

Abstract
There is considerable evidence that many disease are associated with endothelial dysfunction and reduced nitric oxide production such as hypertension, obesity, dyslipidemias, diabetes, heart failure, atherosclerosis. Notably these conditions are also characterized by alteration in the adrenergic tone. Whether these two mechanisms are just epiphenomenal each other or there is a functional link, it is still to be established. A starting ground to establish this issue is that vascular endothelium plays an important role in the function of cardiovascular system and that adrenergic receptors on endothelial cells contribute to the regulation of vasomotor tone. The aim of this excerpt is to review current knowledge on the physiology of endothelial adrenergic receptors to contribute to the basis for newer and better approaches to endothelial dysfunction in the setup of cardiovascular conditions.
Introduction
The endothelium controls several vascular functions, including vasculature tone and permeability, thrombosis, hemostasis and angiogenesis1–4. It is noteworthy that all these functions can be regulated by the activation of receptors and often the same receptor can activate multiple endothelial functions. The adrenergic system is the major regulator of cardiac and vascular function and of endothelial vasorelaxation by means of α and β adrenergic receptors activation. The adrenergic receptors (ARs) are part of a large family of G protein coupled receptors (GPCR) which mediate the functional effects of catecholamines like epinephrine and norepinephrine. The ARs family includes three β (β1, β2, β3), three α1 (α1A, α1B, α1D) and three α2 (α2A, α2B, α2C) receptor subtypes. These receptors actively participate to the release of nitric oxide (NO) in order to regulate endothelial function5. NO plays a crucial role in endothelium homeostasis, with important vasodilatory, anti-thrombotic and anti-atherogenic properties. NO mediates most of the endothelial functions: it has been invoked as a mechanism in vasorelaxation, endothelium permeability and neoangiogenesis3. NO in the endothelium is constitutively produced by the endothelial NO synthase, eNOS6. This latter is then further activated through calcium levels 7 and phosphorylation of various serine residues by a number of protein kinases 8, 9. Indeed, it has been demonstrated that NO is activated by means of the PI3K pathway in response to the stimulation of tyrosine kinase 10,11.
The impaired ability of vascular endothelium to stimulate vasodilation is referred to as “Endothelial Dysfunction” and the major cause is the decreased bioavailability of NO in different conditions which can be due to various mechanisms: reduced eNOS expression, altered NO production and increased NO catabolism. Endothelial dysfunction plays a key role in the development of cardiovascular disease such as hypertension, type 2 diabetes and heart failure. The identification of the underlying pathogenic mechanisms will lead to the discovery of newer and more potent tools to treat such diseases. On this issue, endothelial dysfunction has been associated to signal transduction abnormalities observed in hypertension. In particular, adrenergic vasorelaxation has been demonstrated to be impaired in hypertensive patients, probably due to the presence of increased desensitization and impaired signalling of βAR. Adrenergic receptors on endothelium have been longely not considered functional to the regulation of the vascular tone. On the contrary, it is possible to identify very specific roles for such receptors in several endothelial function. This review will summarize the effects of adrenergic receptors on endothelial functions, focusing on modulation of NO synthesis and angiogenesis.
α adrenergic receptors
αAR are GPCRs that couple to Gαq protein. The Gαq subunit is a primary activator of phospholipase C (PLC). Activation of PLC promotes the cleavage of the inositol substrate phosphatidyl-inositol 4,5 bisphosphate (PIP2) to yield diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). DAG and IP3 promote the activation of a protein kinase C (PKC). α1AR can also activate specific adenylate (adenylyl) cyclases (AC) leading to an increase in cAMP levels. The activation of specific PLCs and ACs requires a complex balance of signals from G-proteins, especially the Gα subunits, within specific cell contexts. DAG and cAMP are second messengers that affect a wide array of cell signaling pathways and responses.
1. α1AR and Nitric oxide
Several reports 12, 13 have produced evidence for the functional presence of vasorelaxant α1AR in the brachial and pulmonary arteries isolated from the rabbit and rat, respectively. According to these reports, the pharmacological stimulation of α1AR located on endothelial cells, is able to generate NO, whereas the stimulation of α2AR releases a relaxing prostanoid12, 13. Filippi demonstrated that nanomolar concentrations of phenylephrine, which are devoid of any contractile effect, induced a slight endothelium-dependent vasorelaxation in the rat mesenteric vascular bed through the stimulation of α1DAR, located on endothelial cells, which act through phospholipase C stimulation, followed by IP1 generation, and nitric-oxide synthase activation. Conversely, the increase in perfusion pressure induced by micromolar concentrations of phenylephrine is attributable to the stimulation of α1AAR14.
2. α1AR and angiogenesis
Neo-angiogenesis has long been known to be a highly ordered multistep molecular process under tight regulation by endothelial cells15 and closely associated with endothelial cell proliferation and migration and to the capability of these cells to modulate the levels of VEGF, the most important cytokine system involved in the formation of new vessels16. A series of biological, chemical, hormonal effectors can interfere with this process. Several data support the notion that α1-adrenergic receptor should also be ranked among these agents. Indeed, it has been demonstrated that the α1A- and the α1B-AR subtypes but not the α1D subtype are expressed in cultured rat aorta endothelial cells. The activation of these α1-AR in endothelial cells provide a negative regulation of angiogenesis17. Indeed, pharmacological antagonism of α1-AR in endothelial cells from WKY rats by doxazosin enhanced, while stimulation of these adrenergic receptors with phenylephrine, inhibited endothelial mechanisms of angiogenesis such as cell proliferation and DNA synthesis, ERK and retinoblastoma protein (Rb) phosphorylation, cell migration and tubule formation17. A similar phenotype can be observed in vivo, since an increased α1-adrenergic receptor density in the ischaemic hindlimb, compared to non-ischaemic hindlimb, suggested an enhanced α1-adrenergic receptor tone in the ischaemic tissue. Treatment with doxazosin did not alter systemic blood pressure but enhanced neo-angiogenesis in the ischaemic hindlimb17.
3. α2ARand Nitric oxide
It has been demonstrated that α2 adrenergic agonists cause endothelium dependent relaxation, that is reduced or abolished by inhibitors of L-arginine/NO pathway. It depends on the activation of α2AR on endothelial cells which stimulates the release of NO, an action that would tend to attenuate vasoconstriction produced by the activation of post-junctional vascular α1AR18–20. The α2AR subtype that cause endothelium dependent relaxation belongs to the α2A/D subtype, despite the prominent presence of α2CAR (77% of α2C versus 23% of α2A/D)21. It appears that this ratio may not be constant, since it varies within the vascular bed. Indeed, Bockman demonstrated that in the rat mesenteric artery the α2AR is coupled to endothelium dependent NO-mediated relaxations and belongs to the α2A/D subtype appearing in its α2D version 22. It has been demonstrated that endothelium dependent relaxation to α2 adrenergic agonists is prevented by pertussis toxin 23–28, suggesting the involvement of Gi proteins in the signal transduction from the receptor to the activation of nitric oxide synthase 29, 30. Indeed, α2 adrenergic agonists cause activation of Gi proteins in endothelial cells and stimulate NO synthase activity 31, 32. Contrary to what expected, cAMP is not involved in the signal transduction pathway for α2A/DAR mediated NO formation 22. Indeed, the use of forskolin to oppose α2 adrenergic receptor mediated inhibition of cAMP formation in endothelium did not affect the relaxant response to α2AR agonists, suggesting that cAMP is not involved in the coupling of α2AR to NO. There are physiological modulation of endothelium dependent relaxation to α2 adrenergic agonists. Such relaxation is upregulated by chronic increase in blood flow 33 or exercise training 34. Insulin enhances NO mediated vasorelaxation both in animal 25 and human 32 vasculature.
β-adrenergic receptors
βARs signal by coupling to the stimulatory G protein, Gs, leads to the activation of adenylyl cyclase and accumulation of the second messenger cAMP35, 36. However, recent studies indicate that under certain conditions βAR, and particularly β2AR, can couple to Gi as well as to Gs 37–41. It is now widely accepted that βAR exist on endothelial cells 10, 38, 40, 42 and contribute to the regulation of vasomotor tone. βAR are classically known to be present in the vascular smooth muscle cells (VSMC) where they cause vasodilation. The relative relevance of endothelial VSMC in adrenergic vasodilation is demonstrated by the observation that, in presence of intact endothelium, vasorelaxation to βAR agonist, isoproterenol (ISO), is sensitive to low doses of ISO (10−10M-10−8M). On the contrary, in absence of endothelium, the vasorelaxation is sensitive to higher doses of ISO (10−7M-10−5M). This appears to hold true through experimental models (rat or man) and vascular districts (see Figure 1).
Figure 1:
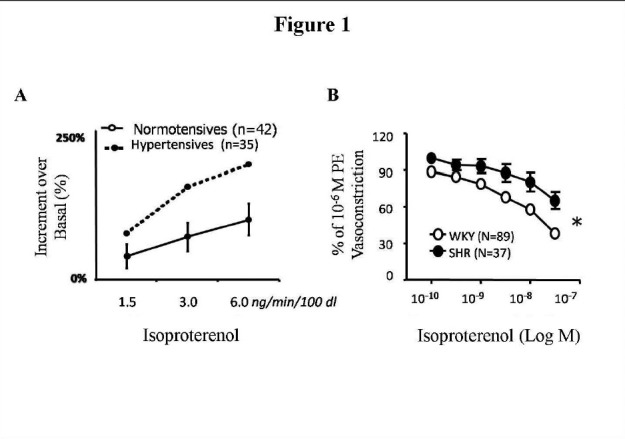
βAR vasodilation is impaired in hypertension: A) In hypertensive patients, forearm vasodilation to ISO yielded an increase in forearm blood flow that was significantly lower to that observed in normotensive patients, at each dose of ISO. B) In hypertensive rats SHR, βAR-induced vasorelaxation to ISO in control-treated carotids was significantly impaired compared with that observed in normotensive WKY(* F= 5.756, p< 0.01, 2-way ANOVA).
1. β1 and β2 adrenergic receptors
It is now recognized that βAR located in the endothelium play an important role in the relaxant response to ISO, since the non selective β1-and β2-adrenergic receptor antagonist propranolol antagonized this relaxant effect43, 44. However, recent studies carried out in humans, in umbilical veins in vitro10 or in the forearm in vivo45, showed that vasorelaxation to ISO is abolished by the selective β2AR antagonist ICI-118551 and remains unchanged in the presence of the β1AR antagonist CGP-20712, indicating that, as in the vascular smooth muscle cells 46, the endothelial βAR are totally or at least predominantly of the β2 subtype 10, 45.
β2AR are seven transmembrane receptors coupled through Gs proteins to a cAMP dependent intracellular pathway47. It has been demonstrated that PKA posphorylation of the third intracellular loop of the β2AR increases the affinity of the receptor for Gi protein48, 49. This switch leads to two consequences: first, it decreases the rate of cAMP generation, since Gi activation inhibits adenylyl cyclase activity. Second, it increases non cAMP dependent signaling through Gi, such as activation of the extracellular signal-regulated kinases ERK1/2 and PI3K50–54. Gi coupled receptors have been shown to regulate non-receptor tyrosine kinases, such as SRC, which acts as an intermediate between Gi and other molecules like RAS and PI3K 53, 55.
2. β2AR and Nitric oxide
For years it has been given for granted that vascular β2AR mediate adrenergic vasorelaxation through direct activation of vascular smooth muscle cells56. However, recent data challenge this vision, and show that β2AR-dependent vasorelaxation is mediated at least in part, by endothelium through nitric oxide (NO) dependent processes10. We have recently demonstrated that the β2AR are expressed on endothelial cells (EC) and their stimulation causes endothelial nitric oxide synthase (eNOS) activation57. In particular, β2AR couple to eNOS and induce NO dependent vasodilation 57. The mechanism of eNOS activation following β2AR stimulation is known to be AKT dependent58.
Indeed, the activity of eNOS is regulated by both a calcium/calmodulin dependent fashion59 and AKT dependent eNOS phosphorylation in Ser 1177 8, 60–63. AKT is primarily activated in response to stimulation of transmembrane receptors with intrinsic tyrosine kinase activity or indirectly coupled to tyrosine kinases or to seven transmembrane G protein-coupled receptor11, 61, 64. Therefore AKT acts as integrator of different signal transduction pathways converging on eNOS, including endothelial β2AR receptor9, 58, 62, 63, 65.
3. β2AR and angiogenesis
In the endothelium βARs control other important endothelial functions like angiogenesis, that is tightly associated to endothelial cell migration and proliferation 57, 65, 66. We demonstrated that β2AR stimulation with ISO and the overexpression of β2AR increases endothelial cell proliferation. Moreover, β2AR stimulation induces ERK phosphorylation and the MEKK inhibitor, U0126, inhibits β2AR induced cell proliferation 66 suggesting that β2AR dependent cell proliferation is dependent on ERK activation. We studied post-ischaemic angiogenesis in the hindlimb (HL) of β2AR knock-out mice (β2AR−/−) in vivo and explored possible molecular mechanisms in vitro. Angiogenesis was severely impaired in β2AR−/− mice subjected to femoral artery resection, but was restored by gene therapy with ADβ2AR. The proangiogenic responses to a variety of stimuli were impaired in β2AR−/− EC in vitro17. Moreover, removal of β2ARs impaired the activation of NFκB, a transcription factor that promotes angiogenesis; ISO did not induce NFκB activation in β2AR(−/−) EC17. ADβp2AR administration restored β2AR membrane density and reinstated the NFκB response to ISO 17. These results suggest that β2ARs control angiogenesis through the tight regulation of nuclear transcriptional activity.
4.
5. α1ARand β2AR differently regulate neo-angiogenesis
α1- and β2-adrenergic receptors mediate opposite effects on neo-angiogenesis, comparable to their regulation of the vascular tone. In particular, the α1-AR is inhibitory, whereas the β2-AR is stimulant to neo-angiogenesis. Interestingly, in ischaemia, the α1-AR are upregulated, thus causing a predominance of α1-adrenergic receptor signalling over that of β2-AR, which is downregulated. Furthermore, in conditions such as hypertension, where the α1-AR tone is higher than that of the β2-AR, there is also an impairment in neo-angiogenesis 66, 67. It is interesting to note that in the ischaemic hindlimb, α1-AR blockade resulted in a normalization of β2-AR density together with improved neo-angiogenesis. α1-AR upregulation, in particular, might be a regulatory mechanism aimed at preventing excessive angiogenesis. This upregulation might be triggered by ischaemia, through regulatory sequences within the gene promoter, which have been demonstrated for both the α1A- and α1B-adrenergic receptor68, 69.
6. β3 adrenergic receptors
In rat thoracic aorta, Trochu showed that β3AR are mainly located on endothelial cells and act in conjunction with β1AR and β2AR to mediate relaxation through activation of NO synthase pathway and subsequent increase in tissue cyclic GMP content and is reduced by endothelium removal or in presence of L-NMMA 70. This β3AR mediated aorta relaxation seems to be independent of Gi proteins stimulation, since the blockage of Gi protein by PTX does not modify β3AR agonists induced relaxation. On the contrary, selective potassium channels blockers of K (Ca), K (ATP) and K (v) decreased β3AR agonists induced relaxation. So it appears that this effect results from the activation of several potassium channels, K (Ca), K (ATP) and K (v) 71.
Pathological implications
It was reported that noradrenaline-induced release of nitric oxide is enhanced in mineralcorticoid hypertension 72 indicating that α2AR may play an important role in the regulation of vascular tone not only in physiological but also in pathological conditions. The implications of impaired βAR signalling in the pathophysiology of several cardiovascular disorders has been studied in animals and humans. Data from these studies indicate that changes in βAR function are induced by heart failure 73, 74 and hypertension 75, 76. Moreover, alteration in βAR function were found also with physiological aging 77, 78, due to receptor downregulation and desensitization. Exercise restored the impaired signalling and βAR dependent vasorelaxation79. We and others have observed that impaired βAR signalling may account for dysfunctional βAR vasorelaxation in hypertension. In this condition, β2AR overexpression in hypertensive rat carotids corrects impaired vasorelaxation to βAR stimulation to levels similar to those seen in normotensive rats57. We proved that impaired endothelium dependent vasorelaxation in spontaneously hypertensive rats (SHR) can be corrected by increasing the signal transduction pathways leading to nitric oxide synthase activation 80. In particular, since eNOS is activated in response to phosphorylation by AKT and impaired AKT activity is involved in endothelial dysfunction, AKT overexpression should result in the correction of impaired phenotype. Indeed, insulin and ISO cause AKT membrane localization and this subcellular localization is impaired in SHR. AKT overexpression, through means of adenovirus mediated AKT gene transfer to the endothelium, increases the amount of AKT localized to the membrane and corrects impaired NO release and endothelium dependent vasodilation to agonists of both the GPCR and tyrosine kinase (TK) dependent pathways.
Conclusions
In the last years great advances have been made in the study of adrenergic receptors signaling and function in the endothelium also thanks to the development of new technologies. Indeed, genetic mouse models have significantly improved our understanding of the mechanisms of action of specific drugs in vivo. The ability to induce transgene expression at defined times or in defined tissues is an important goal as well as the ability to induce or repress the expression of endogenous genes in a developmental or tissue specific fashion. Indeed, deletion of the genes encoding for adrenergic receptor subtypeshas helped to identify the specific subtypes whichmediate in vivo effects of specific drugs. Thus, the combination of molecular biological, genetic, and pharmacological techniques greatly facilitates our understanding of adrenergic receptor function in vivo, and in turn leads to more effective and specific therapeutic treatment in humans. βARs, for instance, are already target of therapeutic intervention in many diseases: βAR stimulation in asthma and obesity or βAR blocking in hypertension and coronary insufficiency. In conclusion, giving the importance of endothelial function in most physiological and pathological conditions, it is clear that the increasing knowledge of adrenergic receptors function in the endothelium is helpful for future progresses in clinical application.
References
- 1.Furchgott RF, Vanhoutte PM. Endothelium-derived relaxing and contracting factors. FASEB J. 1989;3:2007–18. [PubMed] [Google Scholar]
- 2.Lusher JM, Salzman PM. Viral safety and inhibitor development associated with factor VIIIC ultra-purified from plasma in hemophiliacs previously unexposed to factor VIIIC concentrates. The Monoclate Study Group. SeminHematol. 1990;27:1–7. [PubMed] [Google Scholar]
- 3.Schmidt HH, Walter U. NO at work. Cell. 1994;78:919–25. doi: 10.1016/0092-8674(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 4.Vanhoutte PM. Endothelial dysfunction in hypertension. J HypertensSuppl. 1996;14:S83–93. [PubMed] [Google Scholar]
- 5.Guimaraes S, Moura D. Vascular adrenoceptors: an update. Pharmacol Rev. 2001;53:319–56. [PubMed] [Google Scholar]
- 6.Nathan C, Xie QW. Nitric oxide synthases: roles, tolls, and controls. Cell. 1994;78:915–8. doi: 10.1016/0092-8674(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 7.Forstermann U, Pollock JS, Schmidt HH, Heller M, Murad F. Calmodulin-dependent endothelium-derived relaxing factor/nitric oxide synthase activity is present in the particulate and cytosolic fractions of bovine aortic endothelial cells. Proc NatlAcadSci U S A. 1991;88:1788–92. doi: 10.1073/pnas.88.5.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Butt E, Bernhardt M, Smolenski A, et al. Endothelial nitric-oxide synthase (type III) is activated and becomes calcium independent upon phosphorylation by cyclic nucleotide-dependent protein kinases. J BiolChem. 2000;275:5179–87. doi: 10.1074/jbc.275.7.5179. [DOI] [PubMed] [Google Scholar]
- 9.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–5. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 10.Ferro A, Queen LR, Priest RM, et al. Activation of nitric oxide synthase by beta 2-adrenoceptors in human umbilical vein endothelium in vitro. Br J Pharmacol. 1999;126:1872–80. doi: 10.1038/sj.bjp.0702512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kohn AD, Kovacina KS, Roth RA. Insulin stimulates the kinase activity of RAC-PK, a pleckstrin homology domain containing ser/thr kinase. EMBO J. 1995;14:4288–95. doi: 10.1002/j.1460-2075.1995.tb00103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boer C, Scheffer GJ, de Lange JJ, Westerhof N, Sipkema P. Alpha-1-adrenoceptor stimulation induces nitric oxide release in rat pulmonary arteries. J Vasc Res. 1999;36:79–81. doi: 10.1159/000025628. [DOI] [PubMed] [Google Scholar]
- 13.Zschauer AO, Sielczak MW, Smith DA, Wanner A. Norepinephrine-induced contraction of isolated rabbit bronchial artery: role of alpha 1- and alpha 2-adrenoceptor activation. J ApplPhysiol. 1997;82:1918–25. doi: 10.1152/jappl.1997.82.6.1918. [DOI] [PubMed] [Google Scholar]
- 14.Filippi S, Parenti A, Donnini S, Granger HJ, Fazzini A, Ledda F. alpha(1D)-adrenoceptors cause endothelium-dependent vasodilatation in the rat mesenteric vascular bed. J Pharmacol Exp Ther. 2001;296:869–75. [PubMed] [Google Scholar]
- 15.Papetti M, Herman IM. Mechanisms of normal and tumor-derived angiogenesis. Am J Physiol Cell Physiol. 2002;282:C947–70. doi: 10.1152/ajpcell.00389.2001. [DOI] [PubMed] [Google Scholar]
- 16.Carmeliet P, Collen D. Molecular basis of angiogenesis. Role of VEGF and VE-cadherin. Ann N Y Acad Sci. 2000;902:249–62. doi: 10.1111/j.1749-6632.2000.tb06320.x. discussion 62–4. [DOI] [PubMed] [Google Scholar]
- 17.Ciccarelli M, Sorriento D, Cipolletta E, et al. Impaired neoangiogenesis in beta-adrenoceptor gene-deficient mice: restoration by intravascular human beta-adrenoceptor gene transfer and role of NFkappaB and CREB transcription factors. Br J Pharmacol. 162:712–21. doi: 10.1111/j.1476-5381.2010.01078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Angus JA, Cocks TM, Satoh K. The alpha adrenoceptors on endothelial cells. Fed Proc. 1986;45:2355–9. [PubMed] [Google Scholar]
- 19.Richard V, Tanner FC, Tschudi M, Luscher TF. Different activation of L-arginine pathway by bradykinin, serotonin, and clonidine in coronary arteries. Am J Physiol. 1990;259:H1433–9. doi: 10.1152/ajpheart.1990.259.5.H1433. [DOI] [PubMed] [Google Scholar]
- 20.Vanhoutte PM, Miller VM. Alpha 2-adrenoceptors and endothelium-derived relaxing factor. Am J Med. 1989;87:1S–5S. doi: 10.1016/0002-9343(89)90496-8. [DOI] [PubMed] [Google Scholar]
- 21.Bockman CS, Jeffries WB, Abel PW. Binding and functional characterization of alpha-2 adrenergic receptor subtypes on pig vascular endothelium. J Pharmacol Exp Ther. 1993;267:1126–33. [PubMed] [Google Scholar]
- 22.Bockman CS, Gonzalez-Cabrera I, Abel PW. Alpha-2 adrenoceptor subtype causing nitric oxide-mediated vascular relaxation in rats. J Pharmacol Exp Ther. 1996;278:1235–43. [PubMed] [Google Scholar]
- 23.Bryan RM, Jr, Eichler MY, Swafford MW, Johnson TD, Suresh MS, Childres WF. Stimulation of alpha 2 adrenoceptors dilates the rat middle cerebral artery. Anesthesiology. 1996;85:82–90. doi: 10.1097/00000542-199607000-00012. [DOI] [PubMed] [Google Scholar]
- 24.Flavahan NA, Shimokawa H, Vanhoutte PM. Pertussis toxin inhibits endothelium-dependent relaxations to certain agonists in porcine coronary arteries. J Physiol. 1989;408:549–60. doi: 10.1113/jphysiol.1989.sp017475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lembo G, Iaccarino G, Vecchione C, et al. Insulin modulation of an endothelial nitric oxide component present in the alpha2- and beta-adrenergic responses in human forearm. J Clin Invest. 1997;100:2007–14. doi: 10.1172/JCI119732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller VM, Flavahan NA, Vanhoutte PM. Pertussis toxin reduces endothelium-dependent and independent responses to alpha-2- adrenergic stimulation in systemic canine arteries and veins. J Pharmacol Exp Ther. 1991;257:290–3. [PubMed] [Google Scholar]
- 27.Shimokawa H, Flavahan NA, Vanhoutte PM. Natural course of the impairment of endothelium-dependent relaxations after balloon endothelium removal in porcine coronary arteries. Possible dysfunction of a pertussis toxin-sensitive G protein. Circ Res. 1989;65:740–53. doi: 10.1161/01.res.65.3.740. [DOI] [PubMed] [Google Scholar]
- 28.Shimokawa H, Flavahan NA, Vanhoutte PM. Loss of endothelial pertussis toxin-sensitive G protein function in atherosclerotic porcine coronary arteries. Circulation. 1991;83:652–60. doi: 10.1161/01.cir.83.2.652. [DOI] [PubMed] [Google Scholar]
- 29.Boulanger CM, Vanhoutte PM. G proteins and endothelium-dependent relaxations. J Vasc Res. 1997;34:175–85. doi: 10.1159/000159221. [DOI] [PubMed] [Google Scholar]
- 30.Flavahan NA, Vanhoutte PM. G-proteins and endothelial responses. Blood Vessels. 1990;27:218–29. doi: 10.1159/000158813. [DOI] [PubMed] [Google Scholar]
- 31.Freeman JE, Kuo WY, Drenger B, Barnett TN, Levine MA, Flavahan NA. Analysis of lysophophatidylcholine-induced endothelial dysfunction. J CardiovascPharmacol. 1996;28:345–52. doi: 10.1097/00005344-199609000-00001. [DOI] [PubMed] [Google Scholar]
- 32.Lembo G, Iaccarino G, Vecchione C, et al. Insulin enhances endothelial alpha2-adrenergic vasorelaxation by a pertussis toxin mechanism. Hypertension. 1997;30:1128–34. doi: 10.1161/01.hyp.30.5.1128. [DOI] [PubMed] [Google Scholar]
- 33.Miller VM, Barber DA. Modulation of endothelium-derived nitric oxide in canine femoral veins. Am J Physiol. 1996;271:H668–73. doi: 10.1152/ajpheart.1996.271.2.H668. [DOI] [PubMed] [Google Scholar]
- 34.Cheng L, Yang C, Hsu L, Lin MT, Jen CJ, Chen H. Acute exercise enhances receptor-mediated endothelium-dependent vasodilation by receptor upregulation. J Biomed Sci. 1999;6:22–7. doi: 10.1007/BF02256420. [DOI] [PubMed] [Google Scholar]
- 35.Dixon RA, Kobilka BK, Strader DJ, et al. Cloning of the gene and cDNA for mammalian beta-adrenergic receptor and homology with rhodopsin. Nature. 1986;321:75–9. doi: 10.1038/321075a0. [DOI] [PubMed] [Google Scholar]
- 36.Emorine LJ, Marullo S, Briend-Sutren MM, et al. Molecular characterization of the human beta 3-adrenergic receptor. Science. 1989;245:1118–21. doi: 10.1126/science.2570461. [DOI] [PubMed] [Google Scholar]
- 37.Asano T, Katada T, Gilman AG, Ross EM. Activation of the inhibitory GTP-binding protein of adenylatecyclase, Gi, by beta-adrenergic receptors in reconstituted phospholipid vesicles. J BiolChem. 1984;259:9351–4. [PubMed] [Google Scholar]
- 38.Buxton BF, Jones CR, Molenaar P, Summers RJ. Characterization and autoradiographic localization of beta-adrenoceptor subtypes in human cardiac tissues. Br J Pharmacol. 1987;92:299–310. doi: 10.1111/j.1476-5381.1987.tb11324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaudhry A, MacKenzie RG, Georgic LM, Granneman JG. Differential interaction of beta 1- and beta 3-adrenergic receptors with Gi in rat adipocytes. Cell Signal. 1994;6:457–65. doi: 10.1016/0898-6568(94)90093-0. [DOI] [PubMed] [Google Scholar]
- 40.Gauthier C, Tavernier G, Charpentier F, Langin D, Le Marec H. Functional beta3-adrenoceptor in the human heart. J Clin Invest. 1996;98:556–62. doi: 10.1172/JCI118823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiao RP, Ji X, Lakatta EG. Functional coupling of the beta 2-adrenoceptor to a pertussis toxin-sensitive G protein in cardiac myocytes. Mol Pharmacol. 1995;47:322–9. [PubMed] [Google Scholar]
- 42.Molenaar P, Malta E, Jones CR, Buxton BF, Summers RJ. Autoradiographic localization and function of beta-adrenoceptors on the human internal mammary artery and saphenous vein. Br J Pharmacol. 1988;95:225–33. doi: 10.1111/j.1476-5381.1988.tb16568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oriowo MA. Different atypical beta-adrenoceptors mediate isoprenaline-induced relaxation in vascular and non-vascular smooth muscles. Life Sci. 1995;56:PL269–75. doi: 10.1016/0024-3205(95)00076-3. [DOI] [PubMed] [Google Scholar]
- 44.Brawley L, Shaw AM, MacDonald A. Beta 1-, beta 2- and atypical beta-adrenoceptor-mediated relaxation in rat isolated aorta. Br J Pharmacol. 2000;129:637–44. doi: 10.1038/sj.bjp.0703091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dawes M, Chowienczyk PJ, Ritter JM. Effects of inhibition of the L-arginine/nitric oxide pathway on vasodilation caused by beta-adrenergic agonists in human forearm. Circulation. 1997;95:2293–7. doi: 10.1161/01.cir.95.9.2293. [DOI] [PubMed] [Google Scholar]
- 46.Lands AM, Luduena FP, Buzzo HJ. Differentiation of receptors responsive to isoproterenol. Life Sci. 1967;6:2241–9. doi: 10.1016/0024-3205(67)90031-8. [DOI] [PubMed] [Google Scholar]
- 47.Rubenstein RC, Linder ME, Ross EM. Selectivity of the beta-adrenergic receptor among Gs, Gi’s, and Go: assay using recombinant alpha subunits in reconstituted phospholipid vesicles. Biochemistry. 1991;30:10769–77. doi: 10.1021/bi00108a023. [DOI] [PubMed] [Google Scholar]
- 48.Okamoto T, Murayama Y, Hayashi Y, Inagaki M, Ogata E, Nishimoto I. Identification of a Gs activator region of the beta 2-adrenergic receptor that is autoregulated via protein kinase A-dependent phosphorylation. Cell. 1991;67:723–30. doi: 10.1016/0092-8674(91)90067-9. [DOI] [PubMed] [Google Scholar]
- 49.Zamah AM, Delahunty M, Luttrell LM, Lefkowitz RJ. Protein kinase A-mediated phosphorylation of the beta 2-adrenergic receptor regulates its coupling to Gs and Gi. Demonstration in a reconstituted system. J BiolChem. 2002;277:31249–56. doi: 10.1074/jbc.M202753200. [DOI] [PubMed] [Google Scholar]
- 50.Baillie GS, Sood A, McPhee I, et al. beta-Arrestin-mediated PDE4 cAMPphosphodiesterase recruitment regulates beta-adrenoceptor switching from Gs to Gi. Proc NatlAcadSci U S A. 2003;100:940–5. doi: 10.1073/pnas.262787199. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51.Jalali S, Li YS, Sotoudeh M, et al. Shear stress activates p60src-Ras-MAPK signaling pathways in vascular endothelial cells. ArteriosclerThrombVascBiol. 1998;18:227–34. doi: 10.1161/01.atv.18.2.227. [DOI] [PubMed] [Google Scholar]
- 52.Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115:455–65. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- 53.Nagao M, Kaziro Y, Itoh H. The Src family tyrosine kinase is involved in Rho-dependent activation of c-Jun N-terminal kinase by Galpha12. Oncogene. 1999;18:4425–34. doi: 10.1038/sj.onc.1202832. [DOI] [PubMed] [Google Scholar]
- 54.Xiao RP. Beta-adrenergic signaling in the heart: dual coupling of the beta2-adrenergic receptor to G(s) and G(i) proteins. Sci STKE. 2001;2001:re15. doi: 10.1126/stke.2001.104.re15. [DOI] [PubMed] [Google Scholar]
- 55.Ma YC, Huang J, Ali S, Lowry W, Huang XY. Src tyrosine kinase is a novel direct effector of G proteins. Cell. 2000;102:635–46. doi: 10.1016/s0092-8674(00)00086-6. [DOI] [PubMed] [Google Scholar]
- 56.Jazayeri A, Meyer WJ., 3rd Beta-adrenergic receptor binding characteristics and responsiveness in cultured Wistar-Kyoto rat arterial smooth muscle cells. Life Sci. 1988;43:721–9. doi: 10.1016/0024-3205(88)90144-0. [DOI] [PubMed] [Google Scholar]
- 57.Iaccarino G, Cipolletta E, Fiorillo A, et al. Beta(2)-adrenergic receptor gene delivery to the endothelium corrects impaired adrenergic vasorelaxation in hypertension. Circulation. 2002;106:349–55. doi: 10.1161/01.cir.0000022690.55143.56. [DOI] [PubMed] [Google Scholar]
- 58.Isenovic E, Walsh MF, Muniyappa R, Bard M, Diglio CA, Sowers JR. Phosphatidylinositol 3-kinase may mediate isoproterenol-induced vascular relaxation in part through nitric oxide production. Metabolism. 2002;51:380–6. doi: 10.1053/meta.2002.30525. [DOI] [PubMed] [Google Scholar]
- 59.Schneider JC, El Kebir D, Chereau C, et al. Involvement of Ca2+/calmodulin-dependent protein kinase II in endothelial NO production and endothelium-dependent relaxation. Am J Physiol Heart Circ Physiol. 2003;284:H2311–9. doi: 10.1152/ajpheart.00932.2001. [DOI] [PubMed] [Google Scholar]
- 60.Bredt DS, Ferris CD, Snyder SH. Nitric oxide synthase regulatory sites. Phosphorylation by cyclic AMP-dependent protein kinase, protein kinase C, and calcium/calmodulin protein kinase; identification of flavin and calmodulin binding sites. J BiolChem. 1992;267:10976–81. [PubMed] [Google Scholar]
- 61.Franke TF, Yang SI, Chan TO, et al. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–36. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 62.Fulton D, Gratton JP, McCabe TJ, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luo Z, Fujio Y, Kureishi Y, et al. Acute modulation of endothelial Akt/PKB activity alters nitric oxide-dependent vasomotor activity in vivo. J Clin Invest. 2000;106:493–9. doi: 10.1172/JCI9419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- 65.Ciccarelli M, Cipolletta E, Santulli G, et al. Endothelial beta2 adrenergic signaling to AKT: role of Gi and SRC. Cell Signal. 2007;19:1949–55. doi: 10.1016/j.cellsig.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 66.Iaccarino G, Ciccarelli M, Sorriento D, et al. Ischemic neoangiogenesis enhanced by beta2-adrenergic receptor overexpression: a novel role for the endothelial adrenergic system. Circ Res. 2005;97:1182–9. doi: 10.1161/01.RES.0000191541.06788.bb. [DOI] [PubMed] [Google Scholar]
- 67.Emanueli C, Salis MB, Stacca T, et al. Rescue of impaired angiogenesis in spontaneously hypertensive rats by intramuscular human tissue kallikrein gene transfer. Hypertension. 2001;38:136–41. doi: 10.1161/01.hyp.38.1.136. [DOI] [PubMed] [Google Scholar]
- 68.Eckhart AD, Yang N, Xin X, Faber JE. Characterization of the alpha1B-adrenergic receptor gene promoter region and hypoxia regulatory elements in vascular smooth muscle. Proc NatlAcadSci U S A. 1997;94:9487–92. doi: 10.1073/pnas.94.17.9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Michelotti GA, Bauman MJ, Smith MP, Schwinn DA. Cloning and characterization of the rat alpha 1a-adrenergic receptor gene promoter. Demonstration of cell specificity and regulation by hypoxia. J BiolChem. 2003;278:8693–705. doi: 10.1074/jbc.M211986200. [DOI] [PubMed] [Google Scholar]
- 70.Trochu JN, Leblais V, Rautureau Y, et al. Beta 3-adrenoceptor stimulation induces vasorelaxation mediated essentially by endothelium-derived nitric oxide in rat thoracic aorta. Br J Pharmacol. 1999;128:69–76. doi: 10.1038/sj.bjp.0702797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rautureau Y, Toumaniantz G, Serpillon S, Jourdon P, Trochu JN, Gauthier C. Beta 3-adrenoceptor in rat aorta: molecular and biochemical characterization and signalling pathway. Br J Pharmacol. 2002;137:153–61. doi: 10.1038/sj.bjp.0704867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bockman CS, Jeffries WB, Pettinger WA, Abel PW. Enhanced release of endothelium-derived relaxing factor in mineralocorticoid hypertension. Hypertension. 1992;20:304–13. doi: 10.1161/01.hyp.20.3.304. [DOI] [PubMed] [Google Scholar]
- 73.Choi DJ, Rockman HA. Beta-adrenergic receptor desensitization in cardiac hypertrophy and heart failure. Cell BiochemBiophys. 1999;31:321–9. doi: 10.1007/BF02738246. [DOI] [PubMed] [Google Scholar]
- 74.Ungerer M, Parruti G, Bohm M, et al. Expression of beta-arrestins and beta-adrenergic receptor kinases in the failing human heart. Circ Res. 1994;74:206–13. doi: 10.1161/01.res.74.2.206. [DOI] [PubMed] [Google Scholar]
- 75.Gros R, Benovic JL, Tan CM, Feldman RD. G-protein-coupled receptor kinase activity is increased in hypertension. J Clin Invest. 1997;99:2087–93. doi: 10.1172/JCI119381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gros R, Chorazyczewski J, Meek MD, Benovic JL, Ferguson SS, Feldman RD. G-Protein-coupled receptor kinase activity in hypertension : increased vascular and lymphocyte G-protein receptor kinase-2 protein expression. Hypertension. 2000;35:38–42. doi: 10.1161/01.hyp.35.1.38. [DOI] [PubMed] [Google Scholar]
- 77.Davies CH, Ferrara N, Harding SE. Beta-adrenoceptor function changes with age of subject in myocytes from non-failing human ventricle. Cardiovasc Res. 1996;31:152–6. [PubMed] [Google Scholar]
- 78.Marin J. Age-related changes in vascular responses: a review. Mech AgeingDev. 1995;79:71–114. doi: 10.1016/0047-6374(94)01551-v. [DOI] [PubMed] [Google Scholar]
- 79.Leosco D, Iaccarino G, Cipolletta E, et al. Exercise restores beta-adrenergic vasorelaxation in aged rat carotid arteries. Am J Physiol Heart Circ Physiol. 2003;285:H369–74. doi: 10.1152/ajpheart.00019.2003. [DOI] [PubMed] [Google Scholar]
- 80.Iaccarino G, Ciccarelli M, Sorriento D, et al. AKT participates in endothelial dysfunction in hypertension. Circulation. 2004;109:2587–93. doi: 10.1161/01.CIR.0000129768.35536.FA. [DOI] [PubMed] [Google Scholar]