio), was diluted to 4% with aCSF containing the following (in mm): 1.3 CaCl2, 1.2 MgSO4, 3 KCl, 0.4 KH2PO4, 25 NaHCO3, and 122 NaCl, pH 7.35, on the day of use and filtered through 0.1 μm membrane. ISF was collected at 1 μl/min in a refrigerated fraction collector (SciPro).
Lactate assay.
Lactate concentration in ISF was determined by YSI2700 biochemistry analyzer (YSI Life Sciences).
Sensorimotor battery and 1 h locomotor activity.
All mice were evaluated on a battery of sensorimotor tests designed to assess balance (ledge and platform), strength (inverted screen), coordination (pole and inclined screens), and initiation of movement (walking initiation), as described previously (Wozniak et al., 2004; Grady et al., 2006). Locomotor activity was evaluated in all mice over a 1 h period using computerized photobeam instrumentation as described previously (Wozniak et al., 2004, 2007). General activity variables (total ambulations, vertical rearings), along with indices of emotionality, including time spent, distance traveled, and entries made in a 33 × 11 cm central zone, were analyzed.
Elevated plus maze.
As described previously (Schaefer et al., 2000), the elevated plus maze (EPM) apparatus is a four-arm maze shaped like a plus sign. One set of the opposing arms have walls (closed arms) and the other set is not enclosed (open arms). The number of entries made, time spent, and distance traveled in each set of arms were quantified using a computerized, high-resolution photobeam system (Hamilton-Kinder). These three variables were also analyzed after normalizing the values to reflect percentages calculated out of the totals measured in both sets of arms.
Morris water maze.
Spatial learning and memory were evaluated in the Morris water maze using a computerized tracking system (ANY-maze; Stoelting) and procedures that were similar to previously described methods (Wozniak et al., 2004, 2007). The protocol included cued, place, and probe trials and all trials were performed in a 120-cm-diameter pool filled with opaque water. Cued trials were performed to identify nonassociative dysfunctions that might affect performance. This involved conducting four trials (60 s maximum) per day for two consecutive days with very few distal cues being present and with the platform location being moved for each trial to limit spatial learning. Three days later, place trials were initiated to assess spatial learning; mice were required to learn the single location of a submerged (nonvisible) platform in the presence of several salient distal spatial cues. The place trials were conducted for 5 consecutive days, each day consisting of 2 blocks of 2 trials (60 s maximum) separated by 2 h. Escape path length, latency, and swimming speeds were calculated for the cued and place trials. A probe trial lasting 60 s was conducted 1 h after the last trial of the place condition. This involved removing the platform and quantifying the time spent in the pool quadrant that had contained the platform (target) and each of the other quadrants and the number of crossings a mouse made over the exact location of where the platform had been (platform crossings). Spatial bias for the target quadrant was analyzed by comparing the time spent in it versus the times spent in each of the other quadrants.
Picrotoxin seizures and EEG recording.
In vivo picrotoxin (PTX) reverse microdialysis experiments from awake and freely moving mice were developed with modifications to the previously described method (Cirrito et al., 2008). To record EEG activity, bipolar recording electrodes (Teflon-coated, stainless steel wire, 0.0055 inch coated OD, A-M Systems) were attached to the outside of the microdialysis guide cannula shaft using Elmer's Super-Fast Epoxy Resin. The electrodes extended ∼1 mm beyond the tip of guide such that the tips of the electrodes would fall in the center of the 2 mm microdialysis probe membrane once inserted. The guide cannula with attached electrodes (BR-style; Bioanalytical Systems) was stereotaxically implanted in the left hippocampus under isoflurane anesthesia and cemented. Microdialysis probes (2 mm, BR-2, 30 kDa MWCO membrane; Bioanalytical Systems) were inserted into the hippocampus. The perfusion buffer comprised aCSF with 0.15% bovine serum albumin filtered through 0.1 μm membrane on the day of use. Once the microdialysis guide cannula and probe were placed into the left hippocampus, 12 h of basal EEG activity was measured using a P511K A.C. preamplifier (Grass Instruments), digitized with a DigiData 1322A data acquisition system (Molecular Devices), and recorded digitally with pClamp 9.2 (Molecular Devices). PTX (Sigma-Aldrich) was diluted to the indicated concentrations in 0.15% BSA-aCSF perfusion buffer and delivered at a flow rate of 1.0 μl/min, with the lowest dose given first. Each increasing dose was delivered for 90 min and EEG was measured continuously throughout drug delivery. EEG spike frequency was assessed for the last 60 min of each PTX dose and normalized to basal EEG of each mouse.
Experimental pentylenetetrazole seizures.
Pentylenetetrazole (PTZ; Sigma) was dissolved in sterile PBS at a concentration of 5 mg/ml. A dose of 55 or 80 mg/kg was delivered intraperitoneally for the experiments shown in Figures 7 and 8, respectively. A quiet, isolated room was used for all seizures to minimize noise and/or visual distractions. Immediately after PTZ administration, each mouse was videotaped and 15 min later, the mouse was killed and the brain was snap-frozen for biochemical analyses. Seizures recorded on videotapes were scored in a blinded fashion for severity according to published scales (Löscher and Nolting, 1991; Racine, 1972). The seizure severity score used was as follows: 0 = normal behavior; 1 = immobility; 2 = spasm, tremble, or twitch; 3 = tail extension; 4 = forelimb clonus; 5 = generalized clonic activity; 6 = jumping or running seizures; 7 = full tonic extension; and 8 = death.
Statistics.
The data were analyzed for statistical significance using GraphPad Prism 5 software. Two-tailed Student's t tests were used for total tau mRNA/protein and lactate analyses when only one comparison was being made and for analyzing tau mRNA/protein levels in multiple different brain regions. One-way ANOVA with Bonferroni post hoc analyses were used for total tau mRNA and protein expression when more than one comparison was needed and for the inverted screen task. Two-way ANOVA with Bonferroni post hoc analyses were used for the duration of action study and for ISF tau levels in multiple fractions. Two-way repeated-measures ANOVA (rmANOVA) with Bonferroni post hoc analyses were used to analyze Morris water maze, EPM trials, and PTX dose response. Specifically for the Morris water maze and EPM analyses, the Huynh-Feldt adjustment of α levels was used for all within-subjects effects containing more than two levels to protect against violations of sphericity/compound symmetry assumptions underlying rmANOVA models. The Kruskal–Wallis with Dunns post hoc analyses was used for the PTZ seizure severity analysis due to the categorical nature of the seizure severity scale. Linear regression analysis was used to analyze the CSF and brain tau correlations. Linear regression was used to generate the “best-fit” and 95% confidence interval lines seen on the graphs in Figures 6, 7, and 8, and the Spearman correlation was used to generate r and p-values for all total tau and EEG/seizure comparisons. Error bars in the figures represent SEM.
Results
Tau ASOs reduce endogenous tau mRNA and protein expression
To determine the functional effect of reducing tau mRNA and protein in vivo, we developed an ASO that reduces endogenous tau mRNA and protein in adult mice. After screening 80 ASOs for their ability to reduce tau mRNA in murine B16-F10 cells, we selected the three most potent ASOs to screen in vivo. We infused 50 μg of each tau ASO into the right hippocampus of adult nontransgenic (NT) mice using saline and a scrambled ASO as controls. One week after ASO infusion, the hippocampus surrounding the injection site was analyzed for total tau mRNA levels (Fig. 1A). All ASOs screened in the hippocampus provided >75% reduction of tau mRNA (ANOVA F(4,22) = 151.4, p < 0.0001). The ASO TauASO-3 was then tested in a 1 month ICV Alzet osmotic pump infusion at 100 μg/d. Compared with the saline and scrambled ASO controls, those mice treated with TauASO-3 had substantially less total tau mRNA (ANOVA F(2,14) = 291.8, p < 0.0001) and protein levels (ANOVA F(2,13) = 7.578, p = 0.007; Fig. 1B,C). To test for general off-target ASO effects, we normalized tau mRNA to total RNA input and found no difference between tau mRNA results generated through standard GAPDH normalization or total RNA input (two-tailed t test: saline t(12) = 0.000, p = 1.000; scrambled t(10) = 0.297, p = 0.773; TauASO-3 t(6) = 1.317, p = 0.236). To further test specificity for tau, we BLASTED the TauASO-3 sequence against the mouse genome and found that the closest match, neutral sphingomyelinase activation associated factor (Nsmaf), had 5 bp mismatches. When total Nsmaf mRNA levels were measured, no significant difference for saline, 100 μg/d of scrambled, or 100 μg/d of TauASO-3 treated mice was found (ANOVA F(2,14) = 1.914, p = 0.184), reinforcing the specificity of TauASO-3 for murine tau.
To select the optimum dose for behavioral studies, we tested five doses of TauASO-3. There was a dose-dependent decrease in total tau mRNA expression levels (ANOVA F(5,28) = 48.12, p < 0.0001; Fig. 1D). The 25 μg/d dose was the lowest dose that provided >75% tau mRNA reduction and was selected for subsequent studies. In addition, to plan treatment paradigms for behavioral studies, we tested the duration of action of a 1 month ASO infusion. After infusing 25 μg/d TauASO-3 for 1 month, tau mRNA levels were measured in cohorts of mice at 4, 8, 12, and 16 weeks after pump implantation. Four weeks after ASO infusion had stopped (8 weeks after pump implantation), tau mRNA levels remained >80% decreased. Even at 12–16 weeks after pump implantation, tau mRNA remained decreased to >50% of the saline control (ANOVA F(1,34) = 225.7, p < 0.0001; Fig. 1E). The long-term target knock-down results shown here are consistent with what others have reported for RNase-H-activating ASOs (Kordasiewicz et al., 2012), highlighting the long duration of action that RNase-H ASOs exhibit in tissue. This lengthy duration of action is likely due to a long half-life of the ASO itself (Yu et al., 2009; Kordasiewicz et al., 2012), allowing a 1 month infusion of 25 μg/d TauASO-3 to provide 4 months of tau mRNA reduction.
Tau ASOs reduce tau mRNA and protein throughout the brain and spinal cord
To determine the distribution of TauASO-3 after 1 month of ICV infusion of 25 μg/d, we used an antibody that recognizes the backbone chemistry of the ASO (Kordasiewicz et al., 2012). Staining for TauASO-3 demonstrated widespread, diffuse distribution of ASO in the brains of ASO-treated mice (Fig. 2A). Costaining for TauASO-3 and total tau demonstrated a clear link between the presence of ASO and lack of total tau in the contralateral frontal cortex, hippocampus, and cerebellum (Fig. 2A), showing that infusing TauASO-3 using ICV pumps can effectively distribute the ASO throughout the brain and reduce tau protein.
The immunofluorescence results showed a qualitative decrease in tau protein levels. To quantify more precisely the amount of tau mRNA and protein reduction in the CNS, we measured total tau mRNA and protein levels in multiple CNS regions 8 weeks after implantation of 1 month ICV pumps with 25 μg/d TauASO-3 (Fig. 2B). In both the mRNA and protein analyses, total tau levels were decreased in the left and right brain hemispheres and the spinal cord, confirming the widespread reduction of tau expression in the adult mouse CNS (Fig. 2C,D).
Tau protein levels are reduced in the brain ISF and CSF
To better understand the full effects of TauASO-3, we examined tau protein levels in two additional CNS compartments: the brain ISF and CSF. Recently, reports have placed tau in the extracellular space under physiological conditions (Yamada et al., 2011; Pooler et al., 2013). To determine whether tau ASOs can decrease tau that is secreted into the ISF, we treated a cohort of mice with a 75 μg/d concentration of TauASO-3 or saline for 1 month with ICV pumps. At the end of ASO infusion, catheters and pumps were removed and microdialysis probes implanted into the left hippocampus 2 weeks later. We collected ISF for 48 h, with a new fraction being collected every 90 min (Fig. 3A). Immediately after ISF collection, the left hippocampus was dissected out and total tau levels were measured to confirm that brain tau protein was indeed reduced in the TauASO-3 cohort (two-tailed t test t(10) = 8.462, p < 0.0001; Fig. 3B).
Total ISF tau protein levels were steady across time in both the saline and TauASO-3 groups and substantially reduced in the TauASO-3 treated cohort (ANOVA F(1,36) = 48.22, p < 0.0001; Fig. 3C), allowing multiple fractions to be combined from the same mouse. These same eight fractions were pooled for each animal and total tau protein levels were again measured. In the TauASO-3-treated group, ISF total tau protein levels were greatly reduced compared with the saline control (two-tailed t test t(8) = 9.283, p = 0.003). ISF lactate levels were not significantly different between the saline and TauASO-3 groups (two-tailed t test t(8) = 0.6169, p = 0.555). No difference in ISF lactate levels, which usually increase in response to synaptic transmission, suggests that a reduction in endogenous tau does not influence baseline neuronal activity (Bero et al., 2011).
To examine the correlation between brain and CSF tau levels, we treated a cohort of mice with 25 μg/d TauASO-3 or saline for 1 month. Half of the mouse brains were collected 4 weeks after pump implantation and the other 8 weeks after pump insertion. Immediately before collecting the brains, CSF was drawn from the cisterna magna of the mice, averaging 10 μl of CSF per mouse. As predicted, brain tau protein levels were decreased at both the first time point and the second (ANOVA F(2,14) = 20.96, p < 0.0001; Fig. 4A). Interestingly, the CSF total tau was not reduced to the same extent as brain tau at the 4 week collection time, although by the 8 week time point, CSF tau was lower in the TauASO-3-treated mice (ANOVA F(2,24) = 9.720, p = 0.0008; Fig. 4B). Although the reason for this lag in CSF tau reduction is unknown, it may be in part due to a slow turnover of intracellular tau protein to the CSF pool, resulting in a continued decrease in CSF tau while brain tau levels begin to increase. Total brain and CSF tau protein levels were significantly correlated, both in the brain adjacent to the catheter (linear regression R2 = 0.867, F(1,6) = 39.11, p = 0.0008; Fig. 4C) and the contralateral frontal cortex (linear regression R2 = 0.657, F(1,6) = 11.49, p = 0.0147; Fig. 4D). These data, in addition to showing a reduction of extracellular tau, suggest that CSF tau levels may be an excellent predictor of brain tau levels in a TauASO-3 treatment paradigm.
Reducing tau mRNA and protein does not alter baseline behavior
Before assessing whether tau reduction can provide protection in experimental behavioral paradigms, we first analyzed the mice for any gross motor or cognitive behavioral abnormalities. Tau−/− mice appear normal on learning/memory tasks for up to 1 year (Roberson et al., 2007, 2011; Dawson et al., 2010; Ittner et al., 2010), with some minor parkinsonism motor phenotypes developing at ∼12 months of age (Lei et al., 2012; Morris et al., 2013). These largely normal behavior phenotypes, however, could be in part due to developmental compensation. We treated a cohort of NT mice with saline, 25 μg/d scrambled control ASO, or 25 μg/d TauASO-3 for 1 month and conducted behavioral assessments for 1.5 months after pump removal. Total tau mRNA (ANOVA F(2,19) = 101.3, p < 0.0001) and protein (ANOVA F(2,19) = 13.95, p = 0.0002) levels were confirmed to be reduced only in mice treated with TauASO-3 (Fig. 5A,B). The mice with decreased tau levels performed similarly to both the saline and scrambled ASO control groups on all seven measures of the sensorimotor battery, including the inverted screen test (ANOVA F(2,19) = 0.116, p = 0.891; Fig. 5C), suggesting that the TauASO-3 mice did not have any gross sensorimotor dysfunctions. Except for a possible hyperactivity in the TauASO-3 group, TauASO-3 mice also displayed similar behavior during the 1 h locomotor activity test. The TauASO-3 mice did not exhibit any significant performance deficits on the place (rmANOVA F(2,76) = 0.006, p = 0.994) or probe (rmANOVA F(2,76)<0.0001, p = 1.000) trials in the water maze (Fig. 5D,E), thus providing evidence that their spatial learning and memory were intact. Analysis of the EPM data also showed that the TauASO-3 did not differ in levels of anxiety-related behaviors compared with the saline and scrambled ASO control groups (percentage of open arm entries rmANOVA F(2,38) = 0.284, p = 0.756; percentage of open arm time rmANOVA F(2,38) = 0.134, p = 0.875; percentage of open arm distance rmANOVA F(2,38) = 0.211, p = 0.754; Fig. 5F–H). There was also no difference between groups in regard to the total distance traveled in the EPM (ANOVA F(2,19) = 0.639, p = 0.539). Recognizing that the sample size tested was relatively small, the data suggest that, at least in the short term, reducing tau mRNA and protein in the adult mouse does not appear to result in behavioral impairments.
Reducing tau mRNA and protein protects against chemically induced seizures
To determine whether tau reduction is protective in an induced focal seizure model, we used reverse microdialysis to deliver the noncompetitive GABAA receptor antagonist PTX (Olsen, 2006) focally into the left hippocampus of NT male mice treated with saline, 25 μg/d scrambled ASO, or 12–25 μg/d TauASO-3 and simultaneously recorded the EEG activity at the site of PTX delivery. Because this treatment paradigm had not been used previously to study the protective effects of tau reduction, we included a cohort of untreated NT and tau−/− mice to serve as controls. Twelve hours of basal EEG activity were recorded, followed by continuous infusion of PTX into the left hippocampus with a stepwise increase in concentration every 90 min (4, 20, 100, and 500 μm). Untreated NT and saline-treated mice were combined due to no significant difference between the groups. The tau−/− mice showed a reduction in normalized spike frequency compared with the NT/saline group at the 500 μm PTX concentration, confirming the protective effect in tau-null mice in this new excitation paradigm (rmANOVA F(3,72) = 8.634, p = 0.0009; Fig. 6A,B). The TauASO-3-treated group also showed a strong protective effect compared with both the NT/saline and scrambled cohorts (Fig. 6A,B). Furthermore, total tau protein levels in the left hippocampus of pump-treated mice were highly correlated with normalized spike frequency at 500 μm PTX (Spearman correlation, r(12) = 0.670, p = 0.0087; Fig. 6C). These PTX studies in adult mice support a direct correlation between lower tau protein levels and reduced neuronal hyperexcitability.
In addition to the focal increase in EEG activity, we tested the effects of tau reduction in a widely used seizure paradigm-intraperitoneal PTZ injections. PTZ seizures are considered a gold standard when testing the efficacy of anticonvulsant drugs in the early stages of development in vivo (Löscher, 2011). Three-month-old NT male mice were treated with saline, 25 μg/d scrambled ASO, or 25 μg/d TauASO-3 for 1 month and then the pumps were removed. Three weeks later, 55 mg/kg of the GABA antagonist PTZ (Macdonald and Barker, 1977) was administered to the mice by intraperitoneal injection. The mice were videorecorded for 15 min and then immediately collected for total tau analyses (Fig. 7A). Any mouse that had >50% total tau mRNA levels was eliminated from the analysis of the TauASO-3 group. Those mice treated with TauASO-3 had less severe seizures than both the saline and scrambled ASO control groups (Kruskal–Wallis statistic = 16.26, p = 0.0003; Fig. 7B). Total tau mRNA (ANOVA F(2,62) = 281.5, p < 0.0001) and protein (ANOVA F(2,62) = 45.73, p < 0.0001) levels were confirmed to be reduced specifically in the TauASO-3-treated group (Fig. 7C,D). As further confirmation that the effect of TauASO-3 on seizures was secondary to tau reduction and was not an unknown effect of the ASO, we correlated the level of tau protein with seizure severity for individual animals. Indeed, seizure severity and tau protein levels correlated well in all tested mice (Spearman correlations, saline r(24) = 0.5889, p = 0.0016; scrambled r(19) = 0.6795, p = 0.0007; TauASO-3 r(18) = 0.504, p = 0.0236; Fig. 7E–G), providing evidence in a second inducible seizure model that a reduction in tau protein is protective against seizures.
Intrinsic variability in tau protein levels predicts susceptibility to chemically induced seizures
Due to the variability that has been seen with PTZ seizures (Mandhane et al., 2007), we were surprised that the correlation between seizure severity and tau levels persisted even in NT mice treated with only saline (Fig. 7E). This correlation suggests that, among the NT mouse population, normal endogenous tau levels predict susceptibility to neuronal hyperexcitability. It may be, however, that a more severe seizure results in an acute increase in brain tau protein expression. To test this possibility, we induced severe seizures in a separate cohort of untreated NT mice using a high dose of PTZ and analyzed total tau protein levels immediately afterward. There was no difference in tau protein levels in brain homogenate between those mice that underwent severe seizures secondary to PTZ injection compared with those mice that received a saline injection and did not have seizures (two-tailed t test t(12) = 0.354, p = 0.730; Fig. 8A). Interestingly, in the PTZ-injected group, there was an inverse correlation between the time it took to reach a stage 8 seizure and the amount of endogenous tau protein measured. Those mice that had higher levels of endogenous tau protein progressed to severe seizures more quickly than those mice with lower tau (Spearman correlation r(7) = −0.887, p = 0.003; Fig. 8B). These PTZ data show that the seizure itself does not increase tau protein acutely in brain tissues during the period of the seizure and, together with the TauASO-3 PTX and PTZ seizure data, strongly suggest that those mice with higher levels of endogenous tau are inherently more susceptible to neuronal hyperexcitability.
Discussion
Using ASO technology directed against endogenous murine tau, total tau mRNA and protein levels were decreased throughout the brain and spinal cord of adult NT mice (Figs. 1,2). In addition, extracellular tau in the brain ISF (Fig. 3) and CSF (Fig. 4) was also reduced after infusion of TauASO-3 ASO. After tau was reduced in the adult mouse, no significant deviations from baseline were observed in a battery of motor and learning/memory behavior tasks (Fig. 5), demonstrating that short-term tau knock-down is well tolerated in vivo. In the setting of chemically induced seizures, tau reduction protected against seizure severity (Figs. 6,7), consistent with what has been reported with the genetic tau−/− mouse model. Further, we noted a significant correlation between total tau protein levels in the brain and seizure severity both in treated mice (Figs. 6,7) and in untreated mice (Fig. 8). These data strengthen the link between total tau expression levels and neuronal hyperexcitability regulation in vivo and demonstrate that the tau−/− effect on neuronal hyperexcitability is likely a tau-mediated event and not a developmental phenomenon.
The tau−/− genotype has been shown in numerous studies to be protective against excitotoxic insults (Roberson et al., 2007, 2011; Ittner et al., 2010), implicating tau in the physiological regulation of aberrant neuronal excitability. In addition, both a complete reduction and haploinsufficiency of tau significantly reduced seizures and extended survival in a well established genetic mouse model of epilepsy, Kv1.1−/− (Glasscock et al., 2010, 2012; Holth et al., 2013). These reports, in conjunction with previous in vitro data using tau knock-down ASOs to protect cells from glutamate-induced excitotoxicity (Pizzi et al., 1993) and our own in vivo tau knock-down data in two different seizure models, support the application of a tau-lowering therapy to regulate hyperexcitability in human patients. Compounds that provide protection against PTZ seizures in vivo have generally been successful in subsequent human clinical trials (Rogawski, 2006). Although many epilepsy patients respond to one or two anticonvulsants, 20–40% of patients remain untreated (Devinsky, 1999; Brodie et al., 2012). Therefore, a tau reduction approach may be an alternative therapy for this refractory population. Given the previous tau−/− protective findings in multiple seizure paradigms (Roberson et al., 2007, 2011; Holth et al., 2013), we predict that our findings of tau knock-down using two different GABA antagonists will also apply broadly to epilepsy in vivo models and to human epilepsy.
The finding that physiological endogenous tau levels in adult mice can affect susceptibility to hyperexcitability not only lends support to the idea that reducing tau may help lower seizure severity, but also implies that endogenous tau levels in humans may influence the risk of developing seizures. Although the exact reason for variability in tau protein expression between mice is unknown, other groups have shown similar variability in murine total tau levels (Holth et al., 2013). Further, tau mRNA and protein levels in human brains can vary by >2-fold (Lu et al., 2004; Kauwe et al., 2008; Trabzuni et al., 2012) and CSF total tau levels in control human subjects can differ substantially (Clifford et al., 2009; Fagan et al., 2009; Oka et al., 2013), perhaps due to variability in neuronal excretion rates of tau and different baseline tau levels in the brain. Higher levels of tau protein at baseline may not be detrimental, but, upon insult, increased tau expression may predispose human patients to injury-induced seizures. It is well documented that the incidence of seizures increases after different types of brain injury, including ischemic stroke (Camilo and Goldstein, 2004; Kwan, 2010) and traumatic brain injury (Annegers et al., 1998; Vespa et al., 2010). If human patients with higher baseline tau are more prone to developing seizures after an injury, being able to identify such patients through genetic studies of tau polymorphisms (Myers et al., 2007; Kauwe et al., 2008) or tau CSF levels (Palmio et al., 2009; Cruchaga et al., 2013) may help to risk stratify those patients and aid in determining who would benefit from a preventive antiepileptic therapy.
Tau knock-down has also been studied extensively in the presence of Aβ-deposition and proven to be protective against a growing number of Aβ-induced insults, including cognition (Roberson et al., 2007; Andrews-Zwilling et al., 2010; Ittner et al., 2010; Leroy et al., 2012), hyperexcitability (Roberson et al., 2007, 2011; Ittner et al., 2010; Suberbielle et al., 2013), decreased survival (Roberson et al., 2007, 2011; Ittner et al., 2010), axonal transport deficits (Vossel et al., 2010), cell-cycle reentry (Seward et al., 2013), and double-stranded breaks in DNA (Suberbielle et al., 2013). Several human amyloid precursor protein mouse lines and an ApoE4 mouse line have now been generated that display abnormal EEG and increased seizure frequency (Roberson et al., 2007; Ittner et al., 2010; Vogt et al., 2011; Hunter et al., 2012). Similarly, those with familial AD mutations, ApoE4 genotype, and sporadic late-onset AD also experience an increased incidence in seizures (Takao et al., 2001; Mendez and Lim, 2003; Harden, 2004; Velez-Pardo et al., 2004; Amatniek et al., 2006; Kauffman et al., 2010). This AD-associated excitotoxicity has been implicated in the pathogenesis of the disease (Olney et al., 1997; Mattson, 2004) and has recently been linked to an earlier onset of cognitive decline in AD patients (Vossel et al., 2013).
Because of the limitations in detecting abnormal EEG activity in large populations of AD patients, we currently rely heavily on animal models for predictions regarding hyperexcitability in the context of Aβ. In the human amyloid precursor protein J20 Aβ-depositing line, treatment with the anticonvulsant levetiracetam returned the baseline aberrant excitability back to NT levels and restored cognition (Sanchez et al., 2012), similar to what was seen with the tau−/− genetic cross (Roberson et al., 2007, 2011). This rescue in cognition by means of an anticonvulsant suggests that lowering the abnormal neuronal activity alone may have a positive impact on learning and memory. A similar pilot study was performed in human mildly cognitively impaired (MCI) patients, where patients were given either placebo or levetiracetam and then recall memory was tested using functional magnetic resonance imaging methods. Levetiracetam treatment significantly improved the recall performance of the MCI patients, again providing evidence that reducing the aberrant excitability in MCI and AD patients may help to restore cognition (Bakker et al., 2012). To test the hypothesis that decreasing aberrant hyperexcitability by means of tau reduction can in turn rescue cognitive decline, we have initiated tau-lowering ASO therapy in an Aβ-depositing mouse model of AD. We propose that tau may be involved in both AD-associated hyperexcitability and neuronal cell loss by means of tau aggregation and neurofibrillary tangle formation, making tau knock-down a strong therapeutic target for AD.
The human analog of the mouse tau ASO used here may be readily applicable to human patients. ASOs against superoxide dismutase 1 that extended survival in a rat model of amyotrophic lateral sclerosis (Smith et al., 2006) recently finished a phase I clinical trial in human patients. The CSF-delivered ASOs demonstrated excellent safety (Miller et al., 2013). Further, ASOs against survival motor neuron protein that also rescued rodent spinal muscular atrophy models (Hua et al., 2010; Passini et al., 2011; Porensky et al., 2012) are currently being used in phase II studies for children with spinal muscular atrophy. These studies and the growing success of ASOs in preclinical models (Kumar et al., 2000; Yokota et al., 2009; Lanford et al., 2010; DeVos and Miller, 2013b) suggest that the tau reduction strategy outlined here has real potential to be translated to the clinical setting for patients with epilepsy and perhaps tauopathies such as AD, progressive supranuclear palsy, and frontotemporal dementia.
Footnotes
This work was supported by the Tau Consortium (D.M.H. and T.M.M.), The National Institutes of Health (Grant #P50 AG05681 to T.M.M., J.C. Morris, PI; National Institute of Neurological Disorders and Stroke Grant #R01NS078398 to T.M.M.; and National Institute on Aging Grants #R21AG044719-01 to T.M.M. and #R01AG042513 to J.R.C.), Cure PSP (T.M.M.), and a Paul B. Beeson Career Development Award (Grant #NINDS K08NS074194 to T.M.M.) The microscopy work was supported by the Hope Center Alafi Neuroimaging Laboratory and P30 Neuroscience Blueprint Interdisciplinary Center Core award to Washington University (Grant #P30 NS057105). We thank Chengjie Xiong and Mateusz Jasielec (Knight Alzheimer's Disease Center, Washington University) for assistance with statistical analysis and Michael Wong for assistance with induced seizure models.
Isis Pharmaceuticals supplies the authors with the ASOs in the described work. Both Isis Pharmaceuticals and Washington University have filed for patents based on using Tau ASOs to treat CNS disorders. The authors declare no other competing financial interests.
References
- Amatniek JC, Hauser WA, DelCastillo-Castaneda C, Jacobs DM, Marder K, Bell K, Albert M, Brandt J, Stern Y. Incidence and predictors of seizures in patients with Alzheimer's disease. Epilepsia. 2006;47:867–872. doi: 10.1111/j.1528-1167.2006.00554.x. [DOI] [PubMed] [Google Scholar]
- Andrews-Zwilling Y, Bien-Ly N, Xu Q, Li G, Bernardo A, Yoon SY, Zwilling D, Yan TX, Chen L, Huang Y. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J Neurosci. 2010;30:13707–13717. doi: 10.1523/JNEUROSCI.4040-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries. N Engl J Med. 1998;338:20–24. doi: 10.1056/NEJM199801013380104. [DOI] [PubMed] [Google Scholar]
- Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, Yassa MA, Bassett SS, Shelton AL, Gallagher M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012;74:467–474. doi: 10.1016/j.neuron.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barten DM, Cadelina GW, Hoque N, DeCarr LB, Guss VL, Yang L, Sankaranarayanan S, Wes PD, Flynn ME, Meredith JE, Ahlijanian MK, Albright CF. Tau transgenic mice as models for cerebrospinal fluid tau biomarkers. J Alzheimers Dis. 2011;24:127–141. doi: 10.3233/JAD-2011-110161. [DOI] [PubMed] [Google Scholar]
- Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, Holtzman DM. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat Neurosci. 2011;14:750–756. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J. 1997;323:577–591. doi: 10.1042/bj3230577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brion JP, Passareiro H, Nunez J, Flament-Durand J. Mise en evidence immunologique de la proteine tau au niveau des lesions de degenerescence neurofibrillaire de la maladie d'Alzheimer. Archives of Biologie, Bruxelles. 1985;95:229–235. [Google Scholar]
- Brodie MJ, Barry SJ, Bamagous GA, Norrie JD, Kwan P. Patterns of treatment response in newly diagnosed epilepsy. Neurology. 2012;78:1548–1554. doi: 10.1212/WNL.0b013e3182563b19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buée L, Delacourte A. Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick's disease. Brain Pathol. 1999;693:681–693. doi: 10.1111/j.1750-3639.1999.tb00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilo O, Goldstein LB. Seizures and epilepsy after ischemic stroke. Stroke. 2004;35:1769–1775. doi: 10.1161/01.STR.0000130989.17100.96. [DOI] [PubMed] [Google Scholar]
- Cheruvallath ZS, Kumar RK, Rentel C, Cole DL, Ravikumar VT. Solid phase synthesis of phosphorothioate oligonucleotides utilizing diethyldithiocarbonate disulfide (DDD) as an efficient sulfur transfer reagent. Nucleosides Nucleotides Nucleic Acids. 2003;22:461–468. doi: 10.1081/NCN-120022050. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, Holtzman DM. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008;58:42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford DB, Fagan AM, Holtzman DM, Morris JC, Teshome M, Shah AR, Kauwe JS. CSF biomarkers of Alzheimer disease in HIV-associated neurologic disease. Neurology. 2009;73:1982–1987. doi: 10.1212/WNL.0b013e3181c5b445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruchaga C, Kauwe JS, Harari O, Jin SC, Cai Y, Karch CM, Benitez BA, Jeng AT, Skorupa T, Carrell D, Bertelsen S, Bailey M, McKean D, Shulman JM, De Jager PL, Chibnik L, Bennett DA, Arnold SE, Harold D, Sims R, et al. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer's disease. Neuron. 2013;78:256–268. doi: 10.1016/j.neuron.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson HN, Cantillana V, Jansen M, Wang H, Vitek MP, Wilcock DM, Lynch JR, Laskowitz DT. Loss of tau elicits axonal degeneration in a mouse model of AD. Neuroscience. 2010;169:516–531. doi: 10.1016/j.neuroscience.2010.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O. Patients with refractory seizures. N Engl J Med. 1999;340:1565–1570. doi: 10.1056/NEJM199905203402008. [DOI] [PubMed] [Google Scholar]
- DeVos SL, Miller TM. Direct intraventricular delivery of drugs to the rodent central nervous system. J Vis Exp. 2013a;12:75. doi: 10.3791/50326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVos SL, Miller TM. Antisense oligonucleotides: treating neurodegeneration at the level of RNA. Neurotherapeutics. 2013b;10:486–497. doi: 10.1007/s13311-013-0194-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Head D, Shah AR, Marcus D, Mintun M, Morris JC, Holtzman DM. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol. 2009;65:176–183. doi: 10.1002/ana.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasscock E, Yoo JW, Chen TT, Klassen TL, Noebels JL. Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci. 2010;30:5167–5175. doi: 10.1523/JNEUROSCI.5591-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasscock E, Qian J, Kole MJ, Noebels JL. Transcompartmental reversal of single fibre hyperexcitability in juxtaparanodal Kv1.1-deficient vagus nerve axons by activation of nodal KCNQ channels. J Physiol. 2012;590:3913–3926. doi: 10.1113/jphysiol.2012.235606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady RM, Wozniak DF, Ohlemiller KK, Sanes JR. Cerebellar synaptic defects and abnormal motor behavior in mice lacking alpha- and beta-dystrobrevin. J Neurosci. 2006;26:2841–2851. doi: 10.1523/JNEUROSCI.4823-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato-Yoshitake R, Takei Y, Noda T, Hirokawa N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 1994;369:488–491. doi: 10.1038/369488a0. [DOI] [PubMed] [Google Scholar]
- Harden CL. The apolipoprotein E epsilon (epsilon) 4 allele is important for trauma-related epilepsy. Epilepsy currents. 2004;4:29–30. doi: 10.1111/j.1535-7597.2004.04112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holth JK, Bomben VC, Reed JG, Inoue T, Younkin L, Younkin SG, Pautler RG, Botas J, Noebels JL. Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J Neurosci. 2013;33:1651–1659. doi: 10.1523/JNEUROSCI.3191-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Sahashi K, Hung G, Rigo F, Passini MA, Bennett CF, Krainer AR. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010;24:1634–1644. doi: 10.1101/gad.1941310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JM, Cirrito JR, Restivo JL, Kinley RD, Sullivan PM, Holtzman DM, Koger D, Delong C, Lin S, Zhao L, Liu F, Bales K, Paul SM. Emergence of a seizure phenotype in aged apolipoprotein epsilon 4 targeted replacement mice. Brain Res. 2012;1467:120–132. doi: 10.1016/j.brainres.2012.05.048. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Wisniewski HM, Grundke-Iqbal I, Korthals JK, Terry RD. Chemical pathology of neurofibrils–neurofibrillary tangles of Alzheimer's presenile-senile dementia. J Histochem Cytochem. 1975;23:563–569. doi: 10.1177/23.7.1141687. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Kauffman MA, Consalvo D, Moron DG, Lereis VP, Kochen S. ApoE epsilon4 genotype and the age at onset of temporal lobe epilepsy: a case-control study and meta-analysis. Epilepsy Res. 2010;90:234–239. doi: 10.1016/j.eplepsyres.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Kauwe JS, Cruchaga C, Mayo K, Fenoglio C, Bertelsen S, Nowotny P, Galimberti D, Scarpini E, Morris JC, Fagan AM, Holtzman DM, Goate AM. Variation in MAPT is associated with cerebrospinal fluid tau levels in the presence of amyloid-beta deposition. Proc Natl Acad Sci U S A. 2008;105:8050–8054. doi: 10.1073/pnas.0801227105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempf M, Clement A, Faissner A, Lee G, Brandt R. Tau binds to the distal axon early in development of polarity in a microtubule- and microfilament-dependent manner. J Neurosci. 1996;16:5583–5592. doi: 10.1523/JNEUROSCI.16-18-05583.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, Artates JW, Weiss A, Cheng SH, Shihabuddin LS, Hung G, Bennett CF, Cleveland DW. Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis. Neuron. 2012;74:1031–1044. doi: 10.1016/j.neuron.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar VB, Farr SA, Flood JF, Kamlesh V, Franko M, Banks WA, Morley JE. Site-directed antisense oligonucleotide decreases the expression of amyloid precursor protein and reverses deficits in learning and memory in aged SAMP8 mice. Peptides. 2000;21:1769–1775. doi: 10.1016/S0196-9781(00)00339-9. [DOI] [PubMed] [Google Scholar]
- Kwan J. Stroke: predicting the risk of poststroke epilepsy-why and how? Nat Rev Neurol. 2010;6:532–533. doi: 10.1038/nrneurol.2010.140. [DOI] [PubMed] [Google Scholar]
- Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Ørum H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, Wong BX, Adlard PA, Cherny RA, Lam LQ, Roberts BR, Volitakis I, Egan GF, McLean CA, Cappai R, Duce JA, Bush AI. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med. 2012;18:291–295. doi: 10.1038/nm.2613. [DOI] [PubMed] [Google Scholar]
- Leroy K, Ando K, Laporte V, Dedecker R, Suain V, Authelet M, Héraud C, Pierrot N, Yilmaz Z, Octave JN, Brion JP. Lack of tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. Am J Pathol. 2012;181:1928–1940. doi: 10.1016/j.ajpath.2012.08.012. [DOI] [PubMed] [Google Scholar]
- Löscher W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure. 2011;20:359–368. doi: 10.1016/j.seizure.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Löscher W, Nolting B. The role of technical, biological and pharmacological factors in the laboratory evaluation of anticonvulsant drugs. IV. Protective indices. Epilepsy Res. 1991;9:1–10. doi: 10.1016/0920-1211(91)90041-D. [DOI] [PubMed] [Google Scholar]
- Lovestone S, Reynolds CH. The phosphorylation of tau: a critical stage in neurodevelopment and neurodegenerative processes. Science. 1997;78:309–324. doi: 10.1016/S0306-4522(96)00577-5. [DOI] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Barker JL. Pentylenetetrazol and penicillin are selective antagonists of GABA-mediated post-synaptic inhibition in cultured mammalian neurones. Nature. 1977;267:720–721. doi: 10.1038/267720a0. [DOI] [PubMed] [Google Scholar]
- Mandhane SN, Aavula K, Rajamannar T. Timed pentylenetetrazol infusion test: a comparative analysis with s.c.PTZ and MES models of anticonvulsant screening in mice. Seizure. 2007;16:636–644. doi: 10.1016/j.seizure.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay RA, Miraglia LJ, Cummins LL, Owens SR, Sasmor H, Dean NM. Characterization of a potent and specific class of antisense oligonucleotide inhibitor of human protein kinase C-alpha expression. J Biol Chem. 1999;274:1715–1722. doi: 10.1074/jbc.274.3.1715. [DOI] [PubMed] [Google Scholar]
- Mendez M, Lim G. Seizures in elderly patients with dementia: epidemiology and management. Drugs Aging. 2003;20:791–803. doi: 10.2165/00002512-200320110-00001. [DOI] [PubMed] [Google Scholar]
- Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, Andres PL, Mahoney K, Allred P, Alexander K, Ostrow LW, Schoenfeld D, Macklin EA, Norris DA, Manousakis G, Crisp M, Smith R, Bennett CF, Bishop KM, Cudkowicz ME. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12:435–442. doi: 10.1016/S1474-4422(13)70061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris M, Hamto P, Adame A, Devidze N, Masliah E, Mucke L. Age-appropriate cognition and subtle dopamine-independent motor deficits in aged Tau knock-out mice. Neurobiol Aging. 2013;34:1523–1529. doi: 10.1016/j.neurobiolaging.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers AJ, Pittman AM, Zhao AS, Rohrer K, Kaleem M, Marlowe L, Lees A, Leung D, McKeith IG, Perry RH, Morris CM, Trojanowski JQ, Clark C, Karlawish J, Arnold S, Forman MS, Van Deerlin V, de Silva R, Hardy J. The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol Dis. 2007;25:561–570. doi: 10.1016/j.nbd.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Oka M, Hasegawa S, Matsushige T, Inoue H, Kajimoto M, Ishikawa N, Isumi H, Ichiyama T. Tau protein concentrations in the cerebrospinal fluid of children with acute disseminated encephalomyelitis. Brain Dev. 2013 doi: 10.1016/j.braindev.2012.11.013. In Press. [DOI] [PubMed] [Google Scholar]
- Olney JW, Wozniak DF, Farber NB. Excitotoxic neurodegeneration in Alzheimer disease. New hypothesis and new therapeutic strategies. Arch Neurol. 1997;54:1234–1240. doi: 10.1001/archneur.1997.00550220042012. [DOI] [PubMed] [Google Scholar]
- Olsen RW. Picrotoxin-like channel blockers of GABAA receptors. Proc Natl Acad Sci U S A. 2006;103:6081–6082. doi: 10.1073/pnas.0601121103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmio J, Suhonen J, Keränen T, Hulkkonen J, Peltola J, Pirttilä T. Cerebrospinal fluid tau as a marker of neuronal damage after epileptic seizure. Seizure. 2009;18:474–477. doi: 10.1016/j.seizure.2009.04.006. [DOI] [PubMed] [Google Scholar]
- Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, Stanek LM, Hua Y, Rigo F, Matson J, Hung G, Kaye EM, Shihabuddin LS, Krainer AR, Bennett CF, Cheng SH. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011;3:72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzi M, Valerio A, Ribola M, Spano PF, Memo M. A Tau antisense oligonucleotide decreases neurone sensitivity to excitotoxic injury. Neuroreport. 1993;4:823–826. doi: 10.1097/00001756-199306000-00057. [DOI] [PubMed] [Google Scholar]
- Pooler AM, Phillips EC, Lau DH, Noble W, Hanger DP. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013;14:389–394. doi: 10.1038/embor.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porensky PN, Mitrpant C, McGovern VL, Bevan AK, Foust KD, Kaspar BK, Wilton SD, Burghes AH. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum Mol Genet. 2012;21:1625–1638. doi: 10.1093/hmg/ddr600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski MA. Diverse mechanisms of antiepileptic drugs in the development pipeline. Epilepsy Res. 2006;69:273–294. doi: 10.1016/j.eplepsyres.2006.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu GQ, Palop JJ, Mucke L. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc Natl Acad Sci U S A. 2012;109:E2895–E2903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer ML, Wong ST, Wozniak DF, Muglia LM, Liauw JA, Zhuo M, Nardi A, Hartman RE, Vogt SK, Luedke CE, Storm DR, Muglia LJ. Altered stress-induced anxiety in adenylyl cyclase type VIII-deficient mice. J Neurosci. 2000;20:4809–4820. doi: 10.1523/JNEUROSCI.20-13-04809.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seward ME, Swanson E, Norambuena A, Reimann A, Cochran JN, Li R, Roberson ED, Bloom GS. Amyloid-β signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer's disease. J Cell Sci. 2013;26:1278–1286. doi: 10.1242/jcs.1125880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RA, Miller TM, Yamanaka K, Monia BP, Condon TP, Hung G, Lobsiger CS, Ward CM, McAlonis-Downes M, Wei H, Wancewicz EV, Bennett CF, Cleveland DW. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest. 2006;116:2290–2296. doi: 10.1172/JCI25424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suberbielle E, Sanchez PE, Kravitz AV, Wang X, Ho K, Eilertson K, Devidze N, Kreitzer AC, Mucke L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat Neurosci. 2013;16:613–621. doi: 10.1038/nn.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao M, Ghetti B, Murrell JR, Unverzagt FW, Giaccone G, Tagliavini F, Bugiani O, Piccardo P, Hulette CM, Crain BJ, Farlow MR, Heyman A. Ectopic white matter neurons, a developmental abnormality that may be caused by the PSEN1 S169L mutation in a case of familial AD with myoclonus and seizures. J Neuropathol Exp Neurol. 2001;60:1137–1152. doi: 10.1093/jnen/60.12.1137. [DOI] [PubMed] [Google Scholar]
- Trabzuni D, Wray S, Vandrovcova J, Ramasamy A, Walker R, Smith C, Luk C, Gibbs JR, Dillman A, Hernandez DG, Arepalli S, Singleton AB, Cookson MR, Pittman AM, de Silva R, Weale ME, Hardy J, Ryten M. MAPT expression and splicing is differentially regulated by brain region: relation to genotype and implication for tauopathies. Hum Mol Genet. 2012;21:4094–4103. doi: 10.1093/hmg/dds238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker KL, Meyer M, Barde YA. Neurotrophins are required for nerve growth during development. Nat Neurosci. 2001;4:29–37. doi: 10.1038/82868. [DOI] [PubMed] [Google Scholar]
- Velez-Pardo C, Arellano JI, Cardona-Gomez P, Jimenez Del Rio M, Lopera F, De Felipe J. CA1 hippocampal neuronal loss in familial Alzheimer's disease presenilin-1 E280A mutation is related to epilepsy. Epilepsia. 2004;45:751–756. doi: 10.1111/j.0013-9580.2004.55403.x. [DOI] [PubMed] [Google Scholar]
- Vespa PM, McArthur DL, Xu Y, Eliseo M, Etchepare M, Dinov I, Alger J, Glenn TP, Hovda D. Nonconvulsive seizures after traumatic brain injury are associated with hippocampal atrophy. Neurology. 2010;75:792–798. doi: 10.1212/WNL.0b013e3181f07334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt DL, Thomas D, Galvan V, Bredesen DE, Lamb BT, Pimplikar SW. Abnormal neuronal networks and seizure susceptibility in mice overexpressing the APP intracellular domain. Neurobiol Aging. 2011;32:1725–1729. doi: 10.1016/j.neurobiolaging.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, Hegde M, Gornes SB, Henry ML, Nelson AB, Seeley WW, Geschwind MD, Gorno-Tempini ML, Shih T, Kirsch HE, Garcia PA, Miller BL, Mucke L. Seizures and epileptiform activity in the early stages of Alzheimer Disease. JAMA Neurol. 2013;8:1–9. doi: 10.1001/jamaneurol.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, Young C, Olney JW, Muglia LJ. Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol Dis. 2004;17:403–414. doi: 10.1016/j.nbd.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Wozniak DF, Xiao M, Xu L, Yamada KA, Ornitz DM. Impaired spatial learning and defective theta burst induced LTP in mice lacking fibroblast growth factor 14. Neurobiol Dis. 2007;26:14–26. doi: 10.1016/j.nbd.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Cirrito JR, Stewart FR, Jiang H, Finn MB, Holmes BB, Binder LI, Mandelkow EM, Diamond MI, Lee VM, Holtzman DM. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci. 2011;31:13110–13117. doi: 10.1523/JNEUROSCI.2569-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota T, Lu QL, Partridge T, Kobayashi M, Nakamura A, Takeda S, Hoffman E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol. 2009;65:667–676. doi: 10.1002/ana.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu RZ, Lemonidis KM, Graham MJ, Matson JE, Crooke RM, Tribble DL, Wedel MK, Levin AA, Geary RS. Cross-species comparison of in vivo PK/PD relationships for second-generation antisense oligonucleotides targeting apolipoprotein B-100. Biochem Pharmacol. 2009;77:910–919. doi: 10.1016/j.bcp.2008.11.005. [DOI] [PubMed] [Google Scholar]